
Abstract
One clone exhibiting lipolytic activity was selected among 30 positives from a metagenomic library of a microbe consortium specialized in petroleum hydrocarbon degradation. From this clone, a sublibrary was constructed and a metagenome contig was assembled and analyzed using the ORF Finder; thus, it was possible to identify a potential ORF that encodes a lipolytic enzyme, denoted ORF2. This ORF is composed of 1035-bp 345 amino acids and displayed 98 % identity with an alpha/beta hydrolase from Pseudomonas nitroreducens (accession number WP024765380.1). When analyzed against a metagenome database, ORF2 also showed 76 % of sequence identity with a hypothetical protein from a marine metagenome (accession number ECT55726.1). The ProtParam analyses indicated that the recombinant protein ORF2 has a molecular mass approximately 39 kDa, as expected from its amino acid sequence, and based on phylogenetic analysis and molecular modeling, it was possible to suggest that ORF2 is a new member from family V. This enzyme exhibits the catalytic triad and conserved motifs typical from this family, wherein the serine residue is located in the central position of the conserved motif GASMGG. The orf2 gene was cloned in the expression vector pET28a, and the recombinant protein was superexpressed in Escherichia coli BL21(DE3) cells. The lipolytic activity of protein bands presented in a SDS-PAGE gel was confirmed by zymogram analyses, indicating ORF2 activity. These discoveries raise the possibility of employing this protein in biotechnological applications, such as bioremediation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Environmental microorganisms are highly diversified and have countless metabolic activities and products, which can have industrial applications. However, 99 % of these microorganisms cannot be grown under regular laboratory conditions [1]. Thus, their potential remains unexplored and consequently, products resulting from their metabolism are still not investigated [2].
To access the DNA without previous microorganism cultivation, there is the metagenomic approach, in which genomic DNA is extracted directly from soil or any other environment and cloned in vectors, producing metagenomic libraries [1, 3]. These libraries can be used to discover new enzymes and study secondary pathways through the genome of complex microbial communities from natural habitats [4]. This approach and the use of molecular genetics in a noncultivated microbe consortium have an important impact in the search for new industrial enzymes because of the high diversity of genetic material analyzed [5, 6].
The metagenomic approach has been successfully used to find a large variety of new catalysts and secondary metabolites, such as lipases and esterases [7–11], antimicrobials [12], cellulases [13], and xylanases [14]. Certain aspects of lipases and esterases place them among the most industrially used biocatalysts: normally, cofactors are not required; they are stable in organic solvents and have large substrate specificity
The industrial applications of microbial lipolytic enzymes include detergents [15], pharmaceutical products [16–18], biopolymers [16], biodiesel [16, 19], agrochemicals [16], and flavors [16]. Considering the uncountable benefits and industrial applications of lipolytic enzymes, a metagenomic library in fosmid vector was produced at the Biochemistry Laboratory of Microorganisms and Plants (LBMP), São Paulo State University (UNESP), campus of Jaboticabal, São Paulo State, Brazil. The material used to construct this library was extracted from a microbe consortium specialized in petroleum hydrocarbon degradation [20], obtained from soil contaminated for approximately 15 years by chemical residues of an old lubricant industry located in Ribeirão Preto, São Paulo State, Brazil. The library contains 4224 clones and has been used for investigating different genes with biotechnological importance, such as genes encoding lipolytic enzymes. This work was aimed at the search for expression of an esterase/lipase enzyme for later use in biotechnological process.
Materials and Methods
Identification of Clones with Lipolytic Activity
In a previous work (to be published), 4224 clones from the metagenomic library of a microbe consortium specialized in petroleum hydrocarbon degradation were submitted to biochemical assay in Luria-Bertani (LB) media supplemented with 1 % (v/v) tributyrin (Sigma-Aldrich, Saint Louis, MO, USA), 1 % (p/v) arabic gum, and 12.5 μg/mL chloramphenicol. The substrate was added and emulsified [21] with a Branson Sonifier 250 sonicator (Branson, CT, USA), Duty Cycle of 30 % in three 10-s cycles. The clones were incubated and observed for 3 days at 37 °C and 7 days at 4 °C to verify the halo formation, which is characteristic of substrate hydrolysis [22]. Thirty positive clones were found, and in this work, one of them was selected to be subcloned.
Subcloning and Sequence Analysis
The extraction of fosmidial DNA from the selected clone was performed using the kit Wizard® Plus SV Minipreps DNA Purification System (Promega, Madison, WI, USA), following the manufacturer’s instructions. The subcloning was carried out by shotgun technique, and the DNA was cloned into apUC19 vector. The subclones were submitted to the previously described biochemical assay.
The selected positive subclones were sequenced in ABI Prism 3100 (Applied Biosystems, Carlsbad, CA, USA) using the primers M13 forward and reverse, and the BigDye Terminator cycle sequence kit (Applied Biosystems). Sequencing analyses was done using the software Phred/Phrap/Consed [23, 24], and only sequences containing more than 350 bases with quality Phred ≥20 by contGEN were selected. Sequences were prepared, and the resulting contig was analyzed. The open read frames (ORFs) were identified using the program ORF Finder [25] from the National Center for Biotechnology Information (NCBI), in which the amino acid sequences are compared with the NCBI data bank through the BLASTX algorithm [26].
The ProtParam tool was used to calculate the theoretical parameters of the protein (web.expasy.org/protparam) [27]. To proceed with the phylogenetic analysis, sequences of esterases and lipases were extracted from NCBI, cautiously selected to represent the eight bacterial lipolytic families proposed by Arpigny and Jaeger in 1999 [28], which have been used to date. In addition to the phylogenetic tree, two sequences of lipolytic enzymes were added with their three-dimensional structure solved. These sequences were selected from NCBI, using the BLASTP tool versus the Protein Data Bank (PDB). The amino acid sequences were aligned using the ClustalW program present in the BioEdit Sequence Alignment Editor v.7 [29]. The phylogenetic tree was constructed using Mega 4.1 [30] through the neighbor-joining algorithm [31], with a bootstrap of 1000.
Cloning of orf2 Gene into an Expression Vector
To overexpress the ORF2 protein, primers were designed that allow a 6His-Tag insertion at the N-terminal region with sites for the restriction enzymes NdeI and XhoI, respectively, underlined [forward, 5′-GCGTTCCGGATTTTCACTCCATATGCGTG-3′, and reverse, 5′-GATTCCGCAGCTCGAGGATCAGGACCG-3′]. The orf2 gene was amplified by polymerase chain reaction (PCR) using the enzyme Pfu DNA Polymerase (Fermentas, Burlington, Ontario, Canada). The PCR fragment was cloned into pET28a expression vector (Novagen, Gibbstown, NJ, USA), generating the construction pET28a-orf2. This construction was confirmed by sequencing with specific vector primers; thereby, the plasmids were transformed in chemically competent E. coli BL21(D3) cells.
Overexpression, Extraction, and Purification of ORF2
E. coli-transformed cells were cultivated in 2 L of LB medium at 37 °C until DO600 nm of 0.4 to 0.6 and induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The induced culture was incubated at 22 °C, 200 rpm, for 20 h.
The culture medium was centrifuged at 11,953 × g for 20 min at 4 °C. The cellular pellet was resuspended in 10 mL of lysis buffer per liter of culture [50 mM Tris–HCl pH 8.0, 100 mM of NaCl, 10 % (v/v) glycerol] containing 10 mg/mL of lysozyme. After 30 min in the ice bath, the samples were submitted to lysis in a Branson Sonifier 250 sonicator with 30 % of the cyclic ratio for up to 3 min with 20-s intervals. Samples were centrifuged for 20 min at 4 °C and 38,724 × g to separate the soluble and insoluble extracts. To obtain more soluble protein, the insoluble extract was resuspended in buffer B (buffer A containing 2 % Triton X-100), 5 mL/L of culture, followed by a new lysis cycle under the same conditions previously used.
The soluble extracts were pooled and purified using metal affinity chromatography with resin loaded with nickel, Ni-NTA Agarose (Qiagen, Hilden, Germany), allowing the purification of the recombinant His-tagged protein. The polypropylene column (Bio-Rad, Hercules, CA, USA) was washed with buffer A in an imidazole gradient from 10 mM to 1 M concentration. All samples were analyzed in 12 % polyacrylamide gel SDS-PAGE [32].
Zymogram Analyses
To verify which band was in fact active, a zymogram was carried out. Lipolytic activity in the protein bands of the SDS-PAGE gel was detected using tributyrin as substrate, according to the protocol described in the literature [33], but with some modifications: the sample applied in the gel was not previously heated; SDS was removed after the electrophoresis by soft stirring the gel for 10 min in 50 mM Tris–HCl pH 8.0 buffer containing 1 % Triton X-100 (v/v) and twice in the same buffer without detergent. The gel was then placed in an emulsion with 1.3 % agar (w/v), 0.5 % arabic gum (w/v), 25 mM Tris–HCl pH 8.0, and 1 % tributyrin (v/v) for the detection of lipolytic activity and incubated at 30 °C until the appearance of the hydrolysis halo [34].
In Silico Analysis of the ORF2 Protein
The molecular modeling of the ORF2 protein was performed using the structure coordinates of an aclacinomycin methylesterase from Streptomyces purpurascens (PDB code 1Q0R). The comparative modeling was done by spatial restrictions satisfaction using the Modeller program [35], and the final model was selected considering the total stereochemistry quality. All figures and final analysis were developed using the Pymol software [36].
Results and Discussion
In Vitro Analysis of Lipolytic Activity in Clones from the Metagenomic Library
Among the 30 positive previously identified clones (to be published), the clone Pl40.B09 was selected to be subcloned. The sublibrary (approximately 480 subclones) was submitted to tributyrin assay in Petri dishes, and one positive subclone was detected, PL01.A03 (Fig. 1).

In vitro analysis of the Pl40.B09 sublibrary in LB culture medium, supplemented with 1 % tributyrin (v/v), 1 % arabic gum (p/v), 0.001 % arabinose (v/v), and 70 μg/mL ampicillin (v/v). The subclone Pl01.A03 presented halo formation and is indicated with a black arrow
Sequence Analysis
After identification of the subclone showing lipolytic activity, the DNA sequence of this subclone was used as a guide to identify the lipolytic gene on the contig.
The sequences were analyzed, and a contig was assembled. The final contig had 9914 bp and shared 93 % of the nucleotide sequence identity and 95 % query coverage with Pseudomonas denitrificans (accession number CP004143.1). Using the ORF Finder, it was possible to identify 50 ORFs but 14 were selected taking into account the Max score, Query coverage, and E-value. This analysis allowed us to determine for each ORF: the location of the start and stop codon, the nucleotide and amino acid content, and to predict their biological function (Table 1). All selected ORFs were represented in the physical map illustrated in Fig. 2, wherein the ORF2 is detached in black.

Physical map of PL40.B09 partial contig created by Pages’09 program, version 4.04 (Apple Inc., Cupertino, CA, USA). ORF2 is illustrated as a black arrow and is responsible for the lipolytic activity of the clone
The ORF2 amino acid sequence was compared to sequences deposited on NCBI using the BLASTP tool. The highest identity was achieved with an alpha/beta hydrolase from a Pseudomonas nitroreducens (accession number WP024765380.1, 98 % of sequence identity), followed by an alpha/beta hydrolase (accession number WP026079259.1, 95 % of sequence identity) and a hydrolase (accession number WP015476159.1, 94 % of sequence identity) from P. nitroreducens and P. denitrificans, respectively (Table 2).
Although the high amino acid identity with an alpha/beta hydrolase deposited on NCBI, its functional characterization has not been performed yet. A justification for this fact is that despite the interesting discoveries of new lipolytic enzymes through metagenomics, most of them were still not characterized. According to some authors, since the first bioprospection assay in 2000 until 2009, 76 positive clones of esterases or lipases were identified; however, only 11 were superexpressed and purified, aiming for their biochemical characterization [37]. New enzymes are discovered through selection tests, but few are completely characterized because of the necessary effort, which restrains the potential application of these biocatalysts in biotechnological industries [37]. Here, the preliminary features of ORF2 are presented; however, we expect to perform the enzymatic characterization as soon as possible.
In our phylogenetic analysis, it was possible to conclude that ORF2 is a new member of family V of bacterial lipolytic enzymes (Fig. 3a) because the protein was situated in a branch close to the heat-adapted and cold-adapted microorganisms, Sulfolobus acidocaldarius (accession number AF071233) and Psychrobacter immobilis (accession number X67712), respectively. Interestingly, ORF2 showed to be closely related to the aclacinomycin methylesterase from S. purpurascens (PDB code 1Q0R), as the three-dimensional structure of this enzyme was selected for molecular modeling.

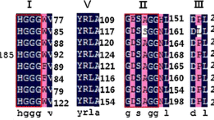
a Dendrogram of the hierarchical grouping based in sequences of lipolytic family members obtained from NCBI database (access numbers indicated), showing the phylogenetic relationship of protein ORF2 (•), found in the partial contig of clone PL40.B09. Bootstrap analysis of 1000 replications was used, and values less than 50 were not shown. b Multiple alignment of amino acid sequences of ORF2 with a representative member from family V (a lipolytic enzyme from Sulfolobus acidocaldarius, accession number AF071233.1) and an aclacinomycin methylesterase from Streptomyces purpurascens (PDB code 1Q0R). Shaded areas represent the conserved motifs of lipolytic enzymes, and the asterisk symbol represents the amino acid residues of the catalytic triad (\( \nabla \))
The representative members of family V display analogous structures to dehalogenases, haloperoxidases, and epoxide hydrolases, presenting an alpha/beta hydrolase fold as the tertiary structure. The conserved blocks GXSXG, PTLV, and GH are typical on members of this family, wherein it is possible to find the catalytic sites in the conserved blocks [28]. To identify these conserved blocks, a sequence alignment was carried out using a member from this family, a lipolytic enzyme from Sulfolobus acidocaldarius (accession number AAC67392). Additionally, it was added the sequence of aclacinomycin methylesterase from S. purpurascens (PDB code 1Q0R).
Through this analysis, it was possible to identify the amino acids that constitute the active site of ORF2 protein: Ser147, Asp273, and His299, through well-established site-directed mutagenesis assays [38, 39]. The serine residue is situated in the central position of the pentapeptide GASMGG, whereas the Asp273 is located after the PTLV motif. Curiously, the histine residue could not be well aligned with the representative sequences, suggesting interesting particularities of ORF2.
Expression and Purification Assays
Initial analysis of the characteristics of ORF2 was performed on ProtParam to determine the molecular weight and isoelectric point (pI). The obtained parameters are shown in Table 3. The recombinant ORF2 protein was overexpressed in different conditions, wherein the temperature, IPTG concentration, and period of induction were varied. ORF2 was obtained as a soluble protein when expressed for 20 h using 0.1 mM IPTG at 22 °C. The low IPTG concentration during induction was used to avoid the formation of inclusion bodies [10].
The molecular mass of the recombinant ORF2 was approximately 39 kDa, as expected from its amino acid sequence and Protparam analysis (Fig. 4a, lane 3). It was possible to obtain soluble protein after the first lysis cycle of protein extraction (Fig. 4a, lane 4); however, a second lysis cycle (Fig. 4a, lane 5) was performed to obtain a significant yield. The materials from both extractions were pooled, and the ORF2 protein was purified by immobilized metal affinity chromatography, being eluted mainly with the elution buffer containing 20 mM imidazole (Fig. 4b, lane 3). After the purification, the pure protein was recovered and shown to be a stable and soluble protein even after prolonged storage at 4 °C, which allowed for the zymogram analysis.

a Expression analysis and solubility of ORF2 demonstrated in SDS-PAGE 10 %: lane 1, molecular weight marker Precision Plus Protein Unstained (Bio-Rad); lane 2, total noninduced cell extract (T0); lane 3, total induced cell extract expressing the protein ORF2 (Tf); lanes 4 and 5, soluble cell extracts; and lane 6, insoluble cell extracts (pellet). b Purification steps demonstrated in SDS-PAGE 10 %: lane 1, molecular weight marker Precision Plus Protein Unstained (Bio-Rad); lane 2, washed fraction regarding to the no-linked column proteins (FT); and lanes 3–8, elution gradient with imidazole (extraction buffer containing 20, 50, 100, 200, and 500 mM and 1 M imidazole, respectively)
Zymogram
After purification, the gel was submitted to a zymogram, a fast and efficient method to test protein activity [33, 34], allowing for the determination of its capacity to hydrolyze the substrate. Here, the zymogram was used to confirm which band in the gel represented the active ORF2 protein. The appearance of a hydrolysis halo revealed that the enzyme was found in its active form (Fig. 5, lanes 5 and 6) and showed its ability to hydrolyze tributyrin as the substrate.

Zymogram: lane 1, molecular weight marker Precision Plus Protein Standards Dual Color (Bio-Rad); lane 2, first soluble extract; lane 3, second soluble extract; lane 4, molecular weight marker Precision Plus Protein Standards Dual Color (Bio-Rad); lane 5, halo formed by the first soluble extract in tributyrin; and lane 6, halo formed by the second soluble extract in tributyrin
Analyses In Silico of ORF2
As mentioned before, the structural coordinates of an aclacinomycin methylesterase from S. purpurascens (PDB code 1Q0R) [40] were used to build the three-dimensional model of ORF2 because this enzyme was the best score obtained through BLASTP tool versus PDB (32 % of sequence identity and 80 % of query coverage with ORF2 amino acid sequence). Interestingly, despite the high identity of ORF2 when compared to the deposited sequences on NCBI, this profile was not observed on PDB analysis, wherein the highest identity [40] with ORF2 was used to build the three-dimensional model.
The final model proposed for the ORF2 protein shows the ellipsoid arrangement formed by alpha-helix and beta-sheet, typical of the alpha/beta hydrolase fold family of which esterases and lipases are members (Fig. 6a). The ORF2 has a mixture of eight beta-sheets in which only the second is antiparallel, surrounded by 12 alpha-helices: Leu83-His85, Asp88-Gln97, Pro135-Ser137, Leu142-Leu156, Met168-Arg183, Gln264-Ile268, Val275-Arg280, Met299-His307, and Gli325-Thr342.

Three-dimensional model of ORF2 protein. a A cartoon representation of the ORF2 model with the alpha-helices (gray), beta-sheet (orange), and loops (purple). The residues of the catalytic triad (Ser147, Asp273, and His299) are shown in green color (in the detail), and the cap domain is in black color. b Localization of the conserved motifs (pink) from family V of bacterial lipolytic enzymes. c Structure model is shown as a surface, and the red arrow indicates the tunnel entrance, which give access to the catalytic triad. d Details of the large tunnel and the catalytic triad (green) localization (color figure online)
Apparently, the ORF2 protein presents a small cap domain composed by three alpha-helices: Glu204-Arg212, Glu218-Gli233, and Arg241-Ala255 (in black color, Fig. 6a–d). The cap domain is located in a region superior to the catalytic triad, which is also observed in other esterases, and it seems to control the substrate entrance and their recognition, keeping the structural integrity around the active site [41].
The enzymatic activity of almost all alpha/beta hydrolase family members depends on a highly preserved catalytic triad, which consists of serine, aspartate or glutamate, and histidine. In the ORF2, it was possible to identify this catalytic triad, Ser147, Asp273, and His299 (sticks in green color, Fig. 6a, b, d), besides the motifs involved in catalysis, 145GASMGGM151, 273DPLLPV278, and 299HG300 (pink color, Fig. 6b). The catalytic triad residues and the characteristics of the conserved motifs corroborate that ORF2 protein is a new member of family V.
Through the analysis of the ORF2 protein model, it was possible to identify the substrate access entrance (Fig. 6c); notwithstanding the low identity (32 %) with the three-dimensional structure used to build the ORF2 model, it is possible to note the large tunnel that almost crosses the protein (Fig. 6d). In view of this result, we speculate that ORF2 will be able to hydrolyze long carbon chains, as well.
The preliminary in silico study of ORF2 provided important details about this protein, such as the location of the catalytic triad, substrate-binding pocket, and the existence of a tunnel toward the active site. Our results indicate that the ORF2 presents great biotechnological potential; thus, we expect in the near future to perform the functional characterization of this enzyme to propose an application for ORF2.
Conclusions
This research indicated that the metagenomic approach is an efficient way to discover enzymes for biotechnological use. Presented here is the in vitro and in silico analyses of ORF2, a new protein of family V of bacterial lipolytic enzymes. This protein was overexpressed and shown to be soluble, stable, and active when tested in a zymogram assay using tributyrin as the substrate. Through sequence analysis and protein modeling, it was possible to identify the catalytic triad, conserved motifs from family V, and an interesting tunnel that gives access to the active site. Our results indicate that ORF2 is a metagenome-derived protein with a high potential for biotechnological applications. Finally, we expect to achieve newsworthy results with the functional characterization of this enzyme.
References
Handelsman, J., Rondon, M. R., Brady, S. F., Clardy, J., & Goodman, R. M. (1998). Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chemistry & Biology, 5, 245–249.
SINGH, B. K. (2009). Exploring microbial diversity for biotechnology: the way forward. Trends Biotechnology Journal, v. xxx, n. x, p. 1-6.
Knight, J. C., Keating, B. J., Rockett, K. A., & Kwiatkowski, D. P. (2003). In vivo characterization of regulatory polymorphisms by allele-specific quantification of RNA polymerase loading. Nature Genetics, 33, 469–475.
Liaw, R. B., Cheng, M. P., Wu, M. C., & Lee, C. Y. (2010). Use of metagenomic approaches to isolate lipolytic genes from activated sludge. Bioresource Technology, 101, 8323–8329.
Lorenz, P., & Eck, J. (2005). Metagenomics and industrial applications. Nature, 3, 510–516.
Jiang, C., Wu, L.-L., Zhao, G.-C., Shen, P.-H., Jin, K., Hao, Z.-Y., Li, S.-X., Ma, G.-F., Luo, F.-F., Hu, G.-Q., Kang, W.-L., Qin, X.-M., Bi, Y.-L., Tang, X.-L., & Wu, B. (2010). Identification and characterization of a novel fumarase gene by metagenome expression cloning from marine microorganisms. Microbial Cell Factories, 9, 91.
Henne, A., Schmitz, R. A., Bomeke, M., Gottschalk, G., & Daniel, R. (2000). Screening of environmental DNA libraries for the presence of genes conferring lipolytic activity on Escherichia coli. Applied Environmental Microbiology, 66, 3113–3116.
Cieslinski, H., et al. (2009). Identification and molecular modeling of a novel lipase from an Antarctic soil metagenomic library. Polish journal of microbiology, 58(3), 199–204.
Jeon, J. H., Kim, J. T., Kang, S. G., Lee, J. H., & Kim, S. J. (2009). Characterization and its potential application of two esterases derived from the arctic sediment metagenome. Journal of Biotechnology, 11, 307–316.
Wu, C., & Sun, B. (2009). Identification of novel esterase from metagenomic library of Yangtze River. Journal of Microbiology and Biotechnology, 19(2), 187–193.
Hu, Y., Fu, C., Huang, Y., Yin, Y., Cheng, G., Lei, F., Lu, N., Li, J., Ashforth, E. J., Zhang, L., & Zhu, B. (2010). Novel lipolytic genes from the microbial metagenomic library of the South China Sea marine sediment. FEMS Microbiology Ecology, 72, 228–237.
Gillespie, D. E., Brady, S. F., Bettermann, A. D., Cianciotto, N. P., Liles, M. R., Rondon, M. R., Clardy, J., Goodman, R. M., & Handelsman, J. (2002). Isolation of antibiotics turbomycin A and B from a metagenomic library of soil microbial DNA. Applied and Environmental Microbiology, 68(9), 4301–4306.
Kim, E. Y., Oh, K. H., Lee, M. H., Kang, C. H., Oh, T. K., & Yoon, J. H. (2009). Novel cold-adapted alkaline lipase from an intertidal flat metagenome and proposal for a new family of bacterial lipases. Applied and Environmental Microbiology, 75(1), 257–260.
Hu, Y., Zhang, G., Li, A., Chen, J., & Ma, L. (2008). Cloning and enzymatic characterization of a xylanase gene from soil derived metagenomic library with an efficient approach. Applied Microbiology Biotechnology, 80, 823–830.
Jaeger, K. E., & Eggert, T. (2002). Lipases for biotechnology. Current Opinion in Biotechnology, 13, 390–397.
Cheetham, P. S. J. (1995). Principles of industrial biocatalysis and bioprocessing. Handbook of enzyme biotechnology. p., 83–234.
Sharma, R., Chisti, Y., & Banerjee, U. C. (2001). Production, purification, characterization and applications of lipases. Biotechnology Advances, 19, 627.
Priya, K., & Chadha, A. (2003). Synthesis of hydrocinnamic esters by Pseudomonas cepacia lipase. Enzyme and Microbial Technology., 32, 485.
Gao, B., Su, E., Lin, J., Jiang, Z., Ma, Y., & Wei, D. (2009). Development of recombinant Escherichia coli whole-cell biocatalyst expressing a novel alkaline lipase-coding gene from Proteus sp. for biodiesel production. Journal of Biotechnology, 139, 169–175.
Paixão, D. A., Dimitrov, M. R., Pereira, R. M., Accorsini, F. R., Vidotti, M. B., & Lemos, E. G. M. (2010). Análise molecular da diversidade bacteriana de um consórcio degradador de óleo diesel. Revista Brasileira de Ciência do Solo, 34, 773–781.
Lee, S. W., Won, K., Lim, H. K., Kim, J. C., Choi, G. J., & Cho, K. Y. (2004). Screening for novel lipolytic enzymes from uncultured soil microorganisms. Applied Microbiology and Biotechnology Journal, 6, 720–726.
Pereira, M. R. (2011). Prospecção de genes codificadores de enzimas lipolíticas em biblioteca metagenômica de consórcio microbiano degradador de óleo diesel. 102 f. Dissertação (Mestrado em Biotecnologia) – Instituto de Ciências Biomédicas, Universidade de São Paulo- São Paulo.
Ewing, B., & Green, P. (1998). Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Research, 8, 186–194.
Ewing, B., Hillier, L., Wendl, M. C., & Green, P. (1998). Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Research, 8, 175–185.
Wheeler D, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, et al. (2003). Database resources of the National Center for Biotechnology. Nucleic Acids Research. n.31, 28–33.
Astschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., Lipman, D. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, n. 25, p. 3389-3402.
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S. E., Wilkins, M. R., Appel, R. D., Bairoch, A. (2005). Protein identification and analysis tools on the ExPASyServer. In: Walker, J. M. (Ed.). The Proteomics Protocols Handbook: Humana Press. p. 571-607.
Arpigny, J. L., & Jaeger, K. E. (1999). Bacterial lipolytic enzymes: classification and properties. The Biochemical journal, 343(Pt 1), 177–183.
Hall, T.A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series n.41, 95-98.
Tamura, K., Dudley, J., Nei, M., & Kumar, S. (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution, 24, 1596–1599.
Saitou, N., & Nei, M. (1987). The neighbor-joining method: a new method for constructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227(5259), 680–685.
Oh, B., Kim, H., Lee, J., Kang, S., & Oh, T. (1999). Staphylococcus haemolyticus lipase: biochemical properties, substrate specificity and gene cloning. FEMS Microbiology Letters, 179(2), 385–392.
Glogauer, A., Martini, V. P., Faoro, H., Couto, G., Santos, M. M., Monteiro, R. A., Mitchell, D. A., Souza, E. M., Pedrosa, F. O., & Krieger, N. (2011). Identification and characterization of a new true lipase isolated through metagenomic approach. Microbial Cell Factories, 10, 54.
Sali, A.; Blundell, T.L. (1993). Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology, n. 234, 779-815.
DeLano, W.L. (2010). The Pymol molecular graphics system, Version 1.2r3pre, Schrödinger, LLC, New York.
Steele, H. L., Jaeger, K. E., Daniel, R., & Streit, W. R. (2009). Advances in recovery of novel biocatalysts from metagenomes. J Mol Microbiol Biotechnol, 16, 25–37.
Elend, C., Schmisser, C., Leggewie, C., Babiak, P., Carballeira, J. D., Steele, H. L., & Reymond, J. (2006). –L.; Jaeger, K. –E.; Streit, W. R. Isolation and biochemical characterization of two novel metagenome-derived esterases. Applied and Environmental Microbiology, 72(5), 3637–3645.
Rao, L., et al. (2013). A novel alkaliphilic bacillus esterase belongs to the 13(th) bacterial lipolytic enzyme family. PloS one, 8(4), e60645.
Jansson, A., Niemi, J., Mäntsälä, P., & Schneider, G. (2003). Crystal structure of aclacinomycin methylesterase with bound product analogues: implications for anthracycline recognition and mechanism. The Journal of Biological Chemistry. v., 278, 39006–39013.
Nardini, M., & Dijkstra, B. W. (1999). α/β Hydrolase fold enzymes : the family keeps growing. Current Opinion in Structural Biology, 9, 732–737.
Acknowledgments
This work was well conducted at the Biochemistry Laboratory of Microorganisms and Plants (LBMP), campus of Jaboticabal, São Paulo State University. We also thank the Agricultural Microbiology postgraduate program, the São Paulo Research Foundation (FAPESP) for financial support, and the Coordination for the Development of Higher Education Personnel (CAPES) for a research scholarship. We thank João Carlos Campanharo for technical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Garcia, R.A.M., Pereira, M.R., Maester, T.C. et al. Investigation, Expression, and Molecular Modeling of ORF2, a Metagenomic Lipolytic Enzyme. Appl Biochem Biotechnol 175, 3875–3887 (2015). https://doi.org/10.1007/s12010-015-1556-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-015-1556-8