
Abstract
A comprehensive understanding of genotype-phenotype links in bacteria is the primary theme of bacterial functional genomics. Transposon sequencing (Tn-seq) or its equivalent methods that combine random transposon mutagenesis and next-generation sequencing (NGS) represent a powerful approach to understand gene functions in bacteria on a genome-wide scale. This approach has been utilized in a variety of bacterial species to provide comprehensive information on gene functions related to various phenotypes or biological processes of significance. With further improvements in the molecular protocol for specific amplification of transposon junction sequences and increasing capacity of next generation sequencing technologies, the applications of Tn-seq have been expanding to tackle questions that are important yet difficult to address in the past. In this review, we will discuss the technical aspects of different Tn-seq methods along with their pros and cons to provide a helpful guidance for those who want to implement or improve Tn-seq for their own research projects. In addition, we also provide a comprehensive summary of recent published studies based on Tn-seq methods to give an updated perspective on the current and emerging applications of Tn-seq.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the major goals in bacterial genetics is to understand the genetic mechanisms underlying the phenotypes of interest. Among various approaches to reveal the underlying genetic mechanism(s) of a phenotype, the most common initial step is to identify the genetic factors involved in or responsible for expression of the phenotype. Traditionally, the gene discovery process has been a rate-limiting step that slows down the entire process of understanding the mechanism. Transposon mutagenesis has been one of the major tools that have contributed significantly to gene discovery in bacteria mainly through loss-of-function screening. However, the necessity to assess the phenotype of each mutant individually required considerable amount of labor and time thus limiting the total number of mutants that could be screened. As interest in high-throughput applications has increased, the methods that allow comprehensive screening of a large number of mutants have been developed and progressed significantly over the past two decades to accelerate the screening process, including signature-tagged mutagenesis (STM) (Mazurkiewicz et al. 2006) followed by microarray-based footprinting of transposon mutants (Sassetti and Rubin 2002) and more recently, transposon sequencing (Tn-seq) (Barquist et al. 2013a; van Opijnen and Camilli 2013). The Tn-seq method is the most recent addition to the transposon tool box, aided mainly by the development of high-throughput NGS technologies. Since the first reports on the development of Tn-seq in 2009 (Gawronski et al. 2009; Goodman et al. 2009; Langridge et al. 2009; van Opijnen et al. 2009), various modifications have been made and applied to facilitate gene discovery in diverse bacterial species. With this more comprehensive approach, high-resolution functional screening of the whole genome can be performed routinely in a small laboratory for various bacterial species, providing remarkably rich information on gene functions for almost every single gene, including both protein-coding genes and noncoding genes, involved in a wide range of biological processes. In this review, we will discuss the recent development in Tn-seq methods and its expanding applications from a rather straightforward fitness profiling, in vitro or in vivo, to implementation of novel experimental designs for discovery of bacterial factors involved in more specific biological processes.
Transposon mutagenesis
Transposons are genetic elements that can move from one genomic location to another. This “mobile nature” of transposons has been harnessed by microbial geneticists for convenient use of transposons as powerful tools for random mutagenesis in bacteria (Hayes 2003). For Tn-seq analysis, Tn5 and mariner transposons have been used most frequently among others due to their simple procedures, broad-host range, and well-characterized near-random nature of those particular transposons (Barquist et al. 2013a). There are various ways to deliver the transposon of choice into the cells for transposon mutagenesis (Maloy 2007). These include the methods based on phage delivery systems (Santiago et al. 2015; Sassetti et al. 2001), plasmid delivery systems (de Lorenzo and Timmis 1994; Martínez-García et al. 2011), in vivo mutagenesis by electroporation of transposon-transposase complex (Goryshin et al. 2000), and in vitro mutagenesis using a purified transposase enzyme followed by natural transformation (Hendrixson et al. 2001; Reid et al. 2008). For more details, readers are encouraged to retrieve the corresponding references.
Development of Tn-seq methods
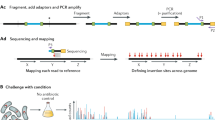
The basic and critical step common to all Tn-seq methods and its variations is to amplify transposon junction sequences in an insertion mutant pool specifically but comprehensively without bias as much as possible (Barquist et al. 2013a; van Opijnen and Camilli 2013). Once transposon junction sequences are amplified, they are sequenced in depth by next-generation sequencing (NGS) to obtain a quantitative profile of all transposon insertions in the library. From the collected DNA sequence data, the DNA sequence of each read is used to precisely locate each transposon insertion in the genome and accordingly, the number of DNA sequence reads originating from the same insertion serve as a measure of relative abundance of the corresponding transposon mutant in the mutant pool. When Tn-seq profiles of a library are quantitatively compared with an appropriate normalization and statistical method between before and after a selection, the genetic factors that are required for optimal growth or survival under the selection process can easily be identified on a genomic scale.
Since the first versions of Tn-seq methods were reported (Gawronski et al. 2009; Goodman et al. 2009; Langridge et al. 2009; van Opijnen et al. 2009), several variations on the method have been described (Christen et al. 2011; Dawoud et al. 2014; Gallagher et al. 2011; Khatiwara et al. 2012; Klein et al. 2012). These variations differ mainly in the manner in which specific amplification of the transposon junction sequences is accomplished. More specifically, these methods employ different strategies to attach the common primer-binding sites to transposon-flanking regions allowing PCR amplification to occur between the binding sites for the transposon-specific primer and the common primer-binding sites on the transposon-flanking regions. The common strategies for amplification of transposon junction sequences used in different Tn-seq methods termed in various names, including INSeq (Goodman et al. 2009), Tn-seq (van Opijnen et al. 2009), TraDIS (Langridge et al. 2009), Tn-seq circle (Gallagher et al. 2011), and HITS (Gawronski et al. 2009) are summarized in (Febrer et al. 2011) (see Fig. 2 in Febrer et al. (2011) for comparative graphical illustration of the different strategies).
Approaches based on C-tailing are the recent technical additions to current Tn-seq methods. The C-tailing procedure uses terminal transferase activity to add poly C tails to 3′ end of either single-stranded or double-stranded DNA. When this reaction is performed in the presence of the mixture of dCTP and dideoxy CTP (ddCTP) at a certain ratio, the average lengths of the C-tail can be efficiently controlled (Lazinski and Camilli 2013). This approach was adopted to attach C-tails to the 3′ ends of randomly sheared gDNA of a transposon insertion library. The C-tails served subsequently as a binding site for poly G primer to amplify transposon junction sequences in conjunction with a transposon-specific primer (Klein et al. 2012). Additional research based on the same Tn-seq method further established the robustness of the method (Carter et al. 2014; Kamp et al. 2013; McDonough et al. 2014; Shan et al. 2015; Valentino et al. 2014). Recently, our lab developed a convenient protocol based on single primer extension of transposon junction sequences using a transposon-specific primer. The single-stranded DNA fragments thus synthesized are subsequently C-tailed using a terminal transferase. The resulting C-tailed transposon junction fragments can thus be easily amplified with transposon-specific primer and poly G primer (Dawoud et al. 2014).
A more recently developed Tn-seq strategy, termed random barcode transposon-site sequencing (RB-TnSeq), is based on incorporating random DNA barcodes into the transposon and utilizing them for fitness profiling in place of transposon junction sequences (Wetmore et al. 2015). This RB-TnSeq method simplifies the steps to prepare the PCR library because the random DNA barcodes located internally inside the transposon can be easily PCR-amplified with two universal primers flanking the barcode region. Consequently, this simplified PCR step increases the throughput of mutant fitness profiling significantly. However, it requires additional steps of random barcode tagging of transposon before construction of a mutant library and initially establishing a database for insertion-barcode pairs (Wetmore et al. 2015).
One issue associated with a transposon mutant library generated using a suicide delivery plasmid is that a significant portion of the mutants could be pseudo-transposon mutants that result from integration of the transposon delivery plasmid into the chromosome. When this type of library is used for Tn-seq analysis, a large number of sequence reads are from transposon junctions in the delivery vector rather than true transposon insertions in chromosome or plasmid of the host cell, resulting in a waste of valuable sequence reads. Santiago et al. (2015) recently described a simple strategy to address this issue by incorporating two recognition sites for a rare-cutting restriction endonuclease (e.g., NotI) on both sides of one inverted repeat (IR) from which the transposon junction sequences are obtained. The genomic DNA from the library are digested with the rare-cutting restriction enzyme, and the resulting small fragments can be efficiently removed by size fractionation before the next step for preparation of the Tn-seq amplicon library.
In Table 1, different Tn-seq methods are grouped according to the strategies used to accomplish amplification of transposon junction sequences accompanied by descriptions of their characteristics.
Comparison of Tn-seq methods
All of the Tn-seq methods that have been described until now have been used successfully to identify genes important for a wide range of biological processes of interest, supporting their utility as a functional genomics tool. However, the sensitivity of the gene discovery can be greatly influenced by the comprehensiveness and quantitative accuracy of the resulting Tn-seq profiles. It is expected that any bias occurring during the preparation of Tn-seq library would negatively influence the accuracy of the resulting Tn-seq profile, leading to false positive or false negative results. There are four theoretical or practical considerations for an ideal Tn-seq method as discussed in detail in the following sections. The potential pros and cons of the currently existing Tn-seq methods based on these criteria are listed in Table 1.
Potential bias in Tn-seq library preparation
The most critical requirement for Tn-seq is minimum bias during Tn-seq library preparation. Theoretically, this type of bias can occur during the preparation of the PCR template or PCR amplification. In the “Tn-seq circle” method, the physically sheared genomic DNA fragments are ligated to an adaptor, digested by restriction enzyme, denatured, and circularized through oligonucleotide-mediated ligation (Gallagher et al. 2011). Therefore, the variable lengths of the fragments can cause bias in the ligation reaction, and the efficiency of ligation itself would be critical in preparing a template library well representative of the transposon mutant pool. However, Gallagher et al. (2015) recently compared the Tn-seq circle method (Gallagher et al. 2011) with the Tn-seq method based on C-tailing (Klein et al. 2012) by analyzing the same genomic DNA from a complex transposon library of Acinetobacter baumannii with the two Tn-seq methods (Gallagher et al. 2015). These two methods provided remarkably similar lists of essential genes, suggesting both methods are robust, and the potential bias, if existed, was insignificant. Bias in the final Tn-seq library could also happen during the PCR amplification step due to variable lengths of the PCR products being amplified. In this aspect, only the methods based on the use of the Type IIS restriction enzymes (restriction enzymes that cleave outside of their recognition sequence to one side) and RB-TnSeq (Goodman et al. 2009; Khatiwara et al. 2012; van Opijnen et al. 2009; Wetmore et al. 2015) can avoid this issue, since all other methods produce PCR products of variable lengths. The Tn-seq method based on nested arbitrary PCR raises concerns for additional bias in PCR amplification due to the nature of primer binding occurring at lower annealing temperatures (Christen et al. 2011). With nested arbitrary PCR, the amplification efficiency could largely be dependent on the nucleotide sequences of the transposon-flanking regions. It is expected that a certain portion of insertions may not allow amplification of transposon junction sequences at all. However, in this particular research, a highly saturating Tn5 library of Caulobacter crescentus was used, focusing only on essential gene discovery (Christen et al. 2011). Since gene essentiality can be assessed only with the information on insertion sites without relying on quantitative information on each insertion mutant in the library (Hutchison et al. 1999), potential bias in Tn-seq library preparation may not have been a major obstacle in essential gene discovery (Christen et al. 2011).
Quantities of genomic DNA
Many Tn-seq methods involve physical shearing during the preparation of the Tn-seq amplicon library. Physical shearing of genomic or metagenomics DNA is a step commonly used to prepare a DNA fragment library for NGS analysis (Knierim et al. 2011). The random nature of physical shearing makes it an attractive choice because it helps to generate a bias-free fragment library. Although effective, it often requires an optimization step, a relatively large quantity of starting DNA materials, and equipment (e.g., sonicator) to perform this step. For the Tn-seq methods that involve physical shearing, the amount of starting DNA (per sample) ranged from 3 to 6 μg (Gallagher et al. 2011; Langridge et al. 2009; Wong et al. 2011). On the contrary, the methods that begin with the PCR to amplify or extend transposon junctions directly from the template DNA require much less amount of starting DNA. In the method based on nested arbitrary PCR (Christen et al. 2011), 1 μl of a bacterial culture (OD 0.1) was directly used as a template, and our lab routinely use 50–100 ng of genomic DNA as a template for the Tn-seq method based on linear PCR followed by C-tailing and PCR (Dawoud et al. 2014; unpublished).
For most Tn-seq applications, collecting a large quantity of bacterial cells representing the entire mutant population is not an issue. However, in certain circumstances where the surviving mutants are recovered from infected host tissues to form a recovered mutant pool, the number of bacterial cell survivors (thus their genomic DNA) can be a limiting factor for performing a physical shearing step, especially when the procedure should be repeated for optimization or due to a mistake. The recovered library can be amplified by bacterial cultivation, but this step may introduce artifacts resulting from differences in mutant in vitro growth rates.
Applicability to any transposon elements
Most Tn-seq methods are universally applicable to a mutant library constructed by any type of transposon elements. However, the Tn-seq methods that utilize Type IIS restriction enzymes (MmeI or BsmFI) require the presence of the restriction sites at the end(s) of the transposon (Goodman et al. 2009; Khatiwara et al. 2012; van Opijnen et al. 2009). This requirement limits this type of Tn-seq methods only to certain transposons. For example, an MmeI site could only be created in the mariner transposon that happened to carry sequence in the inverted repeat region that closely matched the MmeI site except for one nucleotide (Goodman et al. 2009; van Opijnen et al. 2009). Therefore, these methods cannot be applied to any other transposon elements and thus is not applicable to an existing transposon library that is constructed based on wild type mariner transposon or other transposon elements. In the case of RB-TnSeq, the use of barcode regions located within the transposon instead of transposon junction sequences for quantitative profiling of transposon mutants provides multiple advantages (Wetmore et al. 2015). However, it also requires the use of a modified transposon carrying random barcodes within the transposon for library construction.
Precise genome mapping
The length of transposon junction sequence reads should be sufficiently long to allow precise genome mapping of the reads and thus precise determination of the insertion sites. For most Tn-seq methods, the length of transposon junction reads can be adjusted by choosing and purifying an appropriate range of PCR products. The lengths of the Tn-seq amplicons are uniformly fixed to a relatively short length only for the methods based on the use of Type IIS restriction enzymes (Goodman et al. 2009; Khatiwara et al. 2012; van Opijnen et al. 2009). The question then becomes how long should the transposon junction sequences be to serve this purpose? This can be estimated. For example, based on a computer simulation analysis, minimum lengths of 16 bp would be required for unambiguous genome mapping for 98 % of the reads when the genome of Bacteroides thetaiotaomicron was used for the test (Goodman et al. 2009). This fact suggests that the majority of the reads from the Tn-seq method based on MmeI, which produces 16 bp sequence reads, would be sufficient for precise genome mapping (Goodman et al. 2009; van Opijnen et al. 2009). However, Tn-seq method based on the use of BsmFI restriction enzyme suffers from short reads of 11 to 12 bp, for which approximately 50 % of the reads would have to be discarded due to the inability to achieve unambiguous genome mapping (Khatiwara et al. 2012).
Applications of Tn-seq methods
With continuously increasing read numbers for Illumina sequencing (which currently provides approximately 3.0 × 108 reads per lane on HiSeq2500), Tn-seq analysis provides extraordinary opportunities for gene discovery at an accelerated rate to address various biological questions that were impossible to answer in the past before the development of Tn-seq methods. We have highlighted some of the interesting trends in Tn-seq applications in the following sections.
Essential genes
One of the first applications of global transposon mapping data was to discover essential genes of the bacterium Mycoplasma genitalium (Hutchison et al. 1999). An essential gene is defined as the gene that is essential for growth or survival under the optimal growth condition. Therefore, an essential gene set would be expected to change depending on how the optimal condition was initially defined. Conventionally, however, essential genes refer to the genes required for growth or survival of a bacterium in the standard rich media commonly used for routine culture of the bacterial species. When global transposon mapping data became available, essential genes could be identified conceptually by the genomic regions that contain no or very few transposons. By the subtractive nature of the approach for essential gene discovery, the accuracy of prediction would be further enhanced by higher level of genome saturation via transposon insertions. With the Tn-seq method, much higher levels of genome saturation can be accomplished, and therefore, Tn-seq data obtained from various bacterial species under standard growth media have provided high quality data for essential gene discovery. The complete set of essential genes, termed “essential genome” has been defined by Tn-seq data for numerous bacterial species, including Burkholderia pseudomallei (Moule et al. 2014), Campylobacter jejuni (Gao et al. 2014), Pseudomonas aeruginosa (Lee et al. 2015), and Streptococcus pyogenes (Le Breton et al. 2015), and one archaeal species, Methanococcus maripaludis (Sarmiento et al. 2013).
In most studies on essential gene discovery using Tn-seq, viable transposon mutants are usually recovered from a single nutrient-rich condition, and the resulting Tn-seq data is used to identify essential genes. However, in a more recent study, Lee et al. (2015) studied the essential genes in Pseudomonas aeruginosa in six different media and identified 352 general and 199 condition-specific essential genes. This approach allows discernment of “essential genes” specific to different growth conditions from truly essential genes and to define core essential genes that are commonly required for viability under multiple growth conditions.
Conditionally essential genes (in vitro conditions)
Conditionally essential genes should be considered as an extension of the essential genes in the sense that conditionally essential genes are required for growth or survival only under the condition of the interest other than the standard media. Of special interest, for example, would be defining a bacterial gene set conditionally required for growth or survival during specific environmental niches related to the life cycle of the species. For a bacterial pathogen, it would be particularly important to understand which genes are essential to overcome the stressors or immune defenses in the host. By comparing the genetic requirements for growth under different in vitro conditions with the genes required for in vivo growth or survival in the host, the unknown selective pressures that bacterial pathogens encounter in specific host niches can be identified (Khatiwara et al. 2012; Merrell and Camilli 2002; van Opijnen and Camilli 2012). More recently, the potential link between the metabolic capacity of a pathogen and its virulence has been suggested as a critical factor for expression of pathogenic phenotypes (Rohmer et al. 2011). Metabolic genes that enable a pathogen to utilize a nutrient uniquely present in a host niche would play an important role during infection in the host tissues. For example, Griffin et al. (2011) used Tn-seq to define a set of genes in Mycobacterium tuberculosis that are required for in vitro utilization of cholesterol as a sole carbon source. Comparison of the result of this study with previously identified genes in M. tuberculosis required for in vivo survival during mouse infections (Sassetti and Rubin 2003) demonstrated that 10 % of the genes specifically required for bacterial growth in vivo are also required for the utilization of cholesterol in vitro. Until recently, a large portion of Tn-seq studies for gene discovery have focused on screening and characterizing conditionally essential genes under in vitro conditions, largely due to the simplicity of the experimental design, the lack of problems associated with bottlenecks that would occur in animal infection studies, and the resulting in-depth insights that can be gained from the comprehensive sets of genes identified. An extensive list of Tn-seq studies on conditionally essential genes is shown in Table 2.
Genes required for in vivo fitness in the host
Genome-wide identification of bacterial virulence genes required for in vivo fitness during host infection using Tn-seq is an extremely valuable approach in understanding complex mechanisms of virulence. Such applications of Tn-seq for virulence gene discovery using various pathogen-host infection models has been steadily increasing over the years, leading to identification of numerous previously known as well as unknown virulence factors (Table 2). In some animal infection models, however, this approach involving a complex library is not feasible due to the bottlenecks that cause stochastic removal of bacterial cells during establishment of infection (van Opijnen and Camilli 2013). In such cases, multiple transposon libraries of smaller sizes can be used to identify in vivo fitness factors (Chaudhuri et al. 2013). One emerging research area of interest is the comparative analysis of in vivo fitness factors in multiple hosts. Chaudhuri et al. (2013) screened the same collection of Salmonella enterica Typhimurium Tn5 mutants for mutants with reduced gut colonization in three different hosts, chickens, pigs, and calves, and identified a core set of virulence genes as well as host-specific virulence factors. More recently, Weerdenburg et al. (2015) used a similar comparative analysis to identify the factors of a broad-host-range pathogen Mycobacterium marinum that are important for survival in phagocytic cells of five different host species. Finally, Tn-seq method has also been applied to understand the genetic mechanisms associated with Vibrio fischeri’s symbiotic colonization of the light organ of squid (Brooks et al. 2014).
Small RNA genes
Initially, the focus of Tn-seq application was to identify protein-coding genes important for fitness. Although some small RNA (sRNA) (e.g., GlmZ in Escherichia coli) was identified by a phenotypic screening of transposon mutants (Kalamorz et al. 2007), the general applicability of Tn-seq for comprehensive discovery of sRNA genes remained uncertain. However, several Tn-seq studies have demonstrated that Tn-seq can actually be very effective in identifying conditionally essential sRNA genes (Barquist et al. 2013b; Christen et al. 2011; Mann et al. 2012; Zhang et al. 2012). The challenge in analyzing sRNA genes lies simply in the fact that sRNA genes are much smaller than protein-coding genes, thereby reducing the chance to be hit by a transposon, and sRNA knockout mutants usually do not exhibit strong phenotypes (Sharma and Vogel 2009). The utility of Tn-seq in identification of sRNA genes is very significant for high-throughput analysis of sRNA genes in bacteria. Previously, the main approach for sRNA discovery was through detection of sRNA transcripts either by transcriptome analysis (microarray or RNA-seq) or by cloning of reverse transcribed RNA transcripts (RNomics) (Sharma and Vogel 2009). Alternatively, RNA chaperone Hfq protein was used as a bait to capture sRNA transcripts-associated Hfq protein (Sharma and Vogel 2009). However, these approaches seldom reveal critical information regarding biological functions of the sRNAs. To understand sRNA functions, it requires a time-consuming downstream analysis for individual sRNAs (Sharma and Vogel 2009). However, Tn-seq analysis provides the means to identify sRNA genes comprehensively and also valuable insights on the sRNA functions. Two studies using Tn-seq have identified sRNA genes required for growth in rich media for Caulobacter crescentus (Christen et al. 2011) and Mycobacterium tuberculosis (Zhang et al. 2012). More interestingly, Tn-seq analysis has been used in mouse models of colonization to reveal the contribution of the sRNAs in Streptococcus pneumoniae to fitness in vivo (Mann et al. 2012). Collectively, these studies demonstrate the general utility of Tn-seq for global discovery of essential as well as conditionally essential sRNA genes.
Additional genetic elements and features: promoters, operons, and domains
For most Tn-seq analysis, the focus of the research is usually on identification of essential genes or conditionally essential genes, whether it is protein-coding genes or noncoding sRNA genes. Usually, no additional attempts have been made to find more information beyond genetic requirements of the genes. However, Christen et al. (2011) demonstrated that a small change in the design of the transposon itself can provide additional in-depth information beyond genetic requirements. In their study, a Tn5 transposon containing a strong xylose-inducible promoter facing outward was used to construct a genome-saturation library of Caulobacter crescentus. When the mutants were recovered in the presence of the inducer and analyzed for insertions by Tn-seq, the comparative analysis of the insertion groups in two different orientations allowed the identification of the promoter regions of essential genes, the operons with essential functions, and the domains accountable for the essentiality of the corresponding genes (Christen et al. 2011).
Genetic interaction mapping
One powerful approach to tackle functional organization of the genes related to a certain phenotype is to examine genetic interactions among multiple gene products (Dixon et al. 2009). In general, when a double mutant that shows a significant deviation in fitness compared with the expected multiplicative effect of combining two single mutants, it is considered a genetic interaction. Negative genetic interactions refer to a more severe defect in fitness than expected. In extreme cases where the cell is not viable due to mutations in two nonessential genes, it is regarded as a synthetic lethality. Positive interactions refer to double mutants with a less severe fitness defect than expected. Genetic interaction networks can reveal unexpected functional dependencies between genetic loci (i.e., epistasis, wherein the phenotypic effects of mutation in one gene are modified by one or more other genes). For example, negative genetic interactions often result from loss-of-function mutations in pairs of genes in parallel or compensatory pathways that impinge on a common essential process. Conversely, positive interactions can occur between genes in the same pathway if the loss of one gene alone inactivates the pathway such that loss of a second gene confers no additional defect. Genetic interaction networks can be explored by performing Tn-seq analysis in a wild-type strain and its mutant strain counterpart with deletion in a gene of interest (query gene) and comparing the resulting profiles (van Opijnen et al. 2009). This approach was applied to determine genetic interactions in Streptococcus pneumoniae (van Opijnen et al. 2009; van Opijnen and Camilli 2012). Similar genetic interaction mapping based on the use of a microarray-based transposon tracking method was employed previously to uncover genetic interactions important for in vivo fitness of Mycobacterium tuberculosis during infection in mice (Joshi et al. 2006) and for motility of E. coli (Girgis et al. 2007) with a focus on a selected set of query genes.
In a more recent study to understand how trans-translation by transfer-messenger RNA (tmRNA) encoded by ssrA is dispensable in Caulobacter crescentus, Feaga et al. (2014) used Tn-seq method to identify gene(s) that are synthetically lethal with ssrA gene deletion by performing Tn-seq with himar1 transposon libraries in wild-type and ∆ssrA backgrounds, and found that ArfB is a functional homolog of tmRNA that can also release nonstop ribosomes. Genetic interaction mapping can also be performed using an inhibitor that blocks a specific pathway. In a study by Santa Maria et al. (2014), a natural product tunicamycin was used to selectively inhibit TarO, the first enzyme in the wall teichoic acid (WTA) pathway of Staphylococcus aureus. They selected a mariner transposon library in the presence and absence of tunicamycin, and the resulting Tn-seq profiles were compared to identify genes that affect survival in the presence of tunicamycin thus implicating their products in WTA-related activities (Santa Maria et al. 2014). These studies illustrate that Tn-seq has general applicability in mapping genetic interactions for diverse bacterial species (Table 2).
Novel genetic factors involved in specific biological processes
One interesting aspect for Tn-seq application is the development of novel screening strategies that allow genome-wide identification of genetic factors involved in specific biological processes of significance for the bacterial species. These applications require fairly sophisticated experimental designs and optimized experimental conditions to identify the target genes precisely. Some of the examples include identification of genetic factors responsible for (1) Vi capsule expression in Salmonella enterica Typhi (Pickard et al. 2013), (2) immunity against killing by Type VI protein secretion system (T6SS) in Vibrio cholerae (Dong et al. 2013), and (3) in vivo-specific induction of xds gene encoding a secreted exonuclease in Vibrio cholerae (McDonough et al. 2014). For some bacterial pathogens, the genetic factors required to proceed through specific stages in host-pathogen interactions, such as adhesion (de Vries et al. 2013) or invasion to host cells (Gao et al. 2014), have been identified using Tn-seq. More examples of these types of studies are shown in Table 2.
Discovery of adaptive mutations
Before the development of Tn-seq methods, microarray-based footprinting methods were developed to quantitatively track mutants in a complex transposon library (Sassetti and Rubin 2002). One of the unique applications of the microarray-based method was to identify adaptive mutations that contribute to selectable phenotypic variations (Goodarzi et al. 2009). With current NGS technologies, genome sequencing of both wild-type and evolved strains can be easily used to reveal genetic differences (e.g., mutations) between the two strains. However, distinguishing adaptive mutations from neutral mutations is a challenging and labor-intensive process. Goodarzi et al. (2009) described a method termed ADAM (array-based discovery of adaptive mutations) that employs parallel, genome-wide linkage analysis to simultaneously identify all mutated loci with direct contributions to fitness. Although it has not been realized yet, it is quite conceivable that the Tn-seq method could be used in place of a microarray-based transposon mapping approach to advance the strategy currently used in ADAM to identify adaptive mutations at a higher resolution.
Conclusions and perspectives
With the comprehensiveness and sensitivity of Tn-seq, it has emerged as a method of choice to explore genotype-phenotype relationships in bacteria on a genomic scale. Since initial developments of the method in 2009, several variations on Tn-seq have been described with ever increasing applications in numerous bacterial and archaeal species where an efficient random transposon mutagenesis system can be established. The major driving force behind the development of Tn-seq was NGS technologies, more specifically Illumina sequencing. The ability to sequence hundreds of millions of fragments in parallel is the crucial component that provides the comprehensiveness and sensitivity characteristic of Tn-seq methods. With continuous improvements on current NGS platforms (especially increasing read numbers of Illumina sequencing technology), it is expected that the ability to sequence more reads at a reduced cost will occur in the near future (Watson 2014). This will be an advantage in further enhancing the capacity of Tn-seq methods by increasing (1) the number of samples to be analyzed, (2) read depth, or (3) the saturation level of an insertion library.
Although over 70 research articles based on Tn-seq methods have been published within the past 6 years, there are still immense chemical or stress conditions encountered by microorganisms that remain to be explored using straightforward applications of Tn-seq under the corresponding in vitro conditions. For example, the 1144 chemical genomic assays have been performed with the collection of yeast deletion mutants (Hillenmeyer et al. 2008). There has been an increasing number of Tn-seq studies using animal infection models to identify in vivo survival genes. Once such a study is done for a given pathogen using a standard animal infection model under “standard” condition, the next logical step would be to use Tn-seq to understand bacterial genes required for in vivo colonization or survival of the pathogen that would be dependent on the altered host conditions. The altered host conditions could be contributed by genetic factors (e.g., different strains of mice, or transgenic animals) or environmental factors (e.g., modified gut microbiota, co-infection, diets, age, stress, gender etc.) as exemplified in several studies shown in Table 2 (Carter et al. 2014; Goodman et al. 2009; Wong et al. 2013; Wu et al. 2015; Zhang et al. 2013). Knowing the genetic factors required only under specific host or environmental conditions would be extremely helpful in revealing the mechanisms by which the pathogens cope with the dynamically changing microenvironments in the host. Until now, most Tn-seq studies have been conducted to study bacterial species, and only one study was reported in which archaeal species was studied using a Tn-seq method (Sarmiento et al. 2013). Since any haploid microorganisms with an appropriate insertional mutagenesis system can be analyzed by Tn-seq, the method could be applied to the study of more archaeal species and even haploid yeast strains in the future.
References
Ates LS, Ummels R, Commandeur S, van der Weerd R, Sparrius M, Weerdenburg E, Alber M, Kalscheuer R, Piersma SR, Abdallah AM, Abd El Ghany M, Abdel-Haleem AM, Pain A, Jiménez CR, Bitter W, Houben EN (2015) Essential role of the ESX-5 secretion system in outer membrane permeability of pathogenic mycobacteria. PLoS Genet 11(5):e1005190. doi:10.1371/journal.pgen.1005190
Bachman MA, Breen P, Deornellas V, Mu Q, Zhao L, Wu W, Cavalcoli JD, Mobley HL (2015) Genome-wide identification of Klebsiella pneumoniae fitness genes during lung infection. MBio 6(3):e00775. doi:10.1128/mBio.00775-15
Barquist L, Boinett CJ, Cain AK (2013a) Approaches to querying bacterial genomes with transposon-insertion sequencing. RNA Biol 10(7):1161–9. doi:10.4161/rna.24765
Barquist L, Langridge GC, Turner DJ, Phan MD, Turner AK, Bateman A, Parkhill J, Wain J, Gardner PP (2013b) A comparison of dense transposon insertion libraries in the Salmonella serovars typhi and typhimurium. Nucleic Acids Res 41(8):4549–64. doi:10.1093/nar/gkt148
Brooks JF, Gyllborg MC, Cronin DC, Quillin SJ, Mallama CA, Foxall R, Whistler C, Goodman AL, Mandel MJ (2014) Global discovery of colonization determinants in the squid symbiont Vibrio fischeri. Proc Natl Acad Sci U S A 111(48):17284–9. doi:10.1073/pnas.1415957111
Brutinel ED, Gralnick JA (2012) Anomalies of the anaerobic tricarboxylic acid cycle in Shewanella oneidensis revealed by Tn-seq. Mol Microbiol 86(2):273–83. doi:10.1111/j.1365-2958.2012.08196.x
Burghout P, Zomer A, van der Gaast-de Jongh CE, Janssen-Megens EM, Françoijs KJ, Stunnenberg HG, Hermans PW (2013) Streptococcus pneumoniae folate biosynthesis responds to environmental CO2 levels. J Bacteriol 195(7):1573–82. doi:10.1128/JB.01942-12
Byrne RT, Chen SH, Wood EA, Cabot EL, Cox MM (2014) Escherichia coli genes and pathways involved in surviving extreme exposure to ionizing radiation. J Bacteriol 196(20):3534–45. doi:10.1128/JB.01589-14
Carter R, Wolf J, van Opijnen T, Muller M, Obert C, Burnham C, Mann B, Li Y, Hayden RT, Pestina T, Persons D, Camilli A, Flynn PM, Tuomanen EI, Rosch JW (2014) Genomic analyses of pneumococci from children with sickle cell disease expose host-specific bacterial adaptations and deficits in current interventions. Cell Host Microbe 15(5):587–99. doi:10.1016/j.chom.2014.04.005
Chaudhuri RR, Morgan E, Peters SE, Pleasance SJ, Hudson DL, Davies HM, Wang J, van Diemen PM, Buckley AM, Bowen AJ, Pullinger GD, Turner DJ, Langridge GC, Turner AK, Parkhill J, Charles IG, Maskell DJ, Stevens MP (2013) Comprehensive assignment of roles for Salmonella typhimurium genes in intestinal colonization of food-producing animals. PLoS Genet 9(4):e1003456. doi:10.1371/journal.pgen.1003456
Christen B, Abeliuk E, Collier JM, Kalogeraki VS, Passarelli B, Coller JA, Fero MJ, McAdams HH, Shapiro L (2011) The essential genome of a bacterium. Mol Syst Biol 7:528. doi:10.1038/msb.2011.58
Christiansen MT, Kaas RS, Chaudhuri RR, Holmes MA, Hasman H, Aarestrup FM (2014) Genome-wide high-throughput screening to investigate essential genes involved in methicillin-resistant Staphylococcus aureus sequence type 398 survival. PLoS One 9(2):e89018. doi:10.1371/journal.pone.0089018
Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL (2015) Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 347(6218):170–5. doi:10.1126/science.1260580
Dawoud TM, Jiang T, Mandal RK, Ricke SC, Kwon YM (2014) Improving the efficiency of transposon mutagenesis in Salmonella enteritidis by overcoming host-restriction barriers. Mol Biotechnol 56(11):1004–10. doi:10.1007/s12033-014-9779-4
de Lorenzo V, Timmis KN (1994) Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol 235:386–405
de Vries SP, Eleveld MJ, Hermans PW, Bootsma HJ (2013) Characterization of the molecular interplay between Moraxella catarrhalis and human respiratory tract epithelial cells. PLoS One 8(8):e72193. doi:10.1371/journal.pone.0072193
de Vries SP, Rademakers RJ, van der Gaast-de Jongh CE, Eleveld MJ, Hermans PW, Bootsma HJ (2014) Deciphering the genetic basis of Moraxella catarrhalis complement resistance: a critical role for the disulphide bond formation system. Mol Microbiol 91(3):522–37. doi:10.1111/mmi.12475
Dembek M, Barquist L, Boinett CJ, Cain AK, Mayho M, Lawley TD, Fairweather NF, Fagan RP (2015) High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. MBio 6(2):e02383. doi:10.1128/mBio.02383-14
Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C (2009) Systematic mapping of genetic interaction networks. Annu Rev Genet 43:601–25. doi:10.1146/annurev.genet.39.073003.114751
Dong TG, Ho BT, Yoder-Himes DR, Mekalanos JJ (2013) Identification of T6SS-dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc Natl Acad Sci U S A 110(7):2623–8. doi:10.1073/pnas.1222783110
Eckert SE, Dziva F, Chaudhuri RR, Langridge GC, Turner DJ, Pickard DJ, Maskell DJ, Thomson NR, Stevens MP (2011) Retrospective application of transposon-directed insertion site sequencing to a library of signature-tagged mini-Tn5Km2 mutants of Escherichia coli O157:H7 screened in cattle. J Bacteriol 193(7):1771–6. doi:10.1128/JB.01292-10
Feaga HA, Viollier PH, Keiler KC (2014) Release of nonstop ribosomes is essential. MBio 5(6):e01916. doi:10.1128/mBio.01916-14
Febrer M, McLay K, Caccamo M, Twomey KB, Ryan RP (2011) Advances in bacterial transcriptome and transposon insertion-site profiling using second-generation sequencing. Trends Biotechnol 29(11):586–94. doi:10.1016/j.tibtech.2011.06.004
Fu Y, Waldor MK, Mekalanos JJ (2013) Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe 14(6):652–63. doi:10.1016/j.chom.2013.11.001
Gallagher LA, Shendure J, Manoil C (2011) Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. MBio 2(1):e00315–10. doi:10.1128/mBio.00315-10
Gallagher LA, Ramage E, Weiss EJ, Radey M, Hayden HS, Held KG, Huse HK, Zurawski DV, Brittnacher MJ, Manoil C (2015) Resources for genetic and genomic analysis of emerging pathogen Acinetobacter baumannii. J Bacteriol. doi:10.1128/JB.00131-15
Gao B, Lara-Tejero M, Lefebre M, Goodman AL, Galán JE (2014) Novel components of the flagellar system in epsilonproteobacteria. MBio 5(3):e01349–14. doi:10.1128/mBio.01349-14
Gawronski JD, Wong SM, Giannoukos G, Ward DV, Akerley BJ (2009) Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci U S A 106(38):16422–7. doi:10.1073/pnas.0906627106
Girgis HS, Liu Y, Ryu WS, Tavazoie S (2007) A comprehensive genetic characterization of bacterial motility. PLoS Genet 3(9):1644–60. doi:10.1371/journal.pgen.0030154
Goodarzi H, Hottes AK, Tavazoie S (2009) Global discovery of adaptive mutations. Nat Methods 6(8):581–3. doi:10.1038/nmeth.1352
Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI (2009) Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6(3):279–89. doi:10.1016/j.chom.2009.08.003
Goryshin IY, Jendrisak J, Hoffman LM, Meis R, Reznikoff WS (2000) Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat Biotechnol 18(1):97–100. doi:10.1038/72017
Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7(9):e1002251. doi:10.1371/journal.ppat.1002251
Hayes F (2003) Transposon-based strategies for microbial functional genomics and proteomics. Annu Rev Genet 37:3–29. doi:10.1146/annurev.genet.37.110801.142807
Hendrixson DR, Akerley BJ, DiRita VJ (2001) Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol Microbiol 40(1):214–24
Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, Altman RB, Davis RW, Nislow C, Giaever G (2008) The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320(5874):362–5. doi:10.1126/science.1150021
Hutchison CA, Peterson SN, Gill SR, Cline RT, White O, Fraser CM, Smith HO, Venter JC (1999) Global transposon mutagenesis and a minimal Mycoplasma genome. Science 286(5447):2165–9
Johnson CM, Grossman AD (2014) Identification of host genes that affect acquisition of an integrative and conjugative element in Bacillus subtilis. Mol Microbiol 93(6):1284–301. doi:10.1111/mmi.12736
Johnson JG, Livny J, Dirita VJ (2014) High-throughput sequencing of Campylobacter jejuni insertion mutant libraries reveals mapA as a fitness factor for chicken colonization. J Bacteriol 196(11):1958–67. doi:10.1128/JB.01395-13
Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM (2006) Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc Natl Acad Sci U S A 103(31):11760–5. doi:10.1073/pnas.0603179103
Kalamorz F, Reichenbach B, März W, Rak B, Görke B (2007) Feedback control of glucosamine-6-phosphate synthase GlmS expression depends on the small RNA GlmZ and involves the novel protein YhbJ in Escherichia coli. Mol Microbiol 65(6):1518–33. doi:10.1111/j.1365-2958.2007.05888.x
Kamp HD, Patimalla-Dipali B, Lazinski DW, Wallace-Gadsden F, Camilli A (2013) Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog 9(12):e1003800. doi:10.1371/journal.ppat.1003800
Khatiwara A, Jiang T, Sung SS, Dawoud T, Kim JN, Bhattacharya D, Kim HB, Ricke SC, Kwon YM (2012) Genome scanning for conditionally essential genes in Salmonella enterica serotype typhimurium. Appl Environ Microbiol 78(9):3098–107. doi:10.1128/AEM.06865-11
Klein BA, Tenorio EL, Lazinski DW, Camilli A, Duncan MJ, Hu LT (2012) Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics 13:578. doi:10.1186/1471-2164-13-578
Knierim E, Lucke B, Schwarz JM, Schuelke M, Seelow D (2011) Systematic comparison of three methods for fragmentation of long-range PCR products for next generation sequencing. PLoS One 6(11):e28240. doi:10.1371/journal.pone.0028240
Langereis JD, Weiser JN (2014) Shielding of a lipooligosaccharide IgM epitope allows evasion of neutrophil-mediated killing of an invasive strain of nontypeable Haemophilus influenzae. MBio 5(4):e01478–14. doi:10.1128/mBio.01478-14
Langereis JD, Zomer A, Stunnenberg HG, Burghout P, Hermans PW (2013) Nontypeable Haemophilus influenzae carbonic anhydrase is important for environmental and intracellular survival. J Bacteriol 195(12):2737–46. doi:10.1128/JB.01870-12
Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G, Wain J, Parkhill J, Turner AK (2009) Simultaneous assay of every Salmonella typhi gene using one million transposon mutants. Genome Res 19(12):2308–16. doi:10.1101/gr.097097.109
Lazinski DW, Camilli A (2013) Homopolymer tail-mediated ligation PCR: a streamlined and highly efficient method for DNA cloning and library construction. Biotechniques 54(1):25–34. doi:10.2144/000113981
Le Breton Y, Belew AT, Valdes KM, Islam E, Curry P, Tettelin H, Shirtliff ME, El-Sayed NM, McIver KS (2015) Essential genes in the core genome of the human pathogen Streptococcus pyogenes. Sci Rep 5:9838. doi:10.1038/srep09838
Lee SA, Gallagher LA, Thongdee M, Staudinger BJ, Lippman S, Singh PK, Manoil C (2015) General and condition-specific essential functions of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 112(16):5189–94. doi:10.1073/pnas.1422186112
Maloy SR (2007) Use of antibiotic-resistant transposons for mutagenesis. Methods Enzymol 421:11–7. doi:10.1016/S0076-6879(06)21002-4
Mann B, van Opijnen T, Wang J, Obert C, Wang YD, Carter R, McGoldrick DJ, Ridout G, Camilli A, Tuomanen EI, Rosch JW (2012) Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog 8(7):e1002788. doi:10.1371/journal.ppat.1002788
Martínez-García E, Calles B, Arévalo-Rodríguez M, de Lorenzo V (2011) pBAM1: an all-synthetic genetic tool for analysis and construction of complex bacterial phenotypes. BMC Microbiol 11:38. doi:10.1186/1471-2180-11-38
Mazurkiewicz P, Tang CM, Boone C, Holden DW (2006) Signature-tagged mutagenesis: barcoding mutants for genome-wide screens. Nat Rev Genet 7(12):929–39. doi:10.1038/nrg1984
McDonough E, Lazinski DW, Camilli A (2014) Identification of in vivo regulators of the Vibrio cholerae xds gene using a high-throughput genetic selection. Mol Microbiol 92(2):302–15. doi:10.1111/mmi.12557
Merrell DS, Camilli A (2002) Information overload: assigning genetic functionality in the age of genomics and large-scale screening. Trends Microbiol 10(12):571–4
Moule MG, Hemsley CM, Seet Q, Guerra-Assunção JA, Lim J, Sarkar-Tyson M, Clark TG, Tan PB, Titball RW, Cuccui J, Wren BW (2014) Genome-wide saturation mutagenesis of Burkholderia pseudomallei K96243 predicts essential genes and novel targets for antimicrobial development. MBio 5(1):e00926–13. doi:10.1128/mBio.00926-13
Palace SG, Proulx MK, Lu S, Baker RE, Goguen JD (2014) Genome-wide mutant fitness profiling identifies nutritional requirements for optimal growth of Yersinia pestis in deep tissue. MBio 5(4):e01385–14. doi:10.1128/mBio.01385-14
Phan MD, Peters KM, Sarkar S, Lukowski SW, Allsopp LP, Gomes Moriel D, Achard ME, Totsika M, Marshall VM, Upton M, Beatson SA, Schembri MA (2013) The serum resistome of a globally disseminated multidrug resistant uropathogenic Escherichia coli clone. PLoS Genet 9(10):e1003834. doi:10.1371/journal.pgen.1003834
Phan MD, Forde BM, Peters KM, Sarkar S, Hancock S, Stanton-Cook M, Ben Zakour NL, Upton M, Beatson SA, Schembri MA (2015) Molecular characterization of a multidrug resistance IncF plasmid from the globally disseminated Escherichia coli ST131 clone. PLoS One 10(4):e0122369. doi:10.1371/journal.pone.0122369
Pickard D, Kingsley RA, Hale C, Turner K, Sivaraman K, Wetter M, Langridge G, Dougan G (2013) A genomewide mutagenesis screen identifies multiple genes contributing to Vi capsular expression in Salmonella enterica serovar typhi. J Bacteriol 195(6):1320–6. doi:10.1128/JB.01632-12
Reid AN, Pandey R, Palyada K, Whitworth L, Doukhanine E, Stintzi A (2008) Identification of Campylobacter jejuni genes contributing to acid adaptation by transcriptional profiling and genome-wide mutagenesis. Appl Environ Microbiol 74(5):1598–612. doi:10.1128/AEM.01508-07
Roggo C, Coronado E, Moreno-Forero SK, Harshman K, Weber J, van der Meer JR (2013) Genome-wide transposon insertion scanning of environmental survival functions in the polycyclic aromatic hydrocarbon degrading bacterium Sphingomonas wittichii RW1. Environ Microbiol 15(10):2681–95. doi:10.1111/1462-2920.12125
Rohmer L, Hocquet D, Miller SI (2011) Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol 19(7):341–8. doi:10.1016/j.tim.2011.04.003
Roux D, Danilchanka O, Guillard T, Cattoir V, Aschard H, Fu Y, Angoulvant F, Messika J, Ricard JD, Mekalanos JJ, Lory S, Pier GB, Skurnik D (2015) Fitness cost of antibiotic susceptibility during bacterial infection. Sci Transl Med 7(297):297ra114. doi:10.1126/scitranslmed.aab1621
Santa Maria JP, Sadaka A, Moussa SH, Brown S, Zhang YJ, Rubin EJ, Gilmore MS, Walker S (2014) Compound-gene interaction mapping reveals distinct roles for Staphylococcus aureus teichoic acids. Proc Natl Acad Sci U S A 111(34):12510–5. doi:10.1073/pnas.1404099111
Santiago M, Matano LM, Moussa SH, Gilmore MS, Walker S, Meredith TC (2015) A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics 16:252. doi:10.1186/s12864-015-1361-3
Sarmiento F, Mrázek J, Whitman WB (2013) Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc Natl Acad Sci U S A 110(12):4726–31. doi:10.1073/pnas.1220225110
Sassetti C, Rubin EJ (2002) Genomic analyses of microbial virulence. Curr Opin Microbiol 5(1):27–32
Sassetti CM, Rubin EJ (2003) Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100(22):12989–94. doi:10.1073/pnas.2134250100
Sassetti CM, Boyd DH, Rubin EJ (2001) Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci U S A 98(22):12712–7. doi:10.1073/pnas.231275498
Shan Y, Lazinski D, Rowe S, Camilli A, Lewis K (2015) Genetic basis of persister tolerance to aminoglycosides in Escherichia coli. MBio 6(2):e00078–15. doi:10.1128/mBio.00078-15
Sharma CM, Vogel J (2009) Experimental approaches for the discovery and characterization of regulatory small RNA. Curr Opin Microbiol 12(5):536–46. doi:10.1016/j.mib.2009.07.006
Skurnik D, Roux D, Aschard H, Cattoir V, Yoder-Himes D, Lory S, Pier GB (2013) A comprehensive analysis of in vitro and in vivo genetic fitness of Pseudomonas aeruginosa using high-throughput sequencing of transposon libraries. PLoS Pathog 9(9):e1003582. doi:10.1371/journal.ppat.1003582
Subashchandrabose S, Smith SN, Spurbeck RR, Kole MM, Mobley HL (2013) Genome-wide detection of fitness genes in uropathogenic Escherichia coli during systemic infection. PLoS Pathog 9(12):e1003788. doi:10.1371/journal.ppat.1003788
Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M (2015) Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci U S A 112(13):4110–5. doi:10.1073/pnas.1419677112
Valentino MD, Foulston L, Sadaka A, Kos VN, Villet RA, Santa Maria J, Lazinski DW, Camilli A, Walker S, Hooper DC, Gilmore MS (2014) Genes contributing to Staphylococcus aureus fitness in abscess- and infection-related ecologies. MBio 5(5):e01729–14. doi:10.1128/mBio.01729-14
van Opijnen T, Camilli A (2012) A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res. doi:10.1101/gr.137430.112
van Opijnen T, Camilli A (2013) Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat Rev Microbiol 11(7):435–42. doi:10.1038/nrmicro3033
van Opijnen T, Bodi KL, Camilli A (2009) Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6(10):767–72. doi:10.1038/nmeth.1377
Wang N, Ozer EA, Mandel MJ, Hauser AR (2014) Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. MBio 5(3):e01163–14. doi:10.1128/mBio.01163-14
Wang Q, Millet YA, Chao MC, Sasabe J, Davis BM, Waldor MK (2015) A genome-wide screen reveals that the Vibrio cholerae phosphoenolpyruvate phosphotransferase system modulates virulence gene expression. Infect Immun 83(9):3381–95. doi:10.1128/IAI.00411-15
Watson M (2014) Illuminating the future of DNA sequencing. Genome Biol 15(2):108. doi:10.1186/gb4165
Weerdenburg EM, Abdallah AM, Rangkuti F, Abd El Ghany M, Otto TD, Adroub SA, Molenaar D, Ummels R, Ter Veen K, van Stempvoort G, van der Sar AM, Ali S, Langridge GC, Thomson NR, Pain A, Bitter W (2015) Genome-wide transposon mutagenesis indicates that Mycobacterium marinum customizes its virulence mechanisms for survival and replication in different hosts. Infect Immun 83(5):1778–88. doi:10.1128/IAI.03050-14
Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, Blow MJ, Bristow J, Butland G, Arkin AP, Deutschbauer A (2015) Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. MBio 6(3):e00306–15. doi:10.1128/mBio.00306-15
Wong SM, Gawronski JD, Lapointe D, Akerley BJ (2011) High-throughput insertion tracking by deep sequencing for the analysis of bacterial pathogens. Methods Mol Biol 733:209–22. doi:10.1007/978-1-61779-089-8_15
Wong SM, Bernui M, Shen H, Akerley BJ (2013) Genome-wide fitness profiling reveals adaptations required by Haemophilus in coinfection with influenza a virus in the murine lung. Proc Natl Acad Sci U S A 110(38):15413–8. doi:10.1073/pnas.1311217110
Wu M, McNulty NP, Rodionov DA, Khoroshkin MS, Griffin NW, Cheng J, Latreille P, Kerstetter RA, Terrapon N, Henrissat B, Osterman AL, Gordon JI (2015) Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science 350 (6256). doi:10.1126/science.aac5992
Yamaichi Y, Chao MC, Sasabe J, Clark L, Davis BM, Yamamoto N, Mori H, Kurokawa K, Waldor MK (2015) High-resolution genetic analysis of the requirements for horizontal transmission of the ESBL plasmid from Escherichia coli O104:H4. Nucleic Acids Res 43(1):348–60. doi:10.1093/nar/gku1262
Yung MC, Park DM, Overton KW, Blow MJ, Hoover CA, Smit J, Murray SR, Ricci DP, Christen B, Bowman GR, Jiao Y (2015) Transposon mutagenesis paired with deep sequencing of Caulobacter crescentus under uranium stress reveals genes essential for detoxification and stress tolerance. J Bacteriol 197(19):3160–72. doi:10.1128/JB.00382-15
Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, Rubin EJ (2012) Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog 8(9):e1002946. doi:10.1371/journal.ppat.1002946
Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, Trauner A, Wallis D, Galaviz S, Huttenhower C, Sacchettini JC, Behar SM, Rubin EJ (2013) Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 155(6):1296–308. doi:10.1016/j.cell.2013.10.045
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Kwon, Y.M., Ricke, S.C. & Mandal, R.K. Transposon sequencing: methods and expanding applications. Appl Microbiol Biotechnol 100, 31–43 (2016). https://doi.org/10.1007/s00253-015-7037-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7037-8