
Abstract
Trichoderma filamentous fungi have been investigated due to their ability to secrete cellulases which find various biotechnological applications such as biomass hydrolysis and cellulosic ethanol production. Previous studies demonstrated that Trichoderma harzianum IOC-3844 has a high degree of cellulolytic activity and potential for biomass hydrolysis. However, enzymatic, biochemical, and structural studies of cellulases from T. harzianum are scarce. This work reports biochemical characterization of the recombinant endoglucanase I from T. harzianum, ThCel7B, and its catalytic core domain. The constructs display optimum activity at 55 °C and a surprisingly acidic pH optimum of 3.0. The full-length enzyme is able to hydrolyze a variety of substrates, with high specific activity: 75 U/mg for β-glucan, 46 U/mg toward xyloglucan, 39 U/mg for lichenan, 26 U/mg for carboxymethyl cellulose, 18 U/mg for 4-nitrophenyl β-d-cellobioside, 16 U/mg for rye arabinoxylan, and 12 U/mg toward xylan. The enzyme also hydrolyzed filter paper, phosphoric acid swollen cellulose, Sigmacell 20, Avicel PH-101, and cellulose, albeit with lower efficiency. The ThCel7B catalytic domain displays similar substrate diversity. Fluorescence-based thermal shift assays showed that thermal stability is highest at pH 5.0. We determined kinetic parameters and analyzed a pattern of oligosaccharide substrates hydrolysis, revealing cellobiose as a final product of C6 degradation. Finally, we visualized effects of ThCel7B on oat spelt using scanning electron microscopy, demonstrating the morphological changes of the substrate during the hydrolysis. The acidic behavior of ThCel7B and its considerable thermostability hold a promise of its industrial applications and other biotechnological uses under extremely acidic conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Energy consumption has been significantly increasing with the growth of the world population and industrial development of the countries worldwide. Crude oil has been the major natural resource to satisfy this increasing energy demand, but its intensive use result in undesirable environmental consequences (Himmel et al. 2007; Buckeridge et al. 2010; Serpa and Polikarpov 2011; Chundawat et al. 2011). From an environmental point of view, CO2 emissions released into the atmosphere are responsible for climate changes and worsening of the greenhouse effect with potentially irreparable consequences (Himmel et al. 2007; Buckeridge et al. 2010; Serpa and Polikarpov 2011; Chundawat et al. 2011). Jointly, concerns about climate changes, increasing fuel demand and energy insecurity, have motivated the search for alternative forms of energy, especially biomass-based biofuels, which was steadily growing within the last years (Sun and Cheng 2002; Himmel et al. 2007; Chundawat et al. 2011).
Lignocellulosic biomass is the most abundant renewable carbon source on Earth and is readily available in the form of diverse feedstocks, such as forest and agricultural residues. Their annual production worldwide has been estimated in 1 × 1010 MT (Sanchez and Cardona 2008), indicating enormous potential as a low-cost raw material for cellulosic, second-generation bioethanol. A large part of the first-generation ethanol produced in the world is obtained by sugarcane juice fermentation and its residue, sugarcane bagasse, emerges as an exciting opportunity for increasing ethanol production, without increasing cultivated land, and generating very low environmental impacts (Dias et al. 2012).
Bioconversion of lignocellulosic materials to ethanol normally starts with biomass pretreatment. This initial step improves enzymatic hydrolysis by partially removing or relocating lignin and/or hemicellulose on the substrate. It also reduces cellulose crystallinity and increases porosity of the material. The second step for conversion of biomass into simple sugars is the enzymatic hydrolysis of cellulose, which involves the synergistic actions of at least three different classes of enzymes (Sun and Cheng 2002). Canonical set of enzymes includes endoglucanases (E.C. 3.2.1.4), exoglucanases/cellobiohydrolases (E.C. 3.2.1.91), and β-glucosidases (E.C. 3.2.1.21) (Teeri 1997; Schwarz 2001), although recent discoveries strongly suggest that this list is far from being comprehensive (Horn et al. 2012). Fungi of the genus Trichoderma and in particular Trichoderma reesei are widely used for industrial cellulolytic enzyme production (Lynd et al. 2002; Schuster and Schmoll 2010). However, recent studies have revealed potential of the Trichoderma harzianum IOC-3844 strain for cellulase production aimed at cellulosic ethanol industrial applications (de Castro et al. 2010).
Endoglucanases play an important role in biomass conversion by cleaving internal β-1,4-glycosidic bonds in random non-crystalline sites of the cellulose chain (Hasper et al. 2002). In aerobic fungi, endoglucanases are mostly found in the glycoside hydrolase (GH) families GH5, GH6, GH7, GH9, GH12, and GH45 (Cantarel et al. 2009). Cel7B is the major endoglucanase in the cellulase system of T. reesei which corresponds to 10–15 % of secreted protein (García et al. 2001).
In addition to their applications in biofuels industry, endoglucanases are widely used in food industry to increase the yield of fruit juices, beer filtration, oil extraction, for improving the nutritive quality of bakery products and animal feed (Bhat 2000), and also in pulp and paper industry (Hilden et al. 2005). When applied in these industrial processes, the enzymes are frequently exposed to extreme pH and temperature conditions. Thus, the search for stable enzymes capable of functioning in extreme conditions becomes highly relevant. In the present research, we described the biochemical characterization of a recombinant endoglucanase I from T. harzianum IOC-3844 (ThCel7B), a member of the GH7. We analyzed the enzyme specificity, its kinetic parameters, hydrolytic cleavage pattern, themostability, and the morphological changes caused by the ThCel7B enzymatic activity in the oat spell. We demonstrated that this extremely acidic enzyme (pH optimum =3) hydrolyses a wide range of carbohydrates and synthetic substrates and is highly active against β-glucan, xyloglucan, lichenan, and carboxymethyl cellulose (CMC) and has somewhat lower activity against filter paper, pNPC, and rye arabinoxylan.
Materials and methods
Cloning of endoglucanase
The coding sequence of ThCel7B was obtained from JGI’s T. harzianum genome database (http://genome.jgi.doe.gov/Triha1/Triha1.home.html), where it is identified as “transcript ID 20062.” The gene fragment was amplified from a complementary DNA (cDNA) library of T. harzianum IOC-3844 obtained from Instituto Oswaldo Cruz Culture Collection of Filamentous Fungi (CCFF; http://ccff.fiocruz.br/) grown for 3–4 days at 28 °C with stirring using Avicel (Sigma-Aldrich, St. Louis, USA) as carbon source in minimal medium (0.3 g.L−1 urea; 1.4 g.L−1 (NH4)2SO4; 1 ml.L−1) of micronutrients solution 1000× (2.2 % ZnSO4.7H2O; 1.1 % H3BO3; 0.5 % MnCl2.4H2O; 0.5 % FeSO4.7H2O, 0.17 % CoCl2.6H2O; 0.16 % CuSO4.5H2O; 0.15 % Na2MoO4.2H2O; 5 % Na4EDTA (w/v); 0.4 g.L−1 CaCl2; 0.3 g.L−1 MgSO4; 10 mM sodium citrate pH 5.0; 0.6 g.L−1 yeast extract). Total RNA was extracted with TRIzol (Life Technologies, New York, USA) and first strand cDNA synthesis was obtained by “First Strand cDNA Synthesis” kit (Thermo Scientific, Vilnius, LT). The full-length (ThCel7B-full) coding sequence (nucleotides 1–1416) and the catalytic core domain (ThCel7B-CCD) coding sequence (nucleotides 1–1185) were amplified in two steps using the primers listed in Table 1. In the first step, primers Fw1 and Rv1 were used to amplify each fragment from cDNA template. In the second step, primers Fw1 and Rv2 were used and the template was the product of the first step. Amplifications were performed using Phusion High-fidelity DNA polymerase (New England Biolabs, Hitchin, UK). Briefly, a 50-μL reaction mix containing 20 ng of template DNA, 25 pmol of each primer, 0.2 mM dNTP Mix, 3 % of DMSO, 1 unit of Phusion polymerase, and 1× Phusion polymerase buffer was used in a three-step PCR reaction: (1) 98 °C for 30 s, 1 cycle; (2) 98 °C for 10 s, 65 °C for 30 s, followed by 72 °C for 2 min; 35 cycles; (3) 72 °C for 10 min, 1 cycle. The DNA fragments obtained in the second amplification were cloned into ANip7G (Storms et al. 2005) vector using Gateway technology (Hartley et al. 2000; Katzen 2007) and confirmed by sequencing.
Aspergillus niger transformation
Protoplast preparation of Aspergillus niger PY11 (cspA ∆Gla::hiG), derived from strain N593 (cspA − pyrG −), which was obtained from the Centre for Structural and Functional Genomics, Concordia University (http://www.concordia.ca/research/genomics.html), was carried out similarly to the procedure described by Penttilä et al. (1987). Fresh A. niger conidia were inoculated in 200 ml of complete medium (10 g.L−1 glucose, 2 g.L−1 peptone, 1 g.L−1 yeast extract, 1 g.L−1 casamino acids) in a 1000-ml baffled flask reaching the concentration of 2 × 106 spores.mL−1 and then incubated for 16–22 h at 30 °C and 100 rpm. The mycelium was collected on Miracloth (Merck KGaA Darmstadt, DE) and washed with 500 mL sterile solution of MgSO4 0.6 M. About 1 g of mycelium was suspended in 5 mL of protoplasting solution (1.2 M MgSO4, 50 mM NaPO4 pH 5.8), followed by addition of 250 mg of VinoTaste Pro enzymatic cocktail (Novozymes, Bagsvaerd, DK). This reaction was incubated for 3–6 h at 30 °C and 100 rpm, filtered through Miracloth (Merck KGaA Darmstadt, DE) and washed with 200 mL of protoplasting solution. The protoplasts were overlaid with ST buffer (0.6 M sorbitol, 0.1 M Tris–HCl pH 7.5) and centrifuged at 3750 rpm and 4 °C for 20 min. Protoplasts were collected from the interphase and washed three times with cold SC buffer (1.0 M sorbitol, 66.2 mM CaCl2) at 3750 rpm, 4 °C for 5 min. Finally, the sample was suspended in SC buffer at a concentration of approximately 5 × 107 spores.mL−1 and stocked for later use in transformation reactions.
The process of transformation was mediated by polyethylene glycol (PEG) and performed as described by Yelton et al. (1984). Briefly, 200 μL of protoplasts (1 × 107 cells.mL−1), 10 μl plasmid DNA (>5 μg), 100 μL PEG 4000 20 % (w/v) solution (in 1 M Tris-HCl, pH 7.5, and 1 M NaCl2) and 20 μL 0.4 M aurintricarboxylic acid (ATA) were mixed gently and incubated at 25 °C for 10 min. Then, 1.5 ml of PEG 60 % solution (60 % (w/v) PEG 6000, 1 M Tris–HCl, pH 7.5, and 1 M NaCl) was added and the mixture was incubated for 20 min at 25 °C, followed by addition of sorbitol solution to a final concentration of 1.2 M. The sample was centrifuged at 3750 rpm for 10 min at 25 °C. Aliquots of 1 ml of resuspension were plated in recovery agar medium of selective regeneration (34 % sucrose, 70.6 mM NaNO3, 6.7 mM KCl, 11.1 mM KH2PO4, 0.2 mM KOH, 2 mM MgSO4.7H2O, and trace elements) (Kalsner et al. 1995), and incubated at 30 °C for 4 to 6 days.
Expression, purification, and identification of recombinant enzyme
Different colonies of the recombinant A. niger were selected and screened for recombinant protein production in 24-well plates. Each colony was inoculated into 1 mL of MMJ medium (Kalsner et al. 1995) with 15 % (w/v) of maltose and incubated at 30 °C for 7 days. Protein production was monitored between days 5 and 7 by analyzing culture supernatants in SDS-PAGE. Positive transformants were inoculated in 2.0-L Erlenmeyer flasks containing 500 mL of MMJ, at a final concentration of 2 × 106 spores.mL−1. After cultivation for 6 days at 30 °C under static condition, the medium was filtrated to remove spores and the remaining crude extract was precipitated by ammonium sulfate at 80 % saturation. Following 12-h precipitation, the extract was centrifuged and the pellet was diluted in 50 mM sodium citrate buffer (pH 5.0). The protein solution was purified by hydrophobic chromatography on a Phenyl-Sepharose 6 Fast Flow column (GE Healthcare Biosciences, Little Chalfont, UK) previously equilibrated with 50 mM sodium citrate buffer (pH 5.0) supplemented with 1 M ammonium sulfate. The protein fractions were collected and subjected to size exclusion chromatography using a Superdex 75 16/60 column (GE Healthcare Biosciences, Little Chalfont, UK). Purified ThCel7B-full and ThCel7B-CCD samples were analyzed by 15 % SDS-PAGE.
Enzymatic assays and determination of optimal pH and temperature
CMCase activity was determined by a colorimetric method using the 3,5-dinitrosalicylic acid (Miller 1959), using glucose as a standard. All assays were performed in triplicate with 1 % medium-viscosity CMC as a substrate. The reaction mixture containing 10 μL of enzyme solution, 50 μL of 1 % (w/v) CMC and 40 μL of 50 mM sodium citrate buffer at pH 5.0 was incubated at 50 °C for 15 min and reaction was stopped by adding 100 μL of 3,5-dinitrosalicylic acid solution (DNS). The mixture was incubated for 5 min at 100 °C, and its absorbance was measured at 540 nm.
Optimal pH and temperature for activity were determined using purified ThCel7B and its CCD. The optimum temperature was evaluated by incubating the previously described reaction at 25 to 85 °C in 50 mM sodium citrate buffer (pH 5.0). For optimal pH determination, enzyme, and substrate were diluted in 40 mM acetate/borate/phosphate buffer (ABF) with different pH values ranging from 2.0 to 10.0 and incubated at 55 °C for 15 min. Furthermore, the optimal pH was also tested at 50 °C, to verify if the activity profile in different pH values may be affected by the enzyme thermostability.
To test the acidophilic buffer preference of the enzymes, a screening with different acidic buffers was performed. The buffers were diluted to 50 mM and the reaction was incubated as described previously, at the predetermined optimal temperature.
To determine thermal stability, ThCel7B was initially pre-incubated in 50 mM of sodium citrate buffer, pH 3.0 and 5.0 at 55 °C. Aliquots of 10 μL were removed and assayed for activity on CMC 1 % over time.
Thermal stability assays
Thermal stability assay was performed using the fluorescence-based thermal shift method (ThermoFluor), with the purpose of monitoring protein stability at different pH and temperatures. We evaluated the stability of ThCel7B and its catalytic domain in triplicate, in the ABF buffers, pH range from 2 to 10. The proteins’ final concentration was 7 μM. The reaction volume was 20 μl, and the fluorescent stain used was SYPRO Orange (Invitrogen, Carlsbad, USA), 2000 times diluted in water. The reactions were performed in a 96-well thin-wall PCR plate (Bio-Rad, Hercules, USA). For temperature stability studies, the protein samples were heated to the temperatures in the range from 25 to 90 °C, with stepwise increments of 1 °C, holding each temperature for 30 s in a CFX96 Real Time System (Bio-Rad, Hercules, USA). The fluorescence variation of the SYPRO Orange probe was measured using excitation at 490 nm and emission at 530 nm. Analysis of curves and melting temperatures (T m) of each sample were carried out using GraphPad Prism software version 5.0 (San Diego, USA).
Determination of substrate specificity
The substrate specificity of the purified ThCel7B and its CCD were determined using rye arabinoxylan, linear arabinan, debranched arabinan, arabinan, larch arabinogalactomannan, xyloglucan from tamarind, β-glucan from barley, 1-4 β-d-mannan galactomannan, lichenan from Iceland moss, xylan from oat spelt, and xylan beechwood. All these substrates were purchased from Megazyme (Wicklow, IE). We also tested three types of microcrystalline cellulose: Avicel PH-101, cellulose (cat. No 31,069–7; Sigma-Aldrich, St. Louis, USA) and Sigmacell 20 (Sigma-Aldrich, St. Louis, USA) and other substrates: 4-nitrophenyl-β-d-cellobioside (pNPC), 4-nitrophenyl-β-d-glucopyranoside (pNPG), carboxymethyl cellulose (CMC), phosphoric acid swollen cellulose (PASC) (Wood 1988), and filter paper (FP) Whatman No. 1 (GE Healthcare Biosciences, Little Chalfont, UK).
All substrates were diluted in water to a final concentration of 1 % (w/v), and the reaction mixture was composed of 0.1 mL of purified enzyme at a concentration of 0.08 mg.mL−1, 0.4 mL of 50 mM sodium citrate buffer at pH 3.0 and 0.5 mL of 1 % (w/v) substrate aqueous solution. For filter paper assays, 0.7-cm-diameter disc-shaped FP units were produced and the reaction was incubated at 50 °C for 60 min. Other reactions was incubated at 50 °C for 15 min, and after this period of time, 100 μL of DNS was added, followed by incubation at 95 °C for 5 min. The absorbance was measured at 540 nm. For reactions with pNPG and pNPC, after the incubation time of 15 min, 100 μL of 1.0 M Na2CO3 was added, followed by absorbance measurement at 405 nm.
After the tests described above and just with substrates against which the enzyme did not show activity, new assays were performed extending the incubation time to 4 h.
Kinetic assays
Kinetic parameters were determined for the both ThCel7B and its catalytic domain using increasing concentrations of CMC and xyloglucan. Reactions were performed in sodium citrate buffer (pH 3.0) at 50 °C and measured by DNS method as described above. Enzyme unit was defined as the amount of enzyme that produces 1.0 μM of glucose in 1 min for each substrate. Kinect constants were determined by nonlinear regression using OriginPro 8.0 (OriginLab, Northampton, USA).
Circular dichroism studies
Circular dichroism (CD) measurements of the purified enzymes were carried out on a Jasco J-720 spectropolarimeter (JASCO Corporation, Tokyo, Japan). The samples were analyzed at 25 °C using 0.1 cm path length quartz cells and protein far-UV spectra were recorded over a wavelength range of 190–260 nm by signal averaging of 8 spectra. The protein signal was obtained by subtracting buffer spectrum from the sample spectrum. All protein samples, at a concentration of 5 μM, were incubated overnight in 40 mM ABF buffer at pH values 3.0, 4.0, 5.0, 7.0, 8.0, and 9.0 before the spectrum was recorded. All samples were previously centrifuged to avoid any interference of particles in suspension.
Scanning electron microscopy (SEM)
Samples of oat spelt, which are rich in β-glucan, were submitted to hydrolysis by ThCel7B and its catalytic domain for 12 h at 50 °C at two different pH values (pH = 3 and pH = 5). The assays were carried out in triplicate and the reaction mixture was composed of 5.0 μL of enzyme at a concentration of 1.0 mg.mL−1, 95 μL of 50 mM sodium citrate buffer, and 6 mg of oat spelt. In the control sample, the enzyme volume was replaced by 5.0 μL of the same buffer. The remaining solid substrate after enzymatic action was directly dried (without being rinsed) at 30 °C for 12 h prior to SEM analysis.
After drying, samples were gold coated in a SCD 050 sputter coater (Oerlikon-Balzers, Balzers, Lichtenstein) and then imaged using a high-resolution environmental scanning electron microscope, equipped with a field emission gun (FESEM) (FEI, Quanta 650, Hillsboro, USA). Both the coater and the microscope were available at the National Laboratory of Nanotechnology (LNNano) in Campinas-SP, Brazil. Images were obtained under vacuum, using a 5 kV accelerating voltage and a secondary electron detector. A large number of images was obtained for different areas of the samples (at least 20 images per sample) to guarantee the reproducibility of the results.
Hydrolytic cleavage pattern determined by capillary zone electrophoresis of oligosaccharides (CZE)
For CZE analysis, cellohexaose (C6) was labeled with 8-aminopyrene-1,3,6-trisulfonic acid (APTS) by reductive amination (Naran et al. 2007) and used as a substrate. Enzymatic hydrolysis of the labeled substrate was performed using a mix of the protein with the substrate, at citrate buffer at pH 3.0, equilibrated at 50 °C.
The experiment was conducted on a P/ACE MQD instrument (Beckman Coulter, Pasadena, USA), equipped with laser-induced fluorescence detection. A fused-silica capillary (TSP050375, Polymicro Technologies by MOLEX, Lisle, USA) of internal diameter of 50 μm and total length of 31 cm was used as a separation column for oligosaccharides. The capillary column was rinsed with 1 M NaOH followed by running the buffer to prevent carry-over after injection. The electrophoresis conditions were 15 kV/70–100 μA at 20 °C and oligomers with APTS were excited at 488 nm and emission was collected through a 520 nm band pass filter. The combined information obtained from the electrophoretic behavior and co-electrophoresis with mono and oligosaccharides standards were used to identify the degradation products.
Results
Expression and purification of ThCel7B and its catalytic domain
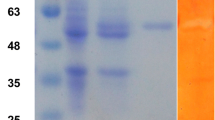
Structurally, T. harzianum endoglucanase I is a typical two domain cellulase, composed of a GH7 family catalytic core domain (CCD), and a CBM1 family carbohydrate binding module connected by a 40-residue linker. Both the full-length enzyme and the isolated catalytic domain of ThCel7B were successfully expressed by A. niger PY11. Culture supernatants were precipitated with 80 % ammonium sulfate, and after solubilization, they were purified by using a Phenyl Sepharose 6 Fast Flow column, followed by size exclusion chromatography in a Superdex 75 16/60 column (GE Healthcare Biosciences, Little Chalfont, UK). The two constructions were obtained with high purity as confirmed by SDS-PAGE (Fig. S1 in the Supplementary Material). Full-length ThCel7B showed an increase in experimentally determined molecular weight (MW ~ 70 kDa), when compared to theoretical prediction based on its amino acid sequence (49.1 kDa). This most probably results from the protein glycosylation, which is generally observed in cellulases expressed by filamentous fungus (Punt et al. 2002). Conversely, the catalytic domain was expressed with the molecular weight close to the predicted value (42.8 kDa), indicating that the glycosylation is mostly restricted to the linker region and/or the CBM (Payne et al. 2013).
Enzymatic characterization of ThCel7B and its CCD
Optimal pH and temperature of purified ThCel7B and its catalytic domain were determined by following their enzymatic activity on CMC in ABF buffer at pH values ranging from 2.0 to 10.0 and temperatures ranging from 25 to 85 °C. The optimal pH studies were performed at two different temperatures: 55 °C, at which the enzyme displays highest activity (Fig. 1a, b) and 50 °C, to ensure that the measurements of optimal pH are not affected by the enzyme thermostability. The activity profiles observed at both temperatures are very similar (Fig. 1c, d). Surprisingly, however, both the full-length enzyme and the catalytic domain of ThCel7B show an optimal activity at pH 3.0 (Fig. 1c, d). Even when pH would decrease to extreme values of 2 and 1.5, the enzyme maintained, respectively, 90 and 80 % of its maximum activity (Fig. 1). For the pH range of 2 to 10, we used ABF buffer, and for pH values below 2, the assays were performed in HCl/KCl buffer.

Effect of pH and temperature in ThCel7B activity on CMC. The temperature influence was measured in sodium citrate buffer pH 5.0 and 1 % CMC as a substrate, for both constructions a CCD and b ThCel7B. Optimal pH identification assay was performed at different pH values (1–2 in HCl/KCl and 2–10 in ABF buffer) for c CCD and d ThCel7B
To investigate the influence of the buffer composition on enzymatic activity of ThCel7B, the catalytic domain was tested in the presence of different acidic buffers (Fig. 2). The enzyme showed a slight preference for citrate buffer, when compared to other buffers at the same pH, and the optimal activity around pH 3 was observed in all the tested buffers (Fig. 3).

CCD activity at different acidic buffers. The graph shows the impact of different acidic buffers in the CMC activity of ThCel7B CCD

Thermal stability curves at different pH. Plots of normalized fluorescence intensity as a function of temperature for a ThCel7B and b CCD to visualize the thermal shift between different pH conditions. c Variation of the melting temperature (T m) determined using the Boltzmann model for all tested pH (2 to 10). The highest T m value was observed for pH 5
Thermal shift (ThermoFluor) analysis
Once the pH dependence of ThCel7B enzymatic activity was determined, we evaluated its thermal stability in the same pH range using thermal shift method. Although both ThCel7B and its CCD showed an optimum activity at pH 3, ThermoFluor experiments revealed that the enzyme thermostability is highest at pH 5.0 (Fig. 3). This suggests that the enzyme requires some conformational flexibility to reach its optimal activity, and this flexibility is not assured by the most stable protein structure conformation. The melting temperature (T m) variation for all tested pH (Fig. 3c) confirms the acidophilic behavior of the enzyme and at pH 7.0 and higher values of T m, the protein stability decreases dramatically.
Residual activity assays
Residual activity of both ThCel7B and its CCD were determined by incubating the enzymes at 55 °C in ABF buffer at pH 3. Enzymatic activity against CMC was followed over time. Both enzymes showed significant stability under such conditions. The full-length enzyme maintained 80 % of its initial activity after approximately 10 h of incubation at 55 °C, whereas its CCD kept at least 80 % of its maximum activity for about 6 h (Fig. 4). Even after 48 h of incubation, ThCel7B CCD still retained about 20 % of its original activity, whereas ThCel7B reached 20 % activity level only after 72 h of continuous incubation. When the same residual activity was determined incubating the enzymes in citrate buffer at pH 5.0, the result was surprising. After 2 months of incubation, the enzyme presented practically the same initial activity (Fig. 4). This result corroborates with the ThermoFluor data, which clearly revealed that the highest enzyme thermostability occurs at pH 5.0.

Thermal resistance of ThCel7B. Residual enzymatic activity was measured at 55 °C and pH 3 and 5, over a time course of 100 days
ThCel7B substrate specificity
The substrate specificity was determined by measuring released reducing sugars from several polysaccharide substrates. Both the catalytic domain and the full-length enzyme displayed the highest activity on β-glucan, followed by xyloglucan, lichenan, CMC, pNPC, rye arabin xylan, and xylan (oat spelt and beechwood) (Fig. 5a). After 4 h of incubation, ThCel7B also showed significant, albeit lower, activity against filter paper, PASC, Sigmacell 20, Avicel, and cellulose (Fig. 5b).

The substrate specificity. Graph of specific activities of ThCel7B and its CCD towards a set of substrates determined with incubation time of a 15 min and b 4 h
ThCel7B kinetic parameters
When xyloglucan was used as a substrate, the K M for ThCel7B and ThCel7B CCD were 1.98 ± 0.47 g.L−1 and 2.9 ± 0.6 g.L−1, while V max were 0.22 ± 0.095 μM.s−1 and 0.012 ± 0.009 μM.s−1, respectively. In this case, the calculated K cat for the full-length enzyme and its CCD were, respectively, 0.45 and 0.054 s−1, which indicated high catalytic efficiency of the full-length enzyme against this substrate. Deletion of the CBM leads to a significant loss of V max and catalytic efficiency of the enzyme, whereas K M was much less affected.
Circular dichroism
We applied circular dichroism spectroscopy to study the ThCel7B and its catalytic domain secondary structures and the influence of pH on their structural stability. Figure 6 shows a general shape of the spectra at 25 °C, which is characteristic of β-class proteins, comparable to the secondary structure of other cellulases (Voutilainen et al. 2009; Colussi et al. 2012). The far UV CD spectra of both constructs did not display a negative band around 222 nm, indicating a relatively low content of α-helices (Fig. 6).

Effect of pH on the secondary structure of ThCel7B at 25 °C monitored by circular dichroism spectroscopy. a ThCel7B CCD and b full-length ThCel7B endoglucanase
These data also show that only for highest pH values (pH 8.0–10.0), for both enzyme constructs, the spectral profiles were progressively altered, indicating a strong effect of high pH values on the secondary structure of the enzyme and their acidophilic behavior (Fig. 6).
Scanning electron microscopy (SEM)
Given elevated enzymatic activity of ThCel7B against β-glucan and xyloglucan, we set out to determine how it would affect substrates rich in these polysaccharides. Scanning electron microscopy images of oat spelt, before and after undergoing enzymatic hydrolysis with ThCel7B and its catalytic domain at two different pH values, are presented in Figs. 7 and 8. Oat spelt is rich in β-glucan, and according to our results, ThCel7B has high specific activity against this substrate.

Scanning electron microscopy images exhibiting the hydrolytic effect of ThCel7B on oat spelt. Images of the reaction control and the effects of the enzyme action after 12 h at pH 3 and 5 analyzed at different magnifications: scale bars of a 20 μm, b 10 μm, and c 3 μm. The arrows indicate some flat regions in the control that becomes to form voids and craters on the surface (white), the formation of eroded spherical particles (yellow) and the degradation of polyhedral grains (red) after enzyme hydrolysis

– Scanning electron microscopy images exhibiting the hydrolytic effect of ThCel7B on oat spelt. Images of the reaction control and the effects of the enzyme action after 12 h at pH 3 and 5 analyzed at different magnifications: scale bars of a 20 μm, b 10 μm, and c 3 μm. The arrows indicate the formation of craters (yellow) and holes (red) as results of enzyme degradation
Oat spelt surface before enzymatic hydrolysis is formed by granules of spherical or polyhedral shapes, as can be observed in Fig. 7. These images were obtained for control samples, kept at the same temperature, reaction time, and reactant concentrations of the hydrolysis media, except for the presence of the enzyme. Spherical shape particles in the non-hydrolyzed samples (Fig. 7, control) have a very smooth surface, while polyhedral grains may present more irregular surfaces, characterized by some particle deposits and small cavities on the surface (Fig. 7). The microscopy analysis of oat spelt subjected to ThCel7B action for 24 h revealed numerous holes on the surfaces of both spherical and polyhedral grains of the oat spelt. Eroded spherical particles are indicated by the yellow arrows, while examples of degraded polyhedral grains are indicated by the red arrows in Fig. 7. Oat substrates also present flat regions, such as the one indicated by the white arrow in Fig. 7a—control, which can also be modified by the formations of voids and craters on the surface after enzymatic hydrolysis (Fig. 7b). The formation of cavities on the substrate surface is clear evidence of the endoglucanase action.
In Fig. 7c (higher magnification), one can observe the degradation effects in more detail, which reveal that the flat surface in the control becomes rough and very irregular in the hydrolyzed samples. The pH effect could not be quantified by these analyses. Though ThCel7B is expected to have higher efficiency at lower pH values (around 2–3), the set of sample images obtained at pH = 3 and pH = 5, were similar.
Figure 8 shows SEM images for the oat spelt substrates after the action of ThCel7B catalytic domain only. The images were also obtained at two different pH values and at different magnifications. The catalytic domain alone shows a similar effect on the oat substrate when compared to the full-length enzyme. The formation of holes and craters on the spherical and polyhedral grains is evident when images after hydrolysis in both pH values are compared to the images of the control (Fig. 8). It is worthwhile highlighting the effect of the ThCel7B CCD resulting in the corrosion of the plates on a huge substrate grain.
Pattern of cello-oligosaccharides enzymatic hydrolysis
To gather detailed information on the pattern of ThCel7B hydrolysis of cello-oligosaccharides, we used capillary electrophoresis to analyze the degradation APTS-labeled cellohexaose—by ThCel7B (Fig. 9). A similar hydrolytic pattern was observed for both the full-length enzyme and its CCD, which is characterized by the release of cellobiose (C2) followed by cellotriose (C3) and cellotetraose (C4). An absence of cellopentaose (C5) and glucose (G1) suggests that ThCel7B is unable (or very inefficient) in releasing glucose. Larger quantity of C4 and residual amount of C6 observed after ThCel7B hydrolysis might indicate that the full-length enzyme was less efficient in APTS-labeled cellohexaose degradation as compared to its CCD (Fig. 9). These results show that ThCel7B, as an endoglucanase, presents a typical random cleavage pattern of oligosaccharides.

Capillary electrophoresis of APTS-labeled oligosaccharide products. Products released by ThCel7B and CCD after hydrolysis of the substrate C6
The capillary electrophoresis was only used to gather qualitative information about the enzyme cleavage profile, and these results cannot be considered to be a quantitative analysis of hydrolysis products. The larger quantity of C4 and the residual amounts of C6, observed after ThCel7B hydrolysis, might indicate that the full-length enzyme has lower efficiency of the APTS-labeled cellohexaose degradation as compared to CCD. However, to be able to compare directly, the differences between the full-length enzyme and the CCD additional set of experiments using different techniques which would allow for quantitative characterization of the products (such as HPLC measurements with amperometric detection, for example) should be conducted.
Discussion
Previous studies demonstrated that endoglucanases from GH7 family are important for lignocellulosic biomass degradation and show synergistic effects with cellobiohydrolases in the process of enzymatic hydrolysis (Momeni et al. 2013). The pH profile for ThCel7B suggests that it has a strongly acidophilic character. The experimentally determined optimum pH for both full-length ThCel7B and its CCD was surprisingly low (pH 3.0), and they both retained about 80 % of their maximum activity at pH down to 1.5 (Fig. 1). On the other hand, the enzymes also maintained about 65 % of its activity at pH 5.0to 5.5, pH range at which ThCel7B was most stable (Fig. 1). The enzyme optimum temperature was similar to the optima temperature of other fungal cellulases (T opt = 55 °C). Most of the fungal endoglucanases has optima activity at a temperature around 50 to 55 °C (Ding et al. 2001; Saha 2004). These biochemical characteristics of ThCel7B might be of biotechnological interest, mainly for second-generation ethanol production, which frequently involves processes run at acidic pH values (Miotto et al. 2014).
Such a strongly acidic behavior is unusual for endoglucanases, which usually requires moderate acidic conditions for optimal activity (Okada et al. 1998; Hasper et al. 2002; Shumiao et al. 2010; Xiang et al. 2014). The GH5 family endoglucanase from the filamentous fungus Gloeophyllum trabeum (GtCel5B) is one of the few described highly acidic endoglucanases and has an optimum pH of 3.5 (Kim et al. 2012). However, when the pH is lowered to 2.0, the activity of GtCel5B decreases more than 80 % (Kim et al. 2012) while ThCel7B retains more than 70 % of its activity down on pH 1.5 (Fig. 1c, d). GtCel12A endoglucanase also has a considerable acidophilic character with pH optimum at 4.5 (Miotto et al. 2014). The enzyme was capable to retain about 40 % of its activity at pH 1.0. At the same time, cellulases from A. niger and closely related T. reesei species display pH optima at slightly acidic pH values (pH 5.0 to 5.5). A wide pH window of ThCel7A enzymatic activity permits its use in a mix with other fungal cellulases active at pH values around 5.0, where ThCel7A is most stable.
The reasons why recombinant T. harzianum Cel7B has such low pH optimum are currently unknown and deserve further investigations. The experimentally determined molecular weight of the recombinant ThCel7B expressed in the A. niger expression system was considerably higher than its theoretically predicted molecular mass computed on the basis of the amino acid sequence, whereas its catalytic domain molecular weight estimated by SDS-PAGE analysis was close to the theoretical value (Fig. S1 in the Supplementary Material). This indicates that the full-length construct is highly glycosylated at the linker peptide region, as it has been observed for other fungal endoglucanases (Sandgren et al. 2005). Other recombinant enzymes produced in the A. niger expression system display low pH optimum (see, for example, Miotto et al. 2014), which could in theory be attributed to the differences in glycosylation profile of the recombinant proteins produced in such system. However, our data also suggest that CCD has few or no post-translational modifications and it has the same highly acidophilic character of the full-length enzyme (Fig. 1). A small number of post-translational modifications of the Cel7B CCD domain is consistent with the previous studies of glycosylation profile of the T. reesei Cel7B CCD (García et al. 2001; Eriksson et al. 2004), and its highly acidophilic character argue against post-translational modifications introduced by the expression system being the main reason for low pH optimum of the enzyme. It is known that Humicola insolens and Fusarium oxysporum Cel7B endoglucanases have considerably higher pH optima and that minor amino acid substitutions in the active site of the enzymes (incorporation of a His residue) could account for the differences in the pH optimum (Becker et al. 2001). Thus, we argue that subtle modifications in the active site of the enzyme might be responsible for its highly acidic character. However, detailed investigations of ThCel7B biochemical features will have to await determination of its three-dimensional structure and comprehensive site-directed mutagenesis analysis.
Our optimum pH studies of ThCel7B were corroborated by thermal displacement experiments under different pH conditions. Results depicted in Fig. 3 show ThCel7B and its CCD maintained tertiary structure integrity against temperature increase better at lower pH values, suggesting a higher temperature stability of these proteins under acid conditions. The maximum stability was observed at pH 5.0, two pH units higher than the pH optimum than the enzyme (Fig. 3c). This might indicate that for the optimum enzymatic activity, ThCel7B must be flexible and that additional enzyme stability might have penalties for its enzymatic activity. A similar difference in pH conditions for the enzyme optimum activity vs. stability has been previously observed for GtCel12A (Miotto et al. 2014). Residual enzymatic activity of ThCel7B at highly acidic conditions was quite considerable, since the enzyme maintained 80 % of its initial activity after more than 8 h of incubation at pH 3.0 and even after 72 h it still retained about 20 % of the initial activity (Fig. 4). When we compare our results with other acidic endoglucanases reported in the literature, ThCel7B is significantly more stable. Cel5A from G. trabeum, for example, lost its total activity in acid conditions with less than 2 h (Kim et al. 2012).
One of the important biotechnological features of ThCel7B is a wide range of substrates it can hydrolyze (Fig. 5). Our substrate specificity studies showed that the enzyme is mostly active against β-glucan, xyloglucan, lichenan, and CMC and displays somewhat lower but still considerable activity against arabinoxylan, PASC, filter paper, Avicel, and Sigmacell. This complies with the enzyme preference toward substrates with β-1,4 linkages and at the same time reveals broad range of ThCel7B hydrolytic activities against main components of the plant cell wall.
T. reesei Cel7B (TrCel7B) has been shown to remove amorphous regions of cellulose substrate exposing crystalline regions for exoglucanase ablation and displaying considerable synergy with the latter enzyme (Ganner et al. 2012). TrCel7B was shown to be a nonspecific endo-β-1,4-glucanase which has enzymatic activity against hydroxyethylcellulose as well as beechwood and grass xylan (Biely et al. 1991), but not homopolymeric mannans (Bailey et al. 1993). In addition, TrCel7B is capable to enzymatically hydrolyse konjac glucomannan resulting in production of glucomannooligosaccharides with non-reducing end mannose (M) and reducing end glucose (G) (Mikkelson et al. 2013). Furthermore, T. reesei Cel7B has been reported to readily cleave xylan, arabinoxylan (Suurnäkki et al. 2000), and xyloglucan (Vlasenko et al. 2010). Our experimental studies of T. harzianum Cel7B substrate specificity indicate a similar broad range of polysaccharide recognition and hydrolysis, indicating that the molecular structure of Cel7B is probably well adapted for recognitions of a wide range of plant cell wall polysaccharides (Fig. 5). Such a wide spectrum of activities against plant biomass polysaccharides is highly attractive for the enzymatic hydrolysis of such heterogeneous and complex substrates.
To allow for efficient enzymatic hydrolysis, lignocellulosic biomass must be pretreated before enzymatic hydrolysis. However, the pretreatments (particularly hydrothermal and steam-explosion pretreatments) often generate inhibitory compounds such as xylooligosaccharides (XOS) and glucooligosaccharides (GOS) as a result of depolymerization of the xylan and mixed linkage β-glucans, respectively (Kont et al. 2013). These oligosaccharides have been shown to be approximately 100 times stronger inhibitors for T. reesei cellobiohydrolases (CBHs) than cellobiose (Kont et al. 2013). Since CBHs are known to be the most important enzymatic component in cellulose degradation, efficient enzymatic hydrolysis of pretreated lignocellulose can be achieved only if XOS and GOS are degraded and their inhibitory effect is reduced. TrCel7B was shown to be the most efficient single enzyme component in this process (Kont et al. 2013). The relative efficiency of TrCel12A and TrCel7B in degrading of XOS and GOS is apparently due to their inherent hemicellulase activity (Biely et al. 1991; Karlsson et al. 2002; Varnai et al. 2011).
Furthermore, Cel7B addition promotes enzymatic hydrolysis of plant biomass and enhances its efficiency. It was shown that enzymatic hydrolysis of alkaline peroxide pretreated alfalfa hay and barley straw by rumen enzymes and commercial cellulases was significantly enhanced by addition of endoglucanase from GH7 family (Badhan et al. 2014). Augmented glucose release as a consequence of the endoglucanase addition was thought to be the result of its broad substrate specificity (Badhan et al. 2014). Cel7B might improve overall cell wall conversion through the hydrolysis of polysaccharides covering the cellulose microfibers and thus making them exposed to cellulase action. In addition, Cel7B are capable of generating more reducing and non-reducing ends, available to Cel7A and Cel7B attack (Badhan et al. 2014). Based on ThCel7B biochemical characterization, we speculate that this enzyme might be equally important for biomass enzymatic hydrolysis, not only directly synergizing with exoglucanases in cellulose microfibers depolymerization but also making them available for cellobiohydrolase action through active hydrolysis of other polysaccharides of the plant cell wall matrix.
To better understand effects of ThCel7B action upon biomass, we provided here the first visualization of Cel7B action on oat substrates (Figs. 7 and 8). In general, very few studies concerning the visualization of cellulase activity by the different microscopy techniques available (electron microscopy or probe microscopy) can be found in literature (Ding et al. 2001; Lee et al. 2000; Wang et al. 2012). Besides, most of the published work deals with exoglucanases, particularly celobiohydrolase I (CBHI) acting on pure cellulose substrates. A comprehensive review including important contributions to the area can be found in (Bubner et al. 2013).
The formation of holes on the substrate, as a consequence of enzyme action, was also observed by Lee and collaborators (Lee et al. 2000), who used atomic force microscopy to image cotton fibers hydrolyzed by hexachloropalladate-inactivated TrCBHI. The holes left by the enzyme on the fibers after incubation had approximately a 13-nm diameter and the authors assigned their formation to the indentation of CBHI binding domains on the cellulose surface. Although the imaging scales are quite different in two cases (the published images had a 400-nm side, while our images have approximately a 10-μm side) and the enzyme types are different as well, our results may represent an important step in the visualization of changes introduced in the sample during enzymatic hydrolysis.
It is also important to highlight the fact that the studied substrates are very different. The complexity of the oat substrate makes the visualization of the enzyme effects more difficult, though by comparing them with the controlling samples the enzyme effect is quite clear. Cellulose substrates free of hemicellulose, lignin, and any other cell wall component are much easier to analyze, which explains why algae and bacterial cellulose are frequently used as model substrates. However, cellulase activity is inherently related to substrate type and morphology and since ThCel7B has relatively low specific activity on the substrates rich in crystalline cellulose, more complex substrates such as oat spelt had to be used in this particular case.
In summary, a β-1,4-endoglucanase from the T. harzianum strain IOC-3844 (ThCel7B) was successfully heterologously expressed in A. niger. Detailed biochemical and biophysical characterization showed that recombinant ThCel7B is highly active on a wide range of plant cell wall polysaccharides including β-glucan, xyloglucan, lichenan, CMC, pNPC, rye arabin xylan, xylan, and also amorphous and crystalline cellulose. The enzyme has an optimum pH of 3.0 and is thermally most stable at pH 5.0. Our SEM analysis shows clear morphological changes caused by the enzyme hydrolytic action on oat spelt. The high stability of ThCel7B in acid pH values and a wide range of the plant cell wall polysaccharides it is active upon make the enzyme a promising candidate for several biotechnological applications, such as plant cell wall depolymerization and cellulosic ethanol production.
References
Badhan A, Wang Y, Gruninger R, Patton D, Powlowski J, Tsang A, McAllister T (2014) Formulation of enzyme blends to maximize the hydrolysis of alkaline peroxide pretreated alfalfa hay and barley straw by rumen enzymes and commercial cellulases. BMC Biotechnol 14:31
Bailey MJ, Siika-aho M, Valkeajärvi A, Penttilä ME (1993) Hydrolytic properties of two cellulases of Trichoderma reesei expressed in yeast. Biotechnol Appl Biochem 17:65–76
Becker D, Braet C, Brumer H, Claeyssens M, Divne C, Fagerström BR, Harris M, Jones TA, Kleywegt GJ, Koivula A, Mahdi S, Piens K, Sinnott ML, Ståhlberg J, Teeri TT, Underwood M, Wohlfahrt G (2001) Engineering of a glycosidase family 7 cellobiohydrolase to more alkaline pH optimum: the pH behaviour of Trichoderma reesei Cel7A and its E223S/A224H/L225V/T226A/D262G mutant. Biochem J 356:19–30
Bhat MK (2000) Cellulases and related enzymes in biotechnology. Biotechnol Adv 18:355–383
Biely P, Vrsanská M, Claeyssens M (1991) The endo-1,4-β-glucanase I from Trichoderma reesei. Eur J Biochem 200:157–163
Bubner P, Plank H, Nidetzky B (2013) Visualizing cellulase activity. Biotechnol Bioeng 110:1529–1549
Buckeridge MS, Dos Santos WD, De Souza AP (2010) Routes for cellulosic ethanol in Brazil. In: LAB C (ed) Sugarcane bioethanol: R&D for productivity and sustainability. Edgard Blucher, Sao Paulo, Brazil, pp. 365–380
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238
Chundawat SPS, Beckham GT, Himmel ME, Dale BE (2011) Deconstruction of lignocellulosic biomass to fuels and chemicals. Annu Rev Chem Biomol Eng 2:121–145
Colussi F, Garcia W, Rosseto FR, de Mello BL, de Oliveira NM, Polikarpov I (2012) Effect of pH and temperature on the global compactness, structure, and activity of cellobiohydrolase Cel7A from Trichoderma harzianum. Eur Biophys J 41:89–98
de Castro AM, de Albuquerque de Carvalho ML, Leite SG, Pereira Jr N (2010) Cellulases from Penicillium funiculosum: production, properties and application to cellulose hydrolysis. J Ind Microbiol Biotechnol 37:151–158
Dias MOS, Junqueira TL, Cavalett O, Cunha MP, Jesus CDF, Rossell CEV, Maciel Filho R, Bonomi A (2012) Integrated versus stand-alone second generation ethanol production from sugarcane bagasse and trash. Bioresour Technol 103(1):152–161
Ding SJ, Ge W, Buswell JA (2001) Endoglucanase I from the edible straw mushroom, Volvariella volvacea. Purification, characterization, cloning and expression. Eur J Biochem 268(22):5687–5695
Eriksson T, Stals I, Collén A, Tjerneld F, Claeyssens M, Stålbrand H, Brumer H (2004) Heterogeneity of homologously expressed Hypocrea jecorina (Trichoderma reesei) Cel7B catalytic module. Eur J Biochem 271:1266–1276
Ganner T, Bubner P, Eibinger M, Mayrhofer C, Plank H, Nidetzky B (2012) Dissecting and reconstructing synergism: in situ visualization of cooperativity among cellulases. J Biol Chem 287:43215–43222
García R, Cremata JA, Quintero O, Montesino R, Benkestock K, Ståhlberg J (2001) Characterization of protein glycoforms with N-linked neutral and phosphorylated oligosaccharides: studies on the glycosylation of endoglucanase 1 (Cel7B) from Trichoderma reesei. Biotechnol Appl Biochem 33:141–152
Hasper AA, Dekkers E, van Mil M, van de Vondervoort PJ, de Graaff LH (2002) EglC, a new endoglucanase from Aspergillus niger with major activity towards xyloglucan. Appl Environ Microbiol 68:1556–1560
Hartley JL, Temple GF, Brasch MA (2000) DNA cloning using in vitro site-specific recombination. Genome Res 10:1788–1795
Hilden L, Valjamae P, Johansson G (2005) Surface character of pulp fibres studied using endoglucanases. J Biotechnol 118:386–397
Himmel ME, Ding SY, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315:804–807
Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VG (2012) Novel enzymes for the degradation of cellulose. Biotechnol Biofuels 5:45
Kalsner I, Hintz W, Reid LS, Schachter H (1995) Insertion into Aspergillus nidulans of functional UDP-GlcNAc: α 3-D-mannoside β-1,2-N-acetylglucosaminyl-transferase I, the enzyme catalysing the first committed step from oligomannose to hybrid and complex N-glycans. Glycoconj J 12:360–370
Karlsson J, Siika-aho M, Tenkanen M, Tjerneld F (2002) Enzymatic properties of the low molecular mass endoglucanases Cel12A (EG III) and Cel45A (EG V) of Trichoderma reesei. J Biotechnol 99:63–78
Katzen F (2007) Gateway(®) recombinational cloning: a biological operating system. Expert Opin Drug Discov 2:571–589
Kim HM, Lee YG, Patel DH, Lee KH, Lee DS, Bae HJ (2012) Characteristics of bifunctional acidic endoglucanase (Cel5B) from Gloeophyllum trabeum. J Ind Microbiol Biotechnol 39:1081–1089
Kont R, Kurasin M, Teugjas H, Valjamae P (2013) Strong cellulase inhibitors from the hydrothermal pretreatment of wheat straw. Biotechnol Biofuels 6:135
Lee I, Evans BR, Woodward J (2000) The mechanism of cellulase action on cotton fibers: evidence from atomic force microscopy. Ultramicroscopy 82:213–221
Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS (2002) Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev 66:506–577
Mikkelson A, Maaheimo H, Hakala TK (2013) Hydrolysis of konjac glucomannan by Trichoderma reesei mannanase and endoglucanases Cel7B and Cel5A for the production of glucomannooligosaccharides. Carbohydr Res 372:60–68
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Miotto LS, de Rezende CA, Bernardes A, Serpa VI, Tsang A, Polikarpov I (2014) The characterization of the endoglucanase Cel12A from Gloeophyllum trabeum reveals an enzyme highly active on β-glucan. PLoS One 9:e108393
Momeni MH, Payne CM, Hansson H, Mikkelsen NE, Svedberg J, Engström A, Sandgren M, Beckham MG, Ståhlberg J (2013) Structural, biochemical, and computational characterization of the glycoside hydrolase family 7 cellobiohydrolase of the tree-killing fungus Heterobasidion irregulare. J Biol Chem 8:5861–5872
Naran R, Pierce ML, Mort AJ (2007) Detection and identification of rhamnogalacturonan lyase activity in intercellular spaces of expanding cotton cotyledons. Plant J 50:95–107
Okada H, Tada K, Sekiya T, Yokoyama K, Takahashi A, Tohda H, Kumagai H, Morikawa Y (1998) Molecular characterization and heterologous expression of the gene encoding a low-molecular-mass endoglucanase from Trichoderma reesei QM9414. Appl Environ Microbiol 64:555–563
Payne CM, Resch MG, Chen L, Crowley MF, Himmel ME, Taylor LE, Sandgren M, Ståhlberg J, Stals I, Tan Z, Beckham GT (2013) Glycosylated linkers in multimodular lignocellulose-degrading enzymes dynamically bind to cellulose. Proc Natl Acad Sci U S A 110:14646–14651
Penttilä M, Nevalainen H, Rättö M, Salminen E, Knowles J (1987) A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene 61:155–164
Punt PJ, van Biezen N, Conesa A, Albers A, Mangnus J, van den Hondel C (2002) Filamentous fungi as cell factories for heterologous protein production. Trends Biotechnol 20:200–206
Saha BC (2004) Production, purification and properties of endoglucanase from a newly isolated strain of Mucor circinelloides. Process Biochem 39:1871
Sanchez OJ, Cardona CA (2008) Trends in biotechnological production of fuel ethanol from different feedstocks. Bioresour Technol 99:5270–5295
Sandgren M, Ståhlberg J, Mitchinson C (2005) Structural and biochemical studies of GH family 12 cellulases: improved thermal stability, and ligand complexes. Prog Biophys Mol Biol 89:246–291
Schuster A, Schmoll M (2010) Biology and biotechnology of Trichoderma. Appl Microbiol Biotechnol 87:787–799
Schwarz W (2001) The cellulosome and cellulose degradation by anaerobic bacteria. Appl Microbiol Biotechnol 56:634–649
Serpa VI, Polikarpov I (2011) Enzymes in bioenergy. In: Buckeridge MS, Goldman GH (eds) Routes to cellulosic ethanol. Springer, New York, London, pp. 97–115
Shumiao Z, Huang J, Zhang C, Deng L, Hu N, Liang Y (2010) High-level expression of an Aspergillus niger endo-β-1,4-glucanase in Pichia pastoris through gene codon optimization and synthesis. J Microbiol Biotechnol 20:467–473
Storms R, Zheng Y, Li H, Sillaots S, Martinez-Perez A, Tsang A (2005) Plasmid vectors for protein production, gene expression and molecular manipulations in Aspergillus niger. Plasmid 53:191–204
Sun Y, Cheng J (2002) Hydrolysis of lignocellulosic materials for ethanol production: a review. Bioresour Technol 83(1):1–11
Suurnäkki A, Tenkanen M, Siika-Aho M, Niku-Paavola ML, Viikari L, Buchert J (2000) Trichoderma reesei cellulases and their core domains in the hydrolysis and modification of chemical pulp. Cellulose 7:189–209
Teeri TT (1997) Crystalline cellulose degradation: new insight into the function of cellobiohydrolases. Trends Biotechnol 15:160–167
Varnai A, Huikko L, Pere J, Siika-Aho M, Viikari L (2011) Synergistic action of xylanase and mannanase improves the total hydrolysis of softwood. Bioresour Technol 102:9096–9104
Vlasenko E, Schülein M, Cherry J, Xu F (2010) Substrate specificity of family 5, 6, 7, 9, 12, and 45 endoglucanases. Bioresour Technol 101:2405–2411
Voutilainen SP, Boer H, Alapuranen M, Janis J, Vehmaanpera J, Koivula A (2009) Improving the thermostability and activity of Melanocarpus albomyces cellobiohydrolase Cel7B. Appl Microbiol Biotechnol 83:261–272
Wang J, Quirk A, Lipkowski J, Dutcher JR, Hill C, Mark A, Clarke AJ (2012) Real-time observation of the swelling and hydrolysis of a single crystalline cellulose fiber catalyzed by cellulase 7B from Trichoderma reesei. Langmuir 28:9664–9672
Wood TM (1988) Preparation of crystalline, amorphous, and dyed cellulase substrates. Methods Enzymol 160:19–25
Xiang L, Li A, Tian C, Zhou Y, Zhang G, Ma Y (2014) Identification and characterization of a new acid-stable endoglucanase from a metagenomic library. Protein Expr Purif 102:20–26
Yelton MM, Hamer JE, Timberlake WE (1984) Transformation of Aspergillus nidulans by using a trpC plasmid. Proc Natl Acad Sci U S A 81:1470–1474
Acknowledgments
We would like to acknowledge support of the Brazilian funding agencies Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) via grants #2008/56255-9, 2009/52840-7, 2009/05328-9, 2010/18773-8, 2011/20977-3, and 2011/05712-3; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) via grants #490022/2009-0, 301981/2011-6 and, 400045/2012–5; CAPES and Universidade de São Paulo via grants “Centro de Instrumentação para estudos avançados de materiais nanoestruturados e biossistemas” and “Núcleo de Apoio à Pesquisa em Bioenergia e Sustentabilidade (NAPBS).”
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 431 kb)
Rights and permissions
About this article

Cite this article
Pellegrini, V.O.A., Serpa, V.I., Godoy, A.S. et al. Recombinant Trichoderma harzianum endoglucanase I (Cel7B) is a highly acidic and promiscuous carbohydrate-active enzyme. Appl Microbiol Biotechnol 99, 9591–9604 (2015). https://doi.org/10.1007/s00253-015-6772-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6772-1