
Abstract
Due to its elevated cellulolytic activity, the filamentous fungus Trichoderma harzianum (T. harzianum) has considerable potential in biomass hydrolysis application. Cellulases from Trichoderma reesei have been widely used in studies of cellulose breakdown. However, cellulases from T. harzianum are less-studied enzymes that have not been characterized biophysically and biochemically as yet. Here, we examined the effects of pH and temperature on the secondary and tertiary structures, compactness, and enzymatic activity of cellobiohydrolase Cel7A from T. harzianum (Th Cel7A) using a number of biophysical and biochemical techniques. Our results show that pH and temperature perturbations affect Th Cel7A stability by two different mechanisms. Variations in pH modify protonation of the enzyme residues, directly affecting its activity, while leading to structural destabilization only at extreme pH limits. Temperature, on the other hand, has direct influence on mobility, fold, and compactness of the enzyme, causing unfolding of Th Cel7A just above the optimum temperature limit. Finally, we demonstrated that incubation with cellobiose, the product of the reaction and a competitive inhibitor, significantly increased the thermal stability of Th Cel7A. Our studies might provide insights into understanding, at a molecular level, the interplay between structure and activity of Th Cel7A at different pH and temperature conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cellulases are important enzymes that can hydrolyze the β-1,4-linkage between the glucose units of the cellulose chain. These enzymes are classified into glycoside hydrolase (GH) families (http://www.cazy.org/Glycoside-Hydrolases.html) (Henrissat and Davies 1997) and can be divided into three types: endo-1,4-β-D-glucanase (EC 3.2.1.4), 1,4-β-D-cellobiohydrolase (EC 3.2.1.91), and β-glucosidase (EC 3.2.1.21). Cellulases are being used in several industrial applications, for example, in the pulp and paper, textile, and detergent industries, and more recently in industrial conversion of cellulose-containing biomass to fermentable sugars as an alternative to fossil fuels (Gray et al. 2006; Kumar et al. 2008).
A large part of currently produced bioethanol is obtained from starch, corn, or sucrose, thus competing directly or indirectly with food. Therefore, biomass utilization offers an alternative carbon source for second-generation biofuel production (Bommarius et al. 2008; Kumar et al. 2008). Cellulose, a polysaccharide composed of β-d-glucose units joined by 1,4-glycosidic bonds, is the major component of the cell wall of plants and the most abundant polymer in nature (Igarashi et al. 2006; Kumar et al. 2008). The degree of crystallinity differs with the source of the cellulose, and crystalline regions are specifically resistant to enzymatic hydrolysis. Thus, degradation of cellulose is one of the most important processes in the carbon cycle, normally involving a combined action of several synergistically functioning enzymes.
The cellulolytic systems of filamentous fungi Trichoderma reesei (Hypocrea jecorina) and Humicola insolens are among the most thoroughly functionally, biochemically, and structurally studied (von Ossowski et al. 2003; Kumar et al. 2008). These enzymatic mixtures consist of at least of two cellobiohydrolases, Cel7A and Cel6A (formerly CBHI and CBHII), that act, respectively, upon reducing and nonreducing ends of crystalline cellulose-releasing cellobiose units, and several endoglucanases that hydrolase amorphous regions of cellulose, thus decreasing the degree of polymerization of the substrate and generating new cellulose chain termini. Enzymatic action of β-glucosidades completes depolymerization by hydrolysis of cellobioses in glucose monomers (Kubicek et al. 1993; Kleywegt et al. 1997). The cellobiohydrolase Cel7A is the major component of total extract produced by Trichoderma and is the key enzyme for efficient degradation of crystalline cellulose. For example, removal of the Cel7A gene reduces overall activity of Trichoderma reesei (T. reesei) on crystalline cellulose by 70%, and inactivation of Cel7A during cellulose hydrolysis is the major factor responsible for the decrease in the hydrolysis rate (Davies and Henrissat 1995; Teeri 1997).
Structural studies show that Cel7A from T. reesei is composed of a large catalytic core domain joined by an O-glycosilated linker peptide to a small cellulose-binding domain that is responsible for anchoring the enzyme on solid substrate surfaces (Davies and Henrissat 1995; Hui et al. 2002; Boraston et al. 2004). Glycosylation plays an important role in protecting the linker peptide against proteolysis. The linker seems to be important for enhanced synergy between the catalytic core domain and the cellulose-binding domain on its natural substrate, since the shortening or the deletion of the linker drastically reduces enzymatic activity on crystalline cellulose (Shen et al. 1991; Srisodsuk et al. 1993). Crystallographic studies reveal that the catalytic core domain has a large enclosed tunnel structure formed by a β-sandwich fold. The tunnel is 50 Å long and contains the active site to mediate the hydrolysis of cellulose from the chain ends (Divne et al. 1994; Davies and Henrissat 1995; Munoz et al. 2001; Grassick et al. 2004; Gray et al. 2006; Parkkinen et al. 2007).
Trichoderma harzianum (T. harzianum or Hypocrea lixii) is a filamentous fungi that secrets a well balanced cellulolytic complex able to efficiently hydrolyze cellulolytic substrates into monomeric glucose. Due to its elevated cellulolytic activity, the fungus T. harzianum has considerable potential in biomass hydrolysis applications (Castro et al. 2010). The mixture of cellulases produced by T. reesei has been widely used as a model in studies of cellulose breakdown. However, cellulases from T. harzianum are less-studied enzymes that have not been characterized biophysically and biochemically as yet. Cel7A from T. harzianum (NCBI accession number AAF36391.1) is 89% homologous to T. reesei Cel7A and presumably also has a bimodular molecular organization composed of a large catalytic domain linked to a small carbohydrate-binding domain by a flexible linker peptide.
Industrial conditions frequently impose restrictions on the pH range applicable at a particular step of the processes. The use of high temperatures generally accelerates rates of enzymatic reactions, reduces bacterial contamination, and in most cases, increases the solubility of substrates and thus is important for biotechnological applications and in bioreactors. High temperature and inadequate pH conditions, on the other hand, will eventually inactivate and/or denature enzymes. To comprehend cellulase inactivation at a molecular level, we studied the effects of pH and temperature on enzymatic activity and secondary and tertiary structures of cellobiohydrolase Cel7A from fungus T. harzianum by employing several biophysical and biochemical tools. This work is the first study that examined the effect of pH and temperature on the structural stability and enzymatic activity of Cel7A from T. harzianum.
Materials and methods
Materials
Protein standards used as sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) markers, Avicel® PH101, and cellobiose were purchased from Sigma Aldrich. DEAE-Sephadex A-50 (5 × 40 cm) column was purchased from GE Healthcare. Sypro Orange dye was purchased from Molecular Probes. All chemicals and reagents used in this study were of the highest purity analytical grade. To cover a pH range of 2–6 and 7–10, phosphate/citric acid and Gly/NaOH were chosen as buffers, respectively, and prepared at a final concentration of 20 mM.
Expression and purification of cellobiohydrolase Cel7A from T. harzianum
The expression and purification of wild-type cellobiohydrolase Cel7A enzyme from T. harzianum (Th Cel7A) was carried out as described previously (Colussi et al. 2010, 2011). The resulting Th Cel7A after purification was exhaustively dialyzed in 50 mM Tris-HCl buffer, pH 7.0 for elimination of NaCl. The purity was checked by SDS-PAGE, and the final concentration of Th Cel7A was determined by UV absorbance at 280 nm using a theoretical extinction coefficient based on the amino acid composition (Gill and von Hippel 1989). The final product Th Cel7A was then frozen and stored at −80°C and was melted on ice before use.
Enzymatic activity measurements
Exoglucanase activity against insoluble substrate was determined using as substrate 1% Avicel dissolved in a buffer. Activity of Th Cel7A was measured in the temperature range between 20 and 70°C and pH values from 3.0 to 7.0 (20 mM phosphate/citric acid or Gly/NaOH buffers adjusted to the different pH values). The final protein concentration for each experiment was 1.5 μM. For measurement of chemical stability, samples and controls were incubated at 50°C for 120 min and agitated every 20 min. The amount of reducing sugars was determined using DNS reagent as described by Wood and Bhat (1988). One unit of the enzyme was defined as 1 μmol of glucose equivalent released per minute under the assay conditions.
Far-UV circular dichroism (CD) spectroscopy
Far-UV circular dichroism (CD) measurements were performed using a Jasco J-815 spectropolarimeter (JASCO, Japan) equipped with a Peltier temperature control unit. All data were measured using quartz cuvettes of 1 mm path length, scanning speed of 20 nm min−1, a spectral bandwidth of 1 nm, and a response time of 1 s. The spectra were recorded over the wavelength range 195–240 nm and were determined as an average of 16 accumulations. The buffer contributions were subtracted in all the analyses to eliminate background effects, and all experiments were carried out in triplicate. The Th Cel7A concentration used was approximately 5 μM in all the experiments. Th Cel7A samples (300 μl) were incubated at pH values ranging from 2 to 10 in 20 mM phosphate/citric acid or Gly/NaOH buffers and adjusted to the different pH values. The incubations took 12 h at 25°C prior to analysis. Thermal denaturation of Th Cel7A, in the presence or absence of cellobiose at pH 5, was also characterized by measuring the ellipticity changes at 212 nm induced by a temperature increase to 90°C from 20°C at a heating rate of 10°C/h. The samples were incubated in each condition, in the presence or absence of cellobiose, for 12 h before the analysis. The fraction of denatured protein (α) was calculated from the relationship: α = (θ n − θ obs)/(θ n − θ d) and α + β = 1, in which θ obs is the ellipticity obtained at a particular temperature and θ d and θ n are the values of the ellipticity characteristic of the denatured and native states, respectively.
Intrinsic fluorescence spectroscopy
Intrinsic fluorescence spectroscopy measurements were performed on a steady-state spectrofluorimeter model K2 ISS, equipped with a refrigerated circulator. The Th Cel7A concentration was 2 μM in all of the experiments. Th Cel7A samples (400 μl) were incubated at pH values ranging from 2 to 10 in 20 mM phosphate/citric acid or Gly/NaOH buffers and adjusted to the different pH values. The samples were incubated at 25°C for 12 h before analysis. Th Cel7A has 10 tryptophans, which were excited at 295 nm, and fluorescence emission spectra were recorded from 300 to 400 nm from samples loaded into a quartz cuvette of 1 cm path length. The buffer contributions were subtracted in all analyses, and all experiments were carried out in triplicate.
Thermofluor method
Solutions of 2 μl of 300× Sypro Orange were added to 18 μl of Th Cel7A at 5 μM, pH 5, in the absence or presence of cellobiose. Subsequently, the mixtures were added to the wells of a 96-well thin-wall PCR plate (Bio-Rad). Water was measured as a control. The plates were sealed with Optical-Quality Sealing Tape (Bio-Rad) and heated in an iCycler iQ Real Time PCR Detection System (Bio-Rad) from 10 to 90°C in increments of 0.5°C. Fluorescence changes in the wells of the plate were monitored simultaneously with a charge-coupled device camera (CCD). The wavelengths for excitation were 470–505 mm and emission 540–700 nm.
Dynamic light scattering (DLS)
Th Cel7A at 0.5 mg/ml and pH 5 was centrifuged at 14,000×g for 15 min at room temperature and immediately loaded into a quartz cuvette (path length of 1 cm) prior to measurements. DLS was performed using a Zetasizer instrument (Malvern Instrument, Worcestershire, UK). The temperature was raised from 25 to 80°C at 5°C intervals every 120 s for equilibration of temperatures. The effect of buffer was subtracted from each recording to eliminate the noise produced by it. The hydrodynamic radius was calculated using the Zetasizer μV Software.
Small-angle X-ray scattering (SAXS)
All data were collected on the D02A-SAXS2 beamline of the Synchrotron Light National Laboratory (LNLS, Campinas, Brazil) (Kellermann et al. 1997). The wavelength of incident monochromatic X-ray beam λ was set at 1.488 Å. The X-ray patterns were recorded using a two-dimensional CCD detector (MarResearch, USA). The sample-detector distance was set at 1,022.5 mm, resulting in a scattering vector (q) range of 0.013–0.33 Å−1, where q = 4 × π × sin(θ)/λ and θ is half the scattering angle. The samples were centrifuged for 15 min at 13,000×g (at 4°C) prior to measurement to remove any aggregates or particles and then placed on ice. The samples were loaded into cells made of two thin parallel mica windows. Two successive frames of 300 s each were recorded for each sample to monitor radiation damage and beam stability. Buffer scattering was recorded before each sample scattering. The SAXS patterns were individually corrected for the detector response and scaled by the incident beam intensity and the sample’s absorption. The parasitic scattering from air and beamline windows was subtracted from the total measured intensities. The integration of SAXS patterns was performed using Fit2D software, and the curves were scaled by protein concentration. The Th Cel7A concentrations used for small-angle X-ray scattering measurements were approximately 4 and 2 mg/ml. Th Cel7A samples (200 μl) were incubated at pH values ranging from 2 to 10 in 20 mM phosphate/citric acid or Gly/NaOH buffers and adjusted to the different pH values. The incubations were prolonged for a further 12 h at 25°C before the analysis. Thermal denaturation of Th Cel7A at pH 5 was also characterized by SAXS induced by a temperature increase from 20 to 70°C.
Results and discussion
Temperature and pH profiles of Th Cel7A enzymatic activity
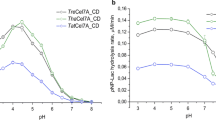
The activity of an enzyme is dependent on pH and temperature, conditions that invoke changes in enzyme folding, conformation, and protonation. As a first step of our studies, the exoglucanase activity of Th Cel7A was studied as a function of pH. Our results showed that, under the conditions of our study (at 50°C), the exoglucanase activity of Th Cel7A increases as pH increases within the pH range of 3–5 and then decreases within the pH range of 5–7 (Fig. 1a). The enzyme reaches its optimum activity at pH 5 and 50°C, conditions similar to those found for other cellobiohydrolases (Bommarius et al. 2008; Hui et al. 2002). Interestingly, even at pH 3, it was found that the remaining activity of the enzyme was 76% and at pH levels above 7 the remaining activity was virtually zero (data not shown). Next, the exoglucanase activity of Th Cel7A was studied as a function of temperature at pH 5. Within the temperature range of 20–50°C (pH 5), the exoglucanase activity of Th Cel7A increased with the increase in temperature reaching its maximum at T = 50°C (Fig. 1b). The exoglucanase activity of Th Cel7A steadily decreased to zero at temperatures above 50°C (Fig. 1b). A better understanding of the biochemistry and biophysics of the processes of conformational change of Th Cel7A is very important to study the relationship of enzyme structure, stability, and activity. Thus, to correlate the enzymatic activity data with the structure of the enzyme, we conducted a series of detailed studies of the unfolding and stability of Th Cel7A as a function of temperature and pH, employing a series of biophysical studies.

Effect of pH (a) and temperature (b) on the enzymatic activity of Th Cel7A
Influence of pH on the structural stability and compactness of Th Cel7A
Influence of pH on the structural stability of Th Cel7A was studied using far-UV circular dichroism (CD) spectroscopy, intrinsic fluorescence spectroscopy, and small-angle X-ray scattering (SAXS). CD spectroscopy is a powerful tool to investigate the conformation of proteins; therefore CD spectroscopy was used to probe the secondary structure of the purified Th Cel7A to assess its folding in terms of secondary structure. The general shape of the spectrum at pH 5 (optimum pH) and 25°C (Fig. 2, solid line), with positive maxima near 195 ± 1 nm and a broad, negative trough at 205–215 ± 1 nm is characteristic of a β-class protein, comparable to the secondary structure of other cellobiohydrolases (Divne et al. 1994; Boer and Koivula 2003; Voutilainen et al. 2009). In addition, the absence of a negative band around 222 ± 1 nm is indicative of a protein with low content of α-helical elements in the secondary structure. Figure 2 shows the influence of pH on the secondary structure of Th Cel7A at 25°C under different pH values (pH = 2, 5, 6, and 10). Our results show that there are no appreciable structural changes in Th Cel7A within the pH range of 2–8 at 25°C. However, when the pH value was increased above 8 (Fig. 2, highly alkaline pH), the spectral profile was progressively altered with a loss of the minimum characteristic at ~210 nm, indicating a strong effect of pH on the secondary structure.

Effect of pH on the secondary structure of Th Cel7A at 25°C monitored by far-UV CD spectroscopy. Far-UV CD spectra as a function of pH. The pH values were 2 (dot), 5 (solid line), 6 (dash dot), and 10 (dash). The experimental conditions are described in “Materials and methods”
As a next step, the tertiary structure of Th Cel7A was investigated by intrinsic fluorescence spectroscopy and SAXS. Figure 3 shows the emission spectra of Th Cel7A after excitation at 295 nm at several pH levels. The spectrum at pH 5 (solid line, optimum pH) is characterized by a maximum emission at 336 ± 1 nm typical of tryptophan residues partially exposed to water environment, indicating that the majority of tryptophan residues present in the Th Cel7A sequence are somewhat exposed to the solvent. Similar results were observed when the protein was incubated at pH 2 and 6 (Fig. 3), indicating that there are no appreciable conformational changes induced in the protein and its tryptophan residue positions. However, the spectrum at pH 10 (dash line) is characterized by a maximum emission at 340 ± 1 nm and a red shift in the spectral center of mass indicating that the tryptophan residues are more exposed to the buffer environment, allowing us to consider the protein unfolded.

Effect of pH on the tertiary structure of Th Cel7A at 25°C monitored by intrinsic fluorescence spectroscopy. The pH values are 2 (dot), 5 (solid line), 6 (dash dot), and 10 (dash). The excitation wavelength was 295 nm, and emission was collected from 300 to 400 nm. The experimental conditions are described in “Materials and methods”
SAXS is a technique applicable to the analysis of the global shapes, dimensions, and oligomeric states of macromolecules in solution. Figure 4a shows the X-ray scattering curves for Th Cel7A at different pH values (pH = 2, 5, 8, and 10). The raw scattering data clearly show that there are no structural alterations of the molecule at pH below 8. However, the raw scattering was altered when the pH value was increased above 8 indicating that there are structural alterations in the tertiary structure of Th Cel7A at highly alkaline pH values. For a more detailed analysis of the effects of pH on the global compactness of Th Cel7A, the SAXS data were analyzed in terms of the Kratky plot (Fig. 4b) (Doniach et al. 1995; Garcia et al. 2008a, b). The Kratky plot, in Fig. 4b, aids in the structural interpretation of such alterations. For a system with a compact shape, this plot shows a well defined curve with an initial rising portion followed by a marked descent thereby forming a well defined maximum (Semisotnov et al. 1996). Nevertheless, the expected curve for a polymer in an extended or random coil conformation rises to a characteristic plateau, with no well defined maximum (Darimont et al. 1998). As can be seen in Fig. 4b all the curves corresponding to measurements made in pH below 8 shows the well defined maxima expected for compact structures. However, in the case of pH values above 8 the Kratky plot indicates an alteration in the peak intensity and its position indicating that alterations to the compactness of the structure have occurred (Fig. 4b). This behavior is typically exhibited by elongated chains (Semisotnov et al. 1996), indicating that at highly alkaline pH values Th Cel7A is unfolded, in contrast to that observed for pH values below 8. Taken together, CD, intrinsic fluorescence, and SAXS results permit us to infer that there were no significant changes in the secondary and tertiary structure of Th Cel7A at pH values below 8, indicating that the protein is a pH acid resistant. However, alterations at both secondary and tertiary structural levels were observed at highly alkaline pH values.

Experimental solution scattering curves as a function of pH. a SAXS curves from Th Cel7A shown as the logarithm of the scattering intensity (log I) versus the momentum transfer q scale. b Kratky plots for Th Cel7A. The pH values are 2 (black filled spheres), 5 (gray filled spheres), 8 (half filled squares), and 10 (triangles). The experimental conditions are given in “Materials and methods” section
Because in the pH range 2–8 there were no appreciable conformational changes in the protein structure that could be detected by the biophysical techniques used in this work, the variation in activity observed was probably due to the variation in ionization of the side chains of the amino acids in the hydrophobic cellulose-binding tunnel, which each had their optimum at pH = 5. However, the observed decrease in activity observed for the pH range 8–10 is most probably mediated by the changes in secondary and tertiary structure of the enzyme, with the formation of molten globule-like state, and its unfolding, provoked by pH variation.
Influence of temperature on Th Cel7A stability and compactness
The nature of the thermal unfolding transition of Th Cel7A at pH 5 was studied by CD spectroscopy, thermofluor method, and SAXS in the range 10–90°C. The CD spectrum of purified Th Cel7A at 25°C was typical of proteins containing elements of β-sheet secondary structure (Fig. 2). The spectrum remains relatively constant below 55°C (data not shown), and this can be readily followed by tracking the ellipticity at 212 nm (Fig. 5a). The spectral profile progressively altered, however, when the temperature increased above 55°C, with a loss of the regular secondary structure (Fig. 5a). In addition, the enzyme could be completely thermally unfolded at temperatures above 80°C.

Thermal unfolding of Th Cel7A. a Thermal denaturation of Th Cel7A at pH 5 in the absence (circles) or presence (triangles) of cellobiose was monitored by measuring the ellipticity at 212 nm as a function of temperature. b Fluorescence intensity emission from Sypro Orange in the presence of Th Cel7A as a function of temperature. Circles Th Cel7A at pH 5 in the absence of cellobiose, triangles Th Cel7A at pH 5 in the presence of cellobiose. The wavelengths for excitation and emission were 490 and 575 nm, respectively
The structural alteration observed by CD spectroscopy was accompanied by an increase in the level of binding of Sypro Orange dye to the protein (Fig. 5b). Sypro Orange is an extrinsic fluorescent probe with low quantum yield in aqueous solution but is highly fluorescent in nonpolar environments with low dielectric constants such as hydrophobic sites in proteins (Ericsson et al. 2006; Garcia et al. 2007). When a protein starts to unfold or melt, the dye binds to exposed hydrophobic regions of the protein, resulting in a significant increase in fluorescence intensity emission. The fluorescence intensity reaches a maximum and then starts to decrease, probably due to precipitation of the complex of the fluorescent probe and the unfolded protein. Sypro Orange is known to bind to hydrophobic surfaces, and the results shown in Fig. 5b therefore indicate that the observed conformational change involves the exposure of such residues.
As can be seen in Fig. 5b, Sypro Orange shows a finite affinity for the protein at temperatures below 55°C, indicating the presence of low exposed hydrophobic regions on the native structure. The catalytic domain structures of Cel7A from T. reesei, for example, reveal tunnel-shaped structures formed by loops (Divne et al. 1994; Davies and Henrissat 1995; Munoz et al. 2001; Grassick et al. 2004; Gray et al. 2006; Parkkinen et al. 2007). A striking feature of the Cel7A structure is an extensive 50-Å-long cellulose-binding tunnel formed by four surface loops, which runs along the concave face of the sandwich and contains the active site at its far end. We advocate that the finite affinity for the native Th Cel7A at temperatures below 55°C is probably due to the interaction between the dye and the hydrophobic cellulose-binding tunnel. When the temperature was increased to values above 55°C, an improved binding of Sypro Orange to the protein was observed (Fig. 5b), implying exposure of hydrophobic regions. The reason for the decrease in the activity of Th Cel7A at temperatures between 55 and 65°C (Fig. 1b) is most likely the formation of a molten globule-like state. While between 55 and 65°C the enzyme activity decreased, the molten globule-like state created exposes hydrophobic surfaces to the external environment and has a tendency to form aggregates. However, when the temperature was further increased to values above 65°C, the fluorescence signal decayed as a result of a reduction in the affinity of the dye to the protein, presumably due to occlusion of the hydrophobic surfaces by aggregation and/or protein unfolding.
To investigate whether Th Cel7A aggregates at high temperatures, we employed dynamic light scattering (DLS) to distinguish different oligomeric protein states and/or amorphous aggregation. When Th Cel7A was analyzed by DLS at 25 and 50°C, the observed profiles were characteristic of monodisperse solutions (data not shown). The hydrodynamic radii determined at 25 and 50°C were 3.11 and 3.19 nm, respectively, consistent, in both cases, with a monomeric macromolecule in solution. However, when Th Cel7A was incubated at 80°C, the profile was altered and showed a hydrodynamic radius of 36.78 nm, which corresponds to high molecular weight aggregates.
To obtain more detailed information about the global compactness of the enzyme, we collected SAXS data as a function of temperature. Figure 6a shows the X-ray scattering curves for Th Cel7A at different temperatures (T = 20, 50, 55, 60, and 65°C). The raw scattering (Fig. 6a) data show that there are no structural alterations in the low resolution structure of Th Cel7A at temperatures between 20 and 55°C. However, the raw scattering was altered when the temperature was increased above 55°C, indicating that Th Cel7A had started to unfold. For a more detailed analysis of the effects of temperature on the global compactness of Th Cel7A, the SAXS data were also analyzed in terms of the Kratky plot (Fig. 6b). As can be seen in Fig. 6b all the curves corresponding to experiments conducted at the temperatures between 20 and 55°C show the well defined maxima expected for compact structures. However for temperatures above 55°C, the Kratky plot clearly shows an alteration in both peak intensity and position indicating that strong alterations to the compactness of the protein structure occurred (Fig. 6b). Taken together, the CD and fluorescence results matched the SAXS experiments and jointly confirmed that the secondary and tertiary structure of Th Cel7A and its compactness are strongly dependent on temperature. In conclusion, the thermal unfolding behavior of Th Cel7A was found to be a two-step process at pH 5. The first step led to the formation of molten globule-like states between 55 and 65°C, followed by a second step that was due to the unfolding of the molten globule-like state at temperatures above 70°C. These molten globule-like states present the following characteristics: they have reduced enzyme activity, they have reduced secondary structure content, they have largely lost their compact core, and they exhibit more hydrophobic surface exposed to the solvent.

Experimental solution scattering curves as a function of temperature. a SAXS curves from Th Cel7A shown as the logarithm of the scattering intensity (log I) versus the momentum transfer q scale. b Kratky plots for Th Cel7A. The temperature values are 20°C (black filled circles), 50°C (half filled squares), 55°C (triangles), 60°C (gray filled circles), and 65°C (circles).
Cellobiose-binding influence on Th Cel7A thermal stability
In the course of cellobiohydrolase action upon cellulose, the polysaccharide chain slides through the substrate binding tunnel, and every second glycosidic bond is correctly presented to the catalytic apparatus, explaining why these enzymes progressively liberate disaccharide units (cellobiose) from the cellulose chain (Wood and Bhat 1988). After cleavage of the β(1→4) glycosidic bond, Th Cel7A might remain bound to the reaction product, cellobiose, which in this case functions as a competitive inhibitor. Thus, we performed thermal unfolding studies monitored by far-UV CD spectroscopy and thermofluor method (Fig. 5) in the absence (apo-Th Cel7A) and presence (cellobiose-Th Cel7A) of cellobiose in order to investigate the influence of this inhibitor on thermal stability of Th Cel7A. The melting temperature value for apo-Th Cel7A, Tm, is equal to 61.5 ± 0.2°C (Fig. 5a). On the other hand, when the Th Cel7A was incubated with 1 mM cellobiose, thermal stability of the complex increased, accompanied by a Tm shift to 65.9 ± 0.2°C. In addition, Sypro Orange fluorescence intensity was monitored as a function of temperature in the absence and presence of cellobiose. In Fig. 5b, Sypro Orange shows a finite affinity for the apo-Th Cel7A at temperatures below 55°C indicating the presence of hydrophobic regions, probably due to the interaction between the dye and the hydrophobic cellulose-binding tunnel. However, the cellobiose addition resulted in a decrease in Sypro Orange intensity emission at temperatures below 50°C, probably now due to the interaction between the cellobiose and the cellulose-binding tunnel. On the other hand, the fluorescence intensity increased only to temperature values above 60°C, indicating an increase in the protein thermal stability in the presence of cellobiose. When the temperature was further increased to values above 60°C, the level of Sypro Orange binding to Th Cel7A further increased (Fig. 5b), indicative of the exposure of hydrophobic regions. However, when the temperature increased above 73°C, a decrease in the fluorescence signal was detected, indicating a reduction in the affinity of the dye, presumably due to the enzyme aggregation and/or protein unfolding. Taken together, the CD and thermofluor method results confirm that the cellobiose significantly increased the thermal stability of Th Cel7A.
Conclusions
Due to its elevated cellulolytic activity, the fungus T. harzianum has considerable potential in several biotechnological applications, such as biomass hydrolysis. In this study, we examined the effects of pH and temperature on the compactness and structure of Th Cel7A and the correlations with enzymatic activity. Variations of pH modify the protonation of the enzyme residues, directly affecting its activity, while leading to structural destabilization only at extreme pH limits. Temperature, however, has a direct influence on mobility, fold, and compactness of the enzyme, causing unfolding just above the optimum temperature limit. Our studies might provide insights into understanding, at the molecular level, the interplay between structure and activity of Th Cel7A at different pH and temperature conditions, which could be useful for biotechnological applications.
References
Boer B, Koivula A (2003) The relationship between thermal stability and pH optimum studied with wild-type and mutant Trichoderma ressei cellobiohydrolase Cel7A. Eur J Biochem 270:841–848
Bommarius AS, Katona A, Cheben SE, Patel AS, Ragauskas AJ, Knudson K, Pu Y (2008) Cellulase kinetics as a function of cellulose pretreatment. Metab Eng 10(6):370–381
Boraston AB, Bolam DN, Gilbert HJ, Davies GJ (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J 382:769–781
Castro AM, Pedro KC, Cruz JC, Ferreira MC, Leite SG, Pereira N (2010) Trichoderma harzianum IOC-4038: a promising strain for the production of a cellulolytic complex with significant β-glucosidase activity from sugarcane bagasse cellulignin. Appl Biochem Biotechnol 162(7):2111–2122
Colussi F, Textor LC, Serpa V, Maeda RN, Pereira N, Polikarpov I (2010) Purification, crystallization and preliminary crystallographic analysis of the catalytic domain of the extracellular cellulase CBHI from Trichoderma harzianum. Acta Crystallogr Sect F Struct Biol Cryst Commun 66:1041–1044
Colussi F, Serpa V, Delabona PS, Manzine LR, Voltatódio ML, Alves R, Mello BL, Pereira JRN, Farinas CS, Golubev AM, Santos MAM, Polikarpov I (2011) Purification, biochemical and biophysical characterization of cellobiohydrolase I from Trichoderma harzianum IOC 3844. J Microbiol Biotech 21:808–817
Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR (1998) Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev 12(21):3343–3356
Davies G, Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3:853–859
Divne C, Stahlberg J, Reinikainen T, Ruohonen L, Pettersson G, Knowles JKC, Teeri TT, Jones TA (1994) The 3-dimensional crystal-structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science 265(5171):524–528
Doniach S, Bascle J, Garel T, Orland H (1995) Partially folded states of proteins—characterization by X-ray-scattering. J Mol Biol 254(5):960–967
Ericsson UB, Hallberg BM, DeTitta GT, Dekker N, Nordlund P (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem 357(2):289–298
Garcia W, Araújo APU, Lara F, Foguel D, Tanaka M, Tanaka T, Garratt RC (2007) An intermediate structure in the thermal unfolding of the GTPase domain of human Sept4 (SEPT4/Bradeion-β) forms amyloid-like filaments in vitro. Biochemistry 46:11101–11109
Garcia W, Figueira ACM, Oliveira Neto M, Guzzi CA, Buzzá HH, Portugal RV, Calgaro MR, Polikarpov I (2008a) Probing conformational changes in orphan nuclear receptor: the NGFI-B intermediate is a partially unfolded dimer. Biophys Chem 137:81–87
Garcia W, Rodrigues NC, Oliveira Neto M, Araújo APU, Polikarpov I, Tanaka M, Tanaka T, Garratt RC (2008b) The stability and aggregation properties of the GTPase domain from human SEPT4. Biochim Biophys Acta 1784:1720–1727
Gill SC, von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 182:319–326
Grassick A, Murray PG, Thompson R, Collins CM, Byrnes L, Birrane G, Higgins TM, Tuohy MG (2004) Three-dimensional structure of a thermostable native cellobiohydrolase, CBH IB, and molecular characterization of the cel7 gene from the filamentous fungus, Talaromyces emersonii. Eur J Biochem 271(22):4495–4506
Gray KA, Zhao LS, Emptage M (2006) Bioethanol. Curr Opin Chem Biol 10(2):141–146
Henrissat B, Davies G (1997) Structural and sequence-based classification of glycoside hydrolases. Curr Opin Chem Biol 7(5):637–644
Hui JPM, White TC, Thibault P (2002) Identification of glycan structure and glycosylation sites in cellobiohydrolase II and endoglucanases I and II from Trichoderma reesei. Glycobiology 12(12):837–849
Igarashi K, Wada M, Hori R, Samejima M (2006) Surface density of cellobiohydrolase on crystalline celluloses—a critical parameter to evaluate enzymatic kinetics at a solid-liquid interface. FEBS J 273(13):2869–2878
Kellermann G, Vicentin F, Tamura E, Rocha M, Tolentino H, Barbosa A, Craievich A, Torriani I (1997) The small-angle X-ray scattering beamline of the Brazilian Synchrotron Light Laboratory. J Appl Crystallogr 30(2):880–883
Kleywegt GJ, Zou JY, Divne C, Davies GJ, Sinning I, Stahlberg J, Reinikainen T, Srisodsuk M, Teeri TT, Jones TA (1997) The crystal structure of the catalytic core domain of endoglucanase I from Trichoderma reesei at 3.6 angstrom resolution, and a comparison with related enzymes. J Mol Biol 272(3):383–397
Kubicek CP, Messner R, Gruber F, Mach RL, Kubicekpranz EM (1993) The Trichoderma cellulase regulatory puzzle—from the interior life of a secretory fungus. Enzyme Micro Technol 15(2):90–99
Kumar R, Singh S, Singh OV (2008) Bioconversion of lignocellulosic biomass: biochemical and molecular perspectives. J Ind Microbiol Biotechnol 35:377–391
Munoz IG, Ubhayasekera W, Henriksson H, Szabo I, Pettersson G, Johansson G, Mowbray SL, Stahlberg J (2001) Family 7 cellobiohydrolases from Phanerochaete chrysosporium: crystal structure of the catalytic module of Cel7D (CBH58) at 1.32 angstrom resolution and homology models of the isozymes. J Mol Biol 314(5):1097–1111
Parkkinen T, Koivula A, Vehmaanpera J, Rouvinen J (2007) Preliminary X-ray analysis of cellobiohydrolase Cel7B from Melanocarpus albomyces. Acta Crystallogr Sect F Struct Biol Cryst Commun 63:754–757
Semisotnov GV, Kihara H, Kotova NV, Kimura K, Amemiya Y, Wakabayashi K, Serdyuk IN, Timchenko AA, Chiba K, Nikaido K et al (1996) Protein globularization during folding. A study by synchrotron small-angle X-ray scattering. J Mol Biol 262(4):559–574
Shen H, Schmuck M, Pilz I, Gilkes NR, Kilburn DG, Miller RC, Warren RAJ (1991) Deletion of the linker connecting the catalytic and cellulose-binding domains of endoglucanase-A (CENA) of cellulomonas-fimi alters its conformation and catalytic activity. J Biol Chem 266(17):11335–11340
Srisodsuk M, Reinikainen T, Penttila M, Teeri TT (1993) Role of the interdomain linker peptide of Trichoderma reesei cellobiohydrolase I in its interaction with the crystalline cellulose. J Biol Chem 268(28):20756–20761
Teeri TT (1997) Crystalline cellulose degradation: new insight into the function of cellobiohydrolases. Trends Biotechnol 15:160–167
von Ossowski I, Stahlberg J, Koivula A, Piens K, Becker D, Boer H, Harle R, Harris M, Divne C, Mahdi S et al (2003) Engineering the exo-loop of Trichoderma reesei cellobiohydrolase, Ce17A. A comparison with Phanerochaete chrysosporium Cel7D. J Mol Biol 333(4):817–829
Voutilainen SP, Boer H, Alapuranen M, Jänis J, Vehmaanperä J, Koivula A (2009) Improving the thermostability and activity of Melanocarpus albomyces cellobiohydrolase Cel7B. Appl Microbiol Biotechnol 83:261–272
Wood TM, Bhat KM (1988) Methods for measuring cellulase activities. Methods Enzymol 160:87–112
Acknowledgments
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) via grant numbers 2008/56255-9, 2009/54035-4, 2010/08370-3, 2010/16542-9; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) via grant number 482166/2010-0; and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). We also thank Laboratório Nacional de Luz Sincrotron (LNLS) for beam time and help with X-ray data collection.
Author information
Authors and Affiliations
Corresponding author
Additional information
Francieli Colussi and Wanius Garcia contributed equally to this work.
Rights and permissions
About this article
Cite this article
Colussi, F., Garcia, W., Rosseto, F.R. et al. Effect of pH and temperature on the global compactness, structure, and activity of cellobiohydrolase Cel7A from Trichoderma harzianum . Eur Biophys J 41, 89–98 (2012). https://doi.org/10.1007/s00249-011-0762-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-011-0762-8