
Abstract
Recent studies have suggested a correlation between genotype groups of Brettanomyces bruxellensis and their source of isolation. To further explore this relationship, the objective of this study was to assess metabolic differences in carbon and nitrogen assimilation between different B. bruxellensis strains from three beverages, including beer, wine, and soft drink, using Biolog Phenotype Microarrays. While some similarities of physiology were noted, many traits were variable among strains. Interestingly, some phenotypes were found that could be linked to strain origin, especially for the assimilation of particular α- and β-glycosides as well as α- and β-substituted monosaccharides. Based upon gene presence or absence, an α-glucosidase and β-glucosidase were found explaining the observed phenotypes. Further, using a PCR screen on a large number of isolates, we have been able to specifically link a genomic deletion to the beer strains, suggesting that this region may have a fitness cost for B. bruxellensis in certain fermentation systems such as brewing. More specifically, none of the beer strains were found to contain a β-glucosidase, which may have direct impacts on the ability for these strains to compete with other microbes or on flavor production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The yeast Brettanomyces bruxellensis (teleomorph Dekkera bruxellensis) is an important contaminant of industrial fermentations, such as those used for biofuel and wine production (de Souza Liberal et al. 2007; Fugelsang 1997; Loureiro and Malfeito-Ferreira 2003). Moreover, B. bruxellensis is considered the major microbial cause of wine spoilage worldwide causing substantial economic losses (Fugelsang 1997; Loureiro and Malfeito-Ferreira 2003). During wine maturation, particularly in wooden barrels, B. bruxellensis can survive for extended periods and convert hydroxycinnamic acids in the wine into odorous ethylphenols (Chatonnet et al. 1992), resulting in the so-called “Brett” character (Licker et al. 1998). The aromas imparted can be described as “burnt plastic,” “barnyard,” “horse sweat,” and “leather,” among some other unpleasant odors, and are therefore considered negative for wine quality. On the other hand, the yeast is also known to be beneficial, or even a crucial contributor in certain fermentation processes, such as the production of lambic and gueuze beers, in which these aromas are highly appreciated (Martens et al. 1997; Vanbeneden et al. 2008). Recently, this species has also been shown to have potential for use in continuous bioethanol production due to its robustness to changes in pH and temperature, its energy-efficient metabolism under oxygen limitation, and its ability to produce high ethanol yields (Passoth et al. 2007; Schifferdecker et al. 2014; Steensels et al. 2015). Apart from isolations from wine, beer, and bioethanol, B. bruxellensis has been detected and isolated in other fermented foods and drinks such as cider, kombucha, and kefir, as well as fruit juices and soft drinks (Cosentino et al. 2001; Coton et al. 2006; Curtin et al. 2007; Laureys and De Vuyst 2014; Morrissey et al. 2004; Oelofse et al. 2009; Teoh et al. 2004).
Genetic diversity studies using classic DNA fingerprinting techniques have revealed significant genotypic interstrain variability within B. bruxellensis (de Barros Lopes et al. 1999; Conterno et al. 2006; Crauwels et al. 2014; de Mitrakul et al. 1999; Martorell et al. 2006; Miot-Sertier and Lonvaud-Funel 2007; Vigentini et al. 2012; Yamada et al. 1994). Additionally, in some of these studies, a correlation was suggested between genotype groups of B. bruxellensis and their source of isolation (Conterno et al. 2006; Crauwels et al. 2014; Vigentini et al. 2012), suggesting niche adaptation. More recently, studies comparing B. bruxellensis genomes have confirmed their considerable interstrain genetic variability (Borneman et al. 2014; Crauwels et al. 2014; Curtin et al. 2012). It was also found that (in addition to diploidy) triploidy may be a relatively common state for B. bruxellensis. In these cases, the triploid genome was found to consist of a core diploid genome of two moderately heterozygous sets of chromosomes and a third haploid complement that is far more divergent (Borneman et al. 2014). Apart from these differences in overall ploidy, chromosomal aberrations such as aneuploidy, strain-specific insertions, and deletions and loss of heterozygosity (LOH) by gene conversion were also commonplace (Borneman et al. 2014; Crauwels et al. 2014; Curtin et al. 2012).
To date, few studies have focused on phenotypic differences between B. bruxellensis strains, probably due to the fact that traditional phenotype assays are labor intensive and time consuming. Moreover, in most of these studies, only a limited set of phenotypic assays were performed, and the strains generally originated from a single niche (i.e., wine) (Conterno et al. 2006). Nevertheless, these studies suggest substantial phenotypic variation between B. bruxellensis strains, such as in carbon and nitrogen assimilation (Conterno et al. 2006), with at least one of these traits, the differential ability to grow on nitrate, being linked to specific genomic deletions (Borneman et al. 2014; Crauwels et al. 2014).
The objective of this study was to assess the phenotypic variation between different B. bruxellensis isolates from three beverages (beer, wine, and soft drink) that represented distinct genotypes as previously determined (Crauwels et al. 2014). More specifically, seven B. bruxellensis isolates were subjected to Phenotype Microarray (PM) technology (Biolog) (Bochner 2003) using three different PM plates. These plates represented 285 phenotypic tests involved in both carbon (PM1 and PM2) and nitrogen metabolism (PM3). In search for genomic signatures of the phenotypic variation, whole genome sequences were compared. Finally, a PCR screening was performed on a set of 30 B. bruxellensis isolates across a genomic region of ~36 kb (harboring 13 genes) that in the sequenced strains displayed variable copy number and LOH, to better understand the genome evolutionary mechanisms underpinning particular phenotypic differences.
Materials and methods
Study strains
Seven B. bruxellensis isolates were used in this study (Table 1). These isolates have been characterized genotypically in previous research using a combination of classic fingerprinting techniques (Crauwels et al. 2014). Isolates were selected from three different origins (beer, wine, and dry ginger ale) and represented four distinct genotypes (Crauwels et al. 2014). All isolates were stored at −80 °C in glycerol-based standard storage medium (yeast extract peptone dextrose (YPD) broth containing 26.1 % glycerol).
Phenotypic analysis
To assess for metabolic differences in carbon and nitrogen metabolism, isolates were subjected to different Phenotype Microarray plates (Biolog, Hayward, CA, USA), including PM1, PM2 (together 190 carbon sources), and PM3 (95 nitrogen sources), following the S. cerevisiae protocol provided by the manufacturer. Substrate concentrations provided in the PM plates are in a typical range that cells prefer (2–20 mM for the carbon sources and 1–5 mM for the nitrogen sources). When the carbon source varied, the nitrogen source was ammonium (PM1 and PM2). When the nitrogen source varied, glucose was used as carbon source (PM3). Briefly, first cryopreserved cultures were streaked on yeast peptone glucose agar (YPG) (10 g/l glucose, 10 g/l peptone, and 5 g/l yeast extract) and incubated for 120 h at 25 °C. After incubation, a single colony was restreaked on YPG and incubated for 96 h under the same conditions. Next, each strain was picked up with a sterile cotton swab and suspended in 15 ml of yeast nutrient supplement (NS) inoculation fluid (Biolog) until an optical cell density of 0.20 was reached, measured using a spectrophotometer at a wavelength of 600 nm. PM plates were then inoculated with 100 μl of a mixture of 10 ml IFY-0 (1.2×), 1000 μl recommended PM additive (12×), 160 μl dye mix D (100×), 375 μl glucose solution (32×) (only for PM3), 590 μl (for PM3) or 965 μl (for PM1 and PM2) autoclaved ultrapure water (IDT, Coralville, IA, USA), and 500 μl yeast cells in NS solution. All microplates were incubated for 8 days at 25 °C in the OmniLog automated incubator-reader (Biolog) and were read every half hour. Each strain was tested in duplicate.
Binary coefficients (1/0) for positive metabolism (1) or no metabolic activity (0) were attributed to each PM well as follows. First, the area under the curve (AUC) was calculated using the OPM package (Vaas et al. 2013) for R version 12.2.1 (R Core Development Team 2006). Next, a strain was scored positive for a particular test source when for each replicate, the AUC of the well exceeded 1.1 times the AUC of the blank. Vice versa, the strain was scored negative if it did not exceed 1.1 times the AUC of the blank for both replicates. If different scores were obtained for both replicates, no value was attributed to that well (and indicated as “unknown”). The robustness of our scoring system was verified with classic tests for a subset of carbon and nitrogen sources, including arbutin, cellobiose, D-galactose, gentiobiose, glucose, and glycerol (carbon sources) and nitrate (nitrogen sources). For carbon assimilation tests, 5-ml yeast nitrogen base (without amino acids and ammonium sulfate) was used supplemented with 0.2 % (w/v) of the tested carbon source. For the nitrate assimilation test, strains were inoculated into 5-ml medium described by Conterno et al. (2006) supplemented with 0.1 % (w/v) nitrate. After 7 days of aerobic incubation under agitation at 25 °C, the growth of the different strains was evaluated by means of visual inspection and confirmed our Biolog results (Tables S1 and S2, supplemental material).
Genome analysis
For three of the seven studied strains (ST05.12/22 (also known as VIB X9085); ST05.12/56 (CBS 2499) and ST05.12/62 (AWRI 1499)), whole genome sequences and annotations were already available and were used for this study (Crauwels et al. 2014; Curtin et al. 2012; Piskur et al. 2012). For the other four strains, genomes were sequenced in this study (Table 1). Genomic DNA was isolated using the phenol/chloroform extraction method described previously (Lievens et al. 2003). Following DNA purification, DNA samples were sequenced using the Illumina HiSeq 2500 sequencing platform (Illumina, San Diego, CA, USA) using the “high-output run” mode. For each strain, the ploidy level was estimated by taking advantage of allele proportions as described previously (Curtin et al. 2012). For single nucleotide polymorphism (SNP) mapping, raw sequencing reads were mapped to the AWRI 1499 genome sequence with SNP positions and whole genome phylogenies constructed as described previously (Borneman et al. 2014). Further, prominent phenotypic differences observed in our dataset were associated with KEGG pathways (Kanehisa and Goto 2000; Kanehisa et al. 2014) and gene presence/absence patterns. To this end, the sequenced raw reads were mapped against the three annotated reference genomes using Bowtie2 (Langmead and Salzberg 2012), and results of the mapping were further analyzed with SAMtools (Li et al. 2009) and BEDTools (Quinlan and Hall 2010). Sequence data obtained in this study have been deposited in the NCBI short-read archive under the Bioproject accession PRNJA281577.
PCR screen of selected genes
In order to assess the distribution of particular genes of interest or genes associated with specific phenotypes within B. bruxellensis, a PCR screen was performed on a collection of 30 B. bruxellensis strains from beer (18 isolates), wine (9 isolates), and soft drinks (3 isolates) (Crauwels et al. 2014). Primers were designed using Primer3 (Rozen and Skaletsky 2000) based on at least two sequences available for each gene (Table S3, supplemental material). Part of the analysis involved an extended screening of what has been done previously in Crauwels et al. (2014) for the genes encoding a galactokinase (GenBank Accession No EIF45409 (AWRI 1499)), a dTDP-glucose dehydratase (EIF45411), a maltase (EIF45413), and a β-glucosidase (EIF45415). PCR amplification was performed in a reaction volume of 20 μl, containing 0.15 μM of each dNTP, 0.5 μM of each primer, 1 U TaKaRa Ex Taq polymerase, 1× Ex Taq buffer (Clontech Laboratories, Palo Alto, CA), and 10 ng genomic DNA (determined using a nanodrop instrument). Amplification was performed using a Bio-Rad T100 thermal cycler according to the following thermal profile: initial denaturation at 94 °C for 10 min, followed by 30 cycles of 94 °C for 1 min, 55 °C for 45 s, and 72 °C for 1 min. A final 10-min extension step at 72 °C concluded the protocol. Presence or absence of the gene was evaluated by agarose gel electrophoresis and, for some amplicons, further confirmed by sequencing of the obtained product using the same reverse primer as used for the amplification.
Results
Phenotypic diversity between B. bruxellensis isolates from different niches
In total, 139 of the 285 sources tested, encompassing 71 carbon sources and 68 nitrogen sources, scored positive for at least one of the tested strains (Tables S1 and S2, supplemental material). One hundred forty-three sources were attributed a binary value for all strains (no “unknown” among the tested strains; meaning that for these 143 sources for each strain both replicates gave identical results (positive or negative for both replicates)) (Tables S1 and S2, supplemental material). Of these 143 sources, 39 % were metabolized by all strains tested, 45 % were not metabolized by any of the strains tested, and 15 % were differentially metabolized among strains. In general, B. bruxellensis strains were found able to oxidize aldoses and disaccharides (Table S1, supplemental material). More specifically, 7 out of 10 and 6 out of 11 tested aldoses and disaccharides, respectively, were oxidized by all strains. In contrast, polysaccharides (with exception of dextrin, laminarin, and pectin) were not oxidized (Table S1, supplemental material). Among nitrogen sources, the majority of amino acids tested were assimilated by all strains. More specifically, whereas L-isomers were commonly oxidized, D-isomers could generally not be used (with the exception of D-alanine) (Table S2, supplemental material). In contrast, when amino acids were used as sole carbon source, most sources yielded a negative response, with exception of D-alanine, L-glutamic acid, and L-proline (Table S1, supplemental material). Further, substrates involved in arginine and proline metabolism as well as dipeptide sources were commonly used (Table S2, supplemental material).
Prominent phenotypic differences were seen in the utilization of particular saccharides and nitrogen sources with regard to the origin of the strains as outlined below. Out of a total of 13 monosaccharides tested, nine sugars were metabolized by all strains and three sugars (D-allose, L-glucose, and L-sorbose) were not oxidized by any of the strains tested. A differential response was observed for D-galactose (Table 2). While the soft drink strain as well as the strains isolated from wine were able to oxidize D-galactose, this carbon source could not be metabolized by the beer strains (with exception of isolate ST05.12/53) (Table 2). Most tested disaccharides could be metabolized by all strains. However, whereas the soft drink isolate ST05.12/59 could not use the α-glycosides maltose and turanose, the other six isolates were able to use both disaccharides. Similarly, the soft drink strain was the only strain that could not use the trisaccharide melezitose and the α-substituted monosaccharide α-methyl-D-glucoside (Table 2). The beer strains (ST05.12/22, ST05.12/26, ST05.12/48), on the other hand, were the only strains that were not capable of consuming the β-disaccharides cellobiose and gentiobiose as well as the β-substituted monosaccharides arbutin and β-methyl-D-glucoside (Table 2). Finally, the soft drink strain and wine strains, along with one beer strain (ST05.12/48), were able to metabolize sodium nitrate and sodium nitrite, whereas the other three beer strains could not utilize this nitrogen source (Table S2, supplemental material).
Genomic features of beer, soft drink, and wine B. bruxellensis
The soft drink isolate and beer isolates sequenced in this study were compared to previously sequenced wine (Borneman et al. 2014; Curtin et al. 2012; Piskur et al. 2012) and beer (Crauwels et al. 2014) isolates, in terms of their genome features (Fig. 1) and overall relatedness (Fig. 2). Beer strains ST05.12/26 and ST05.12/48 were predicted to have diploid genomes based upon heterozygous allele proportions (Table 1; Fig. 1), and cluster with previously sequenced diploid beer (ST05.12/22) and wine (AWRI 1613, CBS 2499 (ST05.12/56)) strains (Fig. 2). ST05.12/53 also clustered with this group, yet was predicted to harbor a triploid genome (Table 1; Fig. 1). The triploid soft drink isolate ST05.12/59 (Table 1; Fig. 1) was most distant from the diploid strains, which are thought to represent the B. bruxellensis “core” genome (Borneman et al. 2014). An interesting feature previously described for diploid wine B. bruxellensis strains is widespread loss-of-heterozygosity (LOH). Diploid and triploid beer strains also exhibit this feature, with the greatest proportion of LOH evident for strain ST05.12/48. Notably, while there are regions where both beer and wine strains share LOH events, there are some that are conserved across beer but not wine strains and vice versa (Fig. 1). While all newly sequenced strains displayed relatively even sequence depth across their genomes, local copy number variations (CNV) were commonplace (Fig. 1). For example, ST05.12/26 displayed several instances of large-scale heterozygous deletions (Fig. 1).

Whole genome analyses. a Copy number variation analysis. For each strain, the average sequencing read depth was recorded for sliding windows across the genome (5-kb window, 1-kb step) and presented relative to the mean across that strain’s entire genome. b Single nucleotide polymorphism (SNP) analysis. For each strain, heterozygous nucleotides were identified and the proportion of aligned reads containing each of the variant bases recorded. The average major allele frequency was then calculated for sliding windows across the genome (5-kb window, 1-kb step) and plotted central to each window. Any regions that lacked heterozygous bases were classified as regions of loss-of-heterozygosity (LOH) and are indicated by gray bars above each plot. The solid black line represents a major allele frequency of 66 % that would be expected for a triploid genome. For a diploid genome, it is expected that the average frequency of a particular allele at a heterozygous site will be around 50 %. Predicted triploids are indicated in green, diploids in blue. Names of Brettanomyces bruxellensis strains sequenced in this study are indicated in bold

Maximum-likelihood tree based on 9.7 Mb of a concatenated alignment that exceeds minimum coverage level. Names of Brettanomyces bruxellensis strains sequenced in this study are indicated in bold. Predicted triploids are indicated with a green mark, diploids with a blue mark
Targeted genomics to explain phenotypes
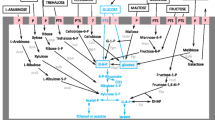
Mapping the raw reads of the four strains sequenced in this study (ST05.12/26, ST05.12/48, ST05.12/53, and ST05.12/59) to the three annotated genomes (ST05.12/22, CBS 2499 (ST05.12/56), and AWRI 1499 (ST05.12/62)) enabled putative identification of the genes involved in shaping the above-mentioned phenotypes. BLAST-derived KEGG annotations were analyzed for genes involved in the galactose pathway (KO00052) and revealed four genes that could be linked to the observed galactose-positive and galactose-negative phenotypes, including genes encoding a galactose-1-phosphate uridyltransferase (K00965, galT; GenBank Accession No EIF45408 (AWRI 1499)), a galactokinase (K00849, galK; EIF45409), a gal10 bifunctional protein (K01784, galE; EIF45410), and a β-galactosidase (K05349, lacZ; EIF45248). None of the genes were found in ST05.12/22, while isolates ST05.12/26 and ST05.12/48 lacked lacZ and galE, and galE, respectively. In all galactose-positive isolates, all four genes were present (Table S4, supplemental material). Additionally, a β-glucosidase (K05349, bglX; GenBank Accession No EIF45415) was found in the genome of strains that could assimilate the β-linked sugars cellobiose, gentiobiose, and arbutin as well as β-methyl-D-glucoside (Table S5, supplemental material). Similarly, a maltase (K01187, malZ; EIF45413) was found only in the genome of strains able to oxidize the α-linked sugars maltose, turanose, and melezitose as well as α-methyl-D-glucoside (Table S5, supplemental material).
In order to assess the distribution of both these α- and β-glucosidase genes, their presence or absence was evaluated by PCR for 30 assorted strains from beer, wine, and soft drinks. Just over half of the beer strains (10 out of 18 tested isolates) and the majority of the wine strains (8 out of 9) were found to contain the maltase gene (X9085_g2204 (ST05.12/22)). In contrast, none of the three tested soft drink isolates were positive for this gene (data not shown). Regarding the β-glucosidase gene, all beer strains scored negative, while all soft drink isolates and almost all wine strains (8 out of 9) scored positive (Table 3). When expanding this PCR screening across a larger genomic region harboring 13 genes (including this β-glucosidase gene; region corresponds to a part of the AWRI 1499 reference contig with GenBank accession number AHIQ01000280), which was found to be absent in a beer strain previously (Crauwels et al. 2014), a systematic pattern of gene loss was observed for the beer strains (Table 3). In contrast, all tested soft drink and wine isolates (except one) were found to contain the complete cluster of genes (Table 3). Interestingly, the region, corresponding to the AWRI 1499 reference contig with GenBank accession number AHIQ01000280, was notable for varying degrees of CNV and LOH between strains (Fig. 3). Indeed, all sequenced strains are predicted to have undergone either heterozygous or homozygous deletion of all or part of the 13-gene cluster, or present with LOH across the same region (Fig. 3).

Analysis of read-mapping against Brettanomyces bruxellensis AWRI 1499 (ST05.12/62) contig AHIQ01000280. a Copy number variation analysis. For each strain, the average sequencing read depth was recorded for sliding windows across the genome (5 -kb window, 1-kb step) and presented relative to the mean across that strain’s entire genome. b Single nucleotide polymorphism (SNP) analysis. For each strain, heterozygous nucleotides were identified and the proportion of aligned reads containing each of the variant bases recorded. The average major allele frequency was then calculated for sliding windows across the genome (5-kb window, 1-kb step) and plotted central to each window. Any regions that lacked heterozygous bases were classified as regions of loss-of-heterozygosity (LOH) and are indicated by gray bars above each plot. The solid black line represents a major allele frequency of 66 % that would be expected for a triploid genome. For a diploid genome, it is expected that the average frequency of a particular allele at a heterozygous site will be around 50 %. Predicted triploids are indicated in green, diploids in blue. Names of Brettanomyces bruxellensis strains sequenced in this study are indicated in bold. Open reading frames are indicated below coverage and SNP plots, with those in red part of the 13-gene cluster referred to in Table 3
Similarly, three genes involved in nitrate assimilation, including nitrate reductase (K10534, NR, GenBank Accession No EIF45249 (AWRI 1499)), nitrite reductase (K00362, nirB, EIF45250), and a nitrate transporter (K02575, NRT, EIF45251), were found to be linked with the nitrate/nitrite phenotypes (Table S6, supplemental material). While all three genes were present in the strains with positive phenotypes, beer isolates ST05.12/22 and ST05.12/53 did not contain any of the three genes, while isolate ST05.12/26 did not contain the reductase genes (NR and nirB) but still contained the nitrate transporter gene (NRT).
Discussion
Genetic diversity studies have revealed substantial genotypic interstrain variability within B. bruxellensis. On the other hand, relatively little is known about phenotypic diversity of this important industrial yeast species. Here, we studied the phenotypic variation between genotypically divergent B. bruxellensis isolates from three beverages (beer (4 isolates), wine (2 isolates), and soft drink (1 isolate)) and attempted to relate this variation to genomic data. Apart from providing novel insights on carbon metabolism in B. bruxellensis, we also confirmed previous findings on nitrogen metabolism, whereby some strains are unable to use nitrate or nitrite due to the lack of one or more essential genes involved in nitrate assimilation (Borneman et al. 2014; Crauwels et al. 2014).
B. bruxellensis strains were able to grow on a variety of carbon and nitrogen sources of which monosaccharides and disaccharides were the most commonly used carbon sources, and L-amino acids, dipeptides, and substrates involved in arginine and proline metabolism the most accessible nitrogen sources. These results are in agreement with the findings of Conterno et al. (2006), who tested a large number of B. bruxellensis isolates, primarily from wine, on several phenotypic parameters. Amino acids such as proline and arginine are the most abundant amino acids in grape juice, wine, and fruit juices (Bell and Henschke 2005; Kliewer 1967; Ting and Rouseff 1979), and may therefore represent an efficient nitrogen source for yeasts such as Brettanomyces in these beverages. In contrast, proline has been found the least-preferred nitrogen source for many lab strains of S. cerevisiae (Magasanik and Kaiser 2002).
Previous studies have suggested a correlation between genotype and source of isolation (Conterno et al. 2006; Crauwels et al. 2014; Vigentini et al. 2012). Our data are in agreement with previous work (Conterno et al. 2006) in that overall phenotypic patterns are not clearly related to sources of isolation; nonetheless, some interesting phenotypes were found that could be linked to strain origin (at least for the strains tested). For example, the tested soft drink isolate as well as the wine strains were capable of oxidizing D-galactose, whereas most beer strains were not. The presence or absence of four genes could be linked to this phenotype, including three clustered genes involved in the Leloir pathway of the galactose metabolism (galactose-1-phosphate uridyltransferase (galT), galactokinase (galK), and gal10 bifunctional protein (UDP-galactose-4-epimerase (galE))) and a β-galactosidase with lactase activity (lacZ). Strains unable to grow on galactose lack at least one of these genes, i.e., galE encoding an UDP-galactose 4-epimerase catalyzing the final step of the Leloir pathway, explaining the observed phenotype. Previous studies on S. cerevisiae have shown that loss of GalE results in yeast growth arrest in response to even trace quantities of environmental galactose despite the presence of an alternate, metabolize-able carbon source (Douglas and Hawthorne 1964). So far, no information is available whether lack of galE would similarly impact Brettanomyces yeasts.
Whereas most disaccharides and trisaccharides were metabolized by all strains, differential responses were found for particular α- and β-glycosides. Additionally, similar observations were made for the α- and β-substituted monosaccharides α-methyl-D-glucoside and β-methyl-D-glucoside, respectively. More specifically, the soft drink isolate was the only isolate that could not use the disaccharides maltose and turanose, consisting of two α-1,4-linked glucose units and a glucose and fructose unit combined by an α-1,3-linkage, respectively. As such, it may be expected that this strain lacks the enzyme(s) that is (are) able to hydrolyze these α-linkages. Each of the seven tested strains was found to contain multiple α-glycosidase (maltase or isomaltase) genes, of which one gene (X9085_g2204) may be correlated with the observed maltose- and turanose-negative phenotypes. Additionally, a β-glucosidase (GenBank Accession No EIF48743) was identified that cocorrelated with these phenotypes (Table S5, supplemental material), but being a β-glucosidase, it is less likely that this gene would be involved. Further research (e.g. using (knock-out) mutants) is needed to examine the exact phenotypic impact of the presence or absence of these genes in B. bruxellensis. Furthermore, the soft drink isolate was unable to hydrolyze the α-substituted monosaccharide α-methyl-D-glucoside and the nonreducing trisaccharide melezitose. Melezitose can be hydrolyzed into turanose and glucose by hydrolyzing the Fru β(2–1)α Glc in melezitose; hydrolysis of the α-1,3 linkage results in glucose and sucrose. Whereas it may be expected that the Fru β(2–1)α Glc bond can be hydrolyzed (as for sucrose) yielding turanose and glucose, and that the glucose can be used by the yeast, it remains to be investigated why a negative response was obtained on melezitose.
Further, the four investigated beer strains were the only strains tested that were not capable of metabolizing the β-glycoside disaccharides cellobiose and gentiobiose as well as the β-substituted monosaccharides β-methyl-glucoside and arbutine. Whether or not this finding represents a general trend for B. bruxellensis beer strains has to be confirmed by investigating more isolates. Our findings also suggest that the beer strains investigated in this study lack the enzyme(s) responsible for the breakage of specific β bonds such as β-1,4 (cellobiose) and β-1,6 (gentiobiose). In this regard, a β-glucosidase (GenBank Accession No EIF45415 (AWRI 1499 (ST05.12/62)) and JGI transcript number 51487 (CBS 2499 (ST05.12/56))) was found to be absent in the beer strains, while it was present in all other strains tested. However, whereas this β-glucosidase gene was absent in the beer strains, the beer strains were found to possess another β-glucosidase gene (homolog of the GenBank Accession No EIF48743 (AWRI 1499 (ST05.12/62)) and JGI transcript number 26490 (CBS 2499 (ST05.12/56))), which may explain the usage of other β-glycosides (e.g. the β-1,3 glucan laminarin). Moreover, this gene was present in all tested strains, except in the soft drink isolate ST05.12/59. β-Glucosidases are well-known for their role in flavor development in beer and wine (Daenen et al. 2004). Many flavor and aroma compounds are locked up as nonvolatile glycosides and are released from their precursors by enzymatic hydrolysis, e.g., mediated by β-glucosidases (Pogorzelski and Wilkowska 2007). As we found different phenotypic profiles for the use of β-glycosides within our set of tested isolates, coinciding with the presence of two different β-glucosidases, it may be expected that these strains will display different flavor profiles. However, further research is needed to investigate the exact role of these β-glucosidases in flavoring capability of B. bruxellensis strains.
Interestingly, the three aforementioned genes involved in the Leloir pathway as well as the β-glucosidase gene absent in the four investigated beer strains belong to a ~36 kb region encompassing 13 clustered genes, the majority of which are involved in carbon metabolism. This cluster was completely absent in beer strain ST15.12/22, while present in the reference genomes of CBS 2499 (ST05.12/56) and AWRI 1499 (ST05.12/62) (Crauwels et al. 2014). Mapping our sequence data against these reference genomes indicated that this cluster is also present in the soft drink isolate ST05.12/59. On the contrary, a portion of this region, encompassing genes encoding a gal10 bifunctional protein, a dtdp-glucose dehydratase, two hexose transporters, a maltase, a multidrug resistance regulator, and the β-glucosidase was lost in the beer strains ST05.12/26 and ST05.12/48. Beer strain ST05.12/53 only lacked the genes encoding the β-glucosidase and a hexose transporter. Notably, this cluster corresponds with a region on the AWRI 1499 reference contig with GenBank accession number AHIQ01000280 that was also found to have varying degrees of CNV and LOH between strains. Altogether, these results show that this genomic region is relatively unstable and prone to (partial) loss in some strains. Indeed, a PCR screening performed for an additional set of isolates (30 in total) on these 13 genes confirmed this scenario (note: in Crauwels et al. (2014), this region of 13 genes was by mistake said to contain 12 genes). More specifically, whereas all tested soft drink and wine isolates (except one) were found to contain the complete cluster of genes, none of the tested beer strains contained the entire cluster. Furthermore, our PCR screening suggests that these genes have been gradually lost over time, as we observe a spectrum of target gene presence, wherein a minority of beer associated strains retain a few target genes, others lack all 13, and all beer strains lack the β-glucosidase gene. Based on these findings, it may be speculated that this gene cluster carries a fitness cost for B. bruxellensis in certain fermentation systems such as brewing, thereby providing a selective pressure for its loss. This phenomenon of loss of genes in specific strains is reminiscent of the concerted loss of the galactose catabolism cluster in Japanese Saccharomyces kudriavzeveii isolates compared to European isolates (Hittinger et al. 2010).
It is interesting to note the possibility that ploidy level reflects the niche from where the strains were isolated. The majority of B. bruxellensis isolates (>90 %) recovered from Australian wineries represent triploid strains (Borneman et al. 2014; Curtin et al. 2007). In addition, wine strains with microsatellite profiles very similar to the sequenced triploid reference strain AWRI 1499 were recently described, which originated from France and South-Africa (Albertin et al. 2014), suggesting that these strains are also triploid. In contrast, the majority of the beer strains investigated in this study (3 out of 4) were found to be diploid. Genome sequencing, or microsatellite analysis, of a large number of B. bruxellensis isolates from different niches will be required to reach a conclusion as to the significance of these observations, but it is tempting to speculate that the additional set of divergent chromosomes may confer a selective advantage in winery and related environments. It was also suggested in other studies that increased levels of resistance to sulphite, the main anti-spoilage agent in wine fermentations, might be at least partially responsible for the common triploidy state in wine strains (Borneman et al. 2014; Curtin et al. 2012). While triploid, phylogenetic analysis based on SNP data positioned beer strain ST05.12/53 closely to the diploid strains. The genetic mechanism by which B. bruxellensis triploids form would, therefore, appear to occur even between relatively closely related strains.
To conclude, we observed substantial phenotypic diversity within B. bruxellensis, some of which could be explained by genomic differences, i.e. presence or abscence of specific genes. However, functional studies using (knock-out) mutants are needed to confirm the involvement of these genes in the phenotypes observed and discussed in this study. Additionally, further genome sequencing is needed to better understand how genome evolutionary mechanisms underpin these phenotypic differences. Moreover, while inferred in the current study, further research using large collections of B. bruxellensis strains from diverse geographical and ecological sources is needed to answer the question whether different ecological niches harbor different B. bruxellensis phenotypes. Such studies may help us investigating whether these phenotypes also have a distinct impact on the production of beer and wine or other beverages, or, more in general, to further understand the ecological behavior of this economically important yeast.
References
Albertin W, Panfili A, Miot-Sertier C, Goulielmakis A, Delcamp A, Salin F, Lonvaud-Funel A, Curtin C, Masneuf-Pomarède I (2014) Development of microsatellite markers for the rapid and reliable genotyping of Brettanomyces bruxellensis at strain level. Food Microbiol 42:188–195
Bell SJ, Henschke PA (2005) Implications of nitrogen nutrition for grapes, fermentation and wine. Aust J Grape Wine Res 11:242–295
Bochner BR (2003) New technologies to assess genotype-phenotype relationships. Nat Rev 4:309–314
Borneman AR, Zeppel R, Chambers PJ, Curtin CD (2014) Insights into the Dekkera bruxellensis genomic landscape: comparative genomics reveals variation in ploidy and nutrient utilization potential amongst wine isolates. PLoS Genet 10, e1004161
Chatonnet P, Dubourdie D, Boidron J, Pons M (1992) The origin of ethylphenols in wines. J Sci Food Agric 60:165–178
Conterno L, Lucy Joseph CM, Arvik TJ, Henick-Kling T, Bisson LF (2006) Genetic and physiological characterization of Brettanomyces bruxellensis strains isolated from wines. Am J Enol Vitic 75:139–147
Cosentino S, Fadda ME, Deplano M, Mulargia AF, Palmas F (2001) Yeasts associated with Sardinian ewe’s dairy products. Int J Food Microbiol 69:53–58
Coton E, Coton M, Levert D, Casaregola S, Sohier D (2006) Yeast ecology in French cider and black olive natural fermentations. Int J Food Microbiol 108:130–135
Crauwels S, Zhu B, Steensels J, Busschaert P, De Samblanx G, Marchal K, Willems KA, Verstrepen K, Lievens B (2014) Assesing genetic diversity among Brettanomyces yeast by DNA fingerprinting and whole genome sequencing. Appl Environ Microbiol 80:4398–4413
Curtin CD, Bellon JR, Henscke PA, Godden PW, de Barros Lopes MA (2007) Genetic diversity of Dekkera bruxellensis yeast isolated from Australian wineries. FEMS Yeast Res 7:471–481
Curtin CD, Borneman AR, Chambers PJ, Pretorius IS (2012) De-novo assembly and analysis of the heterozygous triploid genome of the wine spoilage yeast Dekkera bruxellensis AWRI1499. PLoS ONE 7:1–10
Daenen L, Vanderhaegen B, Verachter H, Derdelinckx G (2004) Flavour enhancement in beer by yeast beta-glucosidase activity. Commun Agric Appl Biol Sci 69:73–76
de Barros Lopes M, Rainieri S, Henschke PA, Langridge P (1999) AFLP fingerprinting for analysis of yeast genetic variation. Int J Syst Bacteriol 49:915–924
de Souza Liberal A, Basilio A, do Monte Resende A, Brasileiro B, da Silva-Filho E, de Morais J, Simoes D, de Morais M (2007) Identification of Dekkera bruxellensis as a major contaminant yeast in continuous fuel ethanol fermentation. J Appl Microbiol 102:538–547
Douglas H, Hawthorne D (1964) Enzymatic expression and genetic linkage of genes controlling galactose utilization in saccharomyces. Genetics 49:837–844.
Fugelsang KC (1997) Wine microbiology. Chapman and Hall, New York
Hittinger CT, Gonçalves P, Sampaio JP, Dover J, Johnston M, Rokas A (2010) Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature 464:54–58
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42:D199–D205
Kliewer WM (1967) Annual cyclic changes in the concentration of free amino acids in grapevines. Am J Enol Vitic 18:126–137
Langmead B, Salzberg S (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Laureys D, De Vuyst L (2014) Microbial species diversity, community dynamics, and metabolite kinetics of water kefir fermentation. Appl Environ Microbiol 80:2564–2572
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The Sequence Alignment/Map (SAM) format and SAMtools. J Bioinforma 25:2078–2079
Licker JL, Acree TE, Henick-Kling T (1998) What is “Brett” (Brettanomyces) flavour? A preliminary investigation. In: Waterhouse AL, Ebeler SE (eds) Chemistry of wine flavour. Symposium series. American Chemical Society, Washington, DC, pp 96–115
Lievens B, Brouwer M, Vanachter ACRC, Lévesque CA, Cammue BPA, Thomma BPHJ (2003) Design and development of a DNA array for rapid detection and identification of multiple tomato vascular wilt pathogens. FEMS Microbiol Lett 223:113–122
Loureiro V, Malfeito-Ferreira M (2003) Spoilage yeasts in the wine industry. Int J Food Microbiol 86:23–50
Magasanik B, Kaiser CA (2002) Nitrogen regulation in Saccharomyces cerevisiae. Gene 290:1–18
Martens H, Iserentant D, Verachtert H (1997) Microbial aspects of a mixed yeast-bacterial fermentation in the production of a special Belgian acidic ale. J Inst Brew 103:85–91
Martorell P, Barata A, Malfeito-Ferreira M, Fernandez-Espinar MT, Loureiro V, Querol A (2006) Molecular typing of the yeast species Dekkera bruxellensis and Pichia guilliermondii recovered from wine related sources. Int J Food Microbiol 106:79–84
Miot-Sertier C, Lonvaud-Funel A (2007) Development of a molecular method for the typing of Brettanomyces bruxellensis (Dekkera bruxellensis) at the strain level. J Appl Microbiol 102:555–562
Mitrakul CM, Henick-Kling T, Egli CM (1999) Discrimination of Brettanomyces/Dekkera yeast isolates from wine by using various DNA fingerprinting methods. Food Microbiol 16:3–4
Morrissey WF, Davenport B, Querol A, Dobson ADW (2004) The role of indigenous yeasts in traditional Irish cider fermentations. J Appl Microbiol 97:647–655
Oelofse A, Lonvaud-Funel A, du Toit M (2009) Molecular identification of Brettanomyces bruxellensis strains isolated from red wines and volatile phenol production. Food Microbiol 26:377–385
Passoth V, Blomqvist J, Schnürer J (2007) Dekkera bruxellensis and Lactobacillus vini form a stable ethanol producing consortium in a commercial alcohol production process. Appl Environ Microbiol 73:4354–4356
Piskur J, Ling Z, Marcet-Houben M, Ishchuk O, Aerts A, LaButti K, Copeland A, Lindquist E, Barry K, Compagno C, Bisson L, Grigoriev I, Gabaldon T, Phister T (2012) The genome of wine yeast Dekkera bruxellensis provides a tool to explore its food-related properties. Int J Food Microbiol 157:202–209
Pogorzelski E, Wilkowska A (2007) Flavour enhancement through the enzymatic hydrolysis of glycosidic aroma precursors in juice and wine beverages: a review. Flavour Fragr J 22:251–254
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842
R Development Core Team (2006) R: a language and environment for statistical computing. R foundation for statistical computing, Vienna
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:265–386
Schifferdecker A, Dashko S, Ishchuk O, Piskur J (2014) The wine and beer yeast Dekkera bruxellensis. Yeast 31:323–332
Steensels J, Daenen L, Malcorps P, Derderlinckx G, Verachtert H, Verstrepen K (2015) Brettanomyces yeast- from spoilage organism to valuable contributors to industrial fermentation. Int J Food Microbiol 206:24–38
Teoh AL, Heard G, Cox J (2004) Yeast ecology of Kombucha fermentation. Int J Food Microbiol 95:119–126
Ting SV, Rouseff RL (1979) Proline content in Florida frozen concentrated orange juice and canned grapefruit juice. Proc Fla State Hortic Soc 92:143–145
Vaas LAI, Sikorsky J, Hofner B, Fiebig A, Buddruhs N, Klenk HPG, Göker M (2013) Opm: an R package for analysing OmniLog® Phenotype MicroArray Data. Bioinformatics 29:1823–1824
Vanbeneden N, Gils F, Delvaux G, Delvaux FR (2008) Formation of 4-vinyl and 4-ethyl derivatives from hydroxycinnamic acids: occurrence of volatile phenolic flavour compounds in beer and distribution of Pad 1-activity among brewing yeasts. Food Chem 107:221–230
Vigentini I, De Lorenzis G, Picozzi C, Imazio S, Merico A, Galafassi S, Piskur J, Foschino R (2012) Intraspecific variations of Dekkera/Brettanomyces bruxellensis genome studied by capillary electrophoresis separation of the intron splice site profiles. Int J Food Microbiol 157:6–15
Yamada Y, Matsuda M, Maeda K, Mikata K (1994) The phylogenetic relationships of species of the genus Dekkera Van Der Walt based on the partial sequences of 18S and 26S ribosomal RNAs (Saccharomycetaceae). Biosci Biotechnol Biochem 58:1803–1808
Acknowledgments
We are grateful to everybody who provided us with Brettanomyces strains. Further, we are grateful to Sofie Malfliet for her help with compiling the tables.
Compliance with ethical standards
Disclosure of potential conflicts of interest/Research involving Human Participants and/or Animals/ Informed consent: not applicable for this study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 684 kb)
Rights and permissions
About this article

Cite this article
Crauwels, S., Van Assche, A., de Jonge, R. et al. Comparative phenomics and targeted use of genomics reveals variation in carbon and nitrogen assimilation among different Brettanomyces bruxellensis strains. Appl Microbiol Biotechnol 99, 9123–9134 (2015). https://doi.org/10.1007/s00253-015-6769-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6769-9