
Abstract
For making a complex synthetic gene network function as designed, the parameters in the network have to be extensively tuned. In this study, a simple and general approach to rapidly tune gene networks in Escherichia coli was developed, which uses the hypermutable simple sequence repeats embedded in the spacer region between the ribosome binding site and the initiation codon. It was found that the change of sequence length and compositions of the repeated base pairs in 5′UTR contributes together to the changeable expression levels of the target gene. The mechanism of this phenomenon is that the transcriptional process makes greater impact on the expression level when compared to the translational process, which is utilized to successfully predict sample gene expression levels over a 50-fold range. The utility of the approach to regulate heterologous ecaA and pepc gene expression in the engineered E. coli for improving succinic acid yield and production has also been demonstrated. When the expression level of ecaA gene was 3.53-fold of the control and the expression level of pepc gene was 1.06-fold of the control, the highest yield of succinic acid and productivity were achieved, which was 0.87 g g−1 and 2.01 g L−1 h−1, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Synthetic biology has emerged as a new branch of life sciences in the twenty-first century (Tong et al. 2001; Benner and Sismour 2005). The great success of constructing artificial life and producing antimalarial drug artemisinin (Gibson et al. 2010; Ro et al. 2006; Paddon and Keasling 2014) has aroused more and more attention on synthetic biology. Recently, the studies of synthetic biology have made great progress in the fields of medicine, e.g., the synthesis of antibiotic, antibody, and new treatments (Yeh et al. 2009; Langer and Tirrell 2004; Leitao and Engutia 2014), biological energy source, e.g., the alcohol and hydrocarbon production (Hawkins et al. 2013; Yang et al. 2012; Bi et al. 2013; Rabinovitch-Deere et al. 2013; Lee et al. 2012; Keasling 2010), bio-based chemicals, e.g., the production of succinic acid and fatty acids by engineered bacteria (Liu et al. 2013; Wang et al. 2014a; Ye and Bhatia 2012) and other industrial circles, environmental protection, and agricultural production (Mossman et al. 2007; Mayfield 2013). While the focus of the synthetic biology in the above fields is to construct artificial gene regulatory networks in biological systems (Feist and Palsson 2008), accordingly, metabolic network reconstructions have significantly expanded over the past two decades.
For the appropriate gene regulation is critical to the implementation of designed multigene networks, it is very important to control the expression levels of the individual gene to operate genetic networks at the desired levels and to achieve the idealized dynamic ranges, particularly when regulating toxic genes or those that may interfere with the host background (Callura et al. 2010; Zhang et al. 2012; Beato 1989; Lu et al. 2009). However, the reliable and predicable gene regulation presents unique challenges as genetic parts such as promoter (Alper et al. 2005; Cox et al. 2007; Lescot et al. 2002), ribosome binding site (RBS) (Barrick et al. 1994; Chen et al. 1994; Zelcbuch et al. 2013; Levin-Karp et al. 2013; Oliver et al. 2014; Mutalik et al. 2012; Salis et al. 2009), and RNA (Sakai et al. 2014; Laetitia and Martin 2006) often behave unexpectedly when they are used in novel designs. A gene regulation system that has been predicted analytically to perform well may perform poorly in practice. Furthermore, even a gene regulation system can be made to work well in a particular strain or environment, there is no guarantee that the same gene regulation system will work well if it is ported to a new strain or environment (Fischbach and Voigt 2010).
The key part in metabolic engineering is the gene regulation by genetic tools, in which, the metabolic networks and gene regulation can be optimized or directly modified to enhance metabolite production or synthesize novel compounds by using metabolic processes (Zhu et al. 2013; Dong et al. 2012; Bailey 1991). One way to improve the performance of a gene network is to introduce focused variability into the design and generate a library with the same genetic components. Egbert and Klavins (2012) have introduced a tuning mechanism for gene regulation in Escherichia coli, in which the expression of a gene was altered by introducing the 5′UTR spacer region between RBS and initiation codon with the high mutation rate and strong bias for insertion/deletion mutations inherent to simple sequence repeats (Vellanoweth and Rabinowitz 1992). The library of regulation sequences is called rbSSR library sequences. However, the selection of the appropriate gene expression level was not explicitly addressed in the study due to its complexity. Moreover, they only tested the target gene expression on protein level and chose the expected gene expression level by microscopy assays. In this study, rbSSR libraries for ecaA and pepc genes have been built and the target gene expression is extensively investigated on both the protein level and mRNA level. Comparing the different rbSSR sequences with the homologous mRNA content and final gene expression level embodied by enzyme activities, a fine-tuning system for gene expression was established.
Moreover, the quantitative regulation system for the gene expression based on the rbSSR library was used for succinic acid production by E .coli. Succinic acid is regarded as one of the top 12 chemical building blocks from biomass by the US Department of Energy (Werpy and Petersen 2004). The conventional production of succinic acid is petrochemical-based, while the bio-based succinic acid has not yet become competitive due to the higher production costs (McKinlay et al. 2007) and relatively lower yield and inefficient productivity (Liu et al. 2008; Balzer et al. 2013). Gene overexpression is considered to be an effective strategy to improve the bio-based succinic acid production by enhancing enzymatic activities and constructing a new pathway, e.g., the phosphoenolpyruvate carboxylase (PEPC), malic enzyme (MaeB), and pyruvate carboxylase (PYC) overexpression (Zhang et al. 2009; Eiteman and Miller 1995; Thakker et al. 2011; Wang et al. 2014b; Liang et al. 2011).
During the production of succinic acid by using E. coli, the catalyzed carboxylation of phosphoenolpyruvate (PEP) to oxaloacetate (OAA) by PEP carboxylase (PEPC) is considered as the most important step (Kai et al. 2003), in which, 1 mol CO2 is assimilated to form the first C4 metabolites OAA (Clark 1989). It has been reported that the overexpression of ecaA gene (encoding for carbonic anhydrase) and pepc gene (encoding for PEPC) in E. coli can improve the efficiency of this reaction (Wang et al. 2009; Liu et al. 2013). The overexpression of ecaA gene can lead to the accumulation of HCO3− as the substrate for PEPC and debilitate the leakage of CO2 in the cytoplasm, while the overexpression of pepc gene can accelerate the carboxylation of phosphoenolpyruvate (PEP) to oxaloacetate (OAA) (Liu et al. 2008). In our preliminary experiments, we investigated the co-expression of these two genes, but the inhibition of cell growth and reduction of succinic acid production were found. Therefore, a new method is necessary to ensure these target genes to express at a desirable level and function positively as expected. For achieving this objective, the rbSSR library was built and used to tune the desirable expression level of ecaA gene and pepc genes with the aim to improve the production of succinic acid in this study. The content of ecaA mRNA and pepc mRNA as well as the enzyme activities of carbonic anhydrase (CA) and PEPC were analyzed, and a quantitative model was developed for helping to choose the appropriate gene expression level. We believed that this is the first study to demonstrate that rbSSR can be used to fine-tune metabolic pathways in engineered strains for the production of bulk chemicals.
Materials and methods
Strains, plasmids, and primers
The strains, plasmids, and primers used for polymerase chain reaction (PCR) amplification in this study were showed in Table S1. E. coli DC1515 [pflA::Cam ldhA::Tn 10 ptsG400::Kan in W1485] was kindly provided by Prof. Clark, Southern Illinois University.
Construction of artificial regulatory rbSSR library
Construction of eight parent plasmids
To understand the effects of expression control with rbSSR library, four rbSSR spacer motifs: (A)16, (AT)8, (AC)8, and (C)16 were experimentally examined to test their effects on the expression of target genes of ecaA and pepc. For each motif is corresponding to one gene, a parent plasmid was constructed with a trc promoter. The backbone for the parent plasmids was generated from a pTrchisB plasmid by digestion with endonucleases BamHI and HindIII, followed by gel extraction and purification. Eight fragments (four ecaA gene fragments and four pepc gene fragments) were amplified from Anabaena sp. PCC 7120 genomic DNA using primers rbSSR-ecaA-up1 and rbSSR-ecaA-down, rbSSR-ecaA-up2 and rbSSR-ecaA-down, rbSSR-ecaA-up3 and rbSSR-ecaA-down, rbSSR-ecaA-up4 and rbSSR-ecaA-down; rbSSR-pepc-up1 and rbSSR-pepc-down, rbSSR-pepc-up2 and rbSSR-pepc-down, rbSSR-pepc-up3 and rbSSR-pepc-down, rbSSR-pepc-up4 and rbSSR-pepc-down, respectively (Table S1). Then, the eight products were isolated and purified. The backbone and eight products were joined respectively using T4 ligase to obtain the eight parent plasmids: pTrc-ecaA1 containing (A)16 sequence in rbSSR region, pTrc-ecaA2 containing (AT)8 sequence in rbSSR region, pTrc-ecaA3 containing (AC)8 sequence in rbSSR region, pTrc-ecaA4 containing (C)16 sequence in rbSSR region; pTrc-pepc1 containing (A)16 sequence in rbSSR region, pTrc-pepc2 containing (AT)8 sequence in rbSSR region, pTrc-pepc3 containing (AC)8 sequence in rbSSR region, pTrc-pepc4 containing (C)16 sequence in rbSSR region. All the engineering plasmids were verified by DNA sequencing at Shanghai Sangon Biological Engineering Technology & Service Co., Ltd.
Generation of rbSSR library
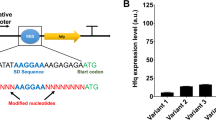
Due to the inherent instability of repeats, PCR—even with a high fidelity polymerase—results in insertions and deletions of rbSSR sequence repeat units with a strong bias for deletion. The generation of rbSSR library via PCR was illustrated in Fig. 1. Spacer variation for the rbSSR library was generated by PCR with 2× Taq PCR Green mix (Dingguo) using the eight parent plasmids as the templates, respectively. rbSSR-ecaA-up and rbSSR-ecaA-down were used as primers when pTrc-ecaA1, pTrc-ecaA2, pTrc-ecaA3, and pTrc-ecaA4 were used as the templates; rbSSR-pepc-up and rbSSR-pepc-down were used as primers when pTrc-pepc1, pTrc-pepc2, pTrc-pepc3, and pTrc-pepc4 were used as the templates (Table S1). PCR amplicons were purified by the gel extraction. The backbone and rbSSR amplicons were joined using T4 ligase and introduced directly into E. coli DC1515. The engineered E. coli strains containing the plasmids which express ecaA gene with rbSSR region were named as KCA1 library, and the engineered E. coli containing the plasmids which express pepc gene with rbSSR region were named as KPC1 library. All the fragments in the plasmids were verified by DNA sequencing at Shanghai Sangon Biological Engineering Technology & Service Co., Ltd.

Construction of the ecaA and pepc gene with rbSSR library sequences. a The simple sequence repeats ((A)16, (AC)8, (AT)8, (C)16) are embedded in the region between ribosome binding site and the initiation codon of the ecaA and pepc gene; b getting the eight parent plasmids: backbone (pTrchisB plasmid by digestion with BamHI and HindIII) and eight products were joined, respectively, using T4 ligase to obtain the eight parent plasmids; c the simple sequence repeats ((A)16, (AC)8, (AT)8, (C)16) mutated for the rbSSR library were generated by PCR using the eight parent plasmids as templates, respectively; d Backbone and mutated fragments were joined by T4 ligase to obtain the plasmids containing target gene with rbSSR library sequences
Growth and fermentation conditions
For the rbSSR library construction, all E. coli strains were cultured in Luria-Bertani (LB) medium at 37 °C including tryptone, 10 g L−1; yeast extract, 5 g L−1; and NaCl, 10 g L−1. Antibiotics were included as necessary at a concentration of 50 μg mL−1 ampicillin. The colonies that contain the recombinant plasmids were selected from LB plates containing the appropriate antibiotic(s). For the succinic acid production, the colonies which contain recombinant plasmids were selected from LB plates containing the appropriate antibiotic(s). Antibiotics were included as necessary at the following concentrations: 50 μg mL−1 ampicillin, 50 μg mL−1 kanamycin, 10 μg mL−1 tetracycline, and 30 μg mL−1 chloramphenicol. The fermentation medium contains tryptone, 20 g L−1; yeast extract, 10 g L−1; MgSO4 · 7H2O, 0.3 g L−1; Na2HPO4 · 12H2O, 0.45 g L−1; NaH2PO4 · 2H2O, 6 g L−1; (NH4)2SO4 · 7H2O, 3 g L−1; CaCl2, 0.2 g L−1. After being inoculated into 5 × 100 mm tubes containing 3 mL LB, the colonies were grown at 37 °C and 220 rpm overnight. Two hundred microliters seed inoculum from an overnight 5-mL culture was added to a 100-mL flask containing 20 mL of LB medium with the appropriate antibiotic(s) to facilitate the aerobic growth. When OD600 reached 1.0, the culture (1 mL) was inoculated into a 50-mL sealed serum tube, which contained 20 mL fermentation medium supplemented with MgCO3, the appropriate antibiotic(s), 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and approximately 20 g L−1 glucose. The initial pH of the medium was controlled to be 7.0. The tubes were sealed airtight for not allowing gas exchange with the outside atmosphere. Since the headspace of the tube was not replaced with CO2, this setup allows the cells to grow faster in the presence of oxygen and, therefore, was preferred in the fermentation experiments because it can help generating biomass before the anaerobic fermentation occurs. The cultures were grown in a rotary shaker at 37 °C and 180 rpm for 48 h.
mRNA analysis and enzyme assays
Messenger RNA analysis was performed by reverse transcription polymerase chain reaction (RT-PCR) and real-time PCR. Total RNA was isolated using an E.Z.N.A Total RNA Kit I (Omega), 10 μg total RNA was reversely transcribed with reverse transcriptase (Vazyme) to obtain the cDNA. The real-time PCR was performed in an iCycler (Bio-Rad) monitoring double-stranded DNA assayed continuously with SYBR-Green (Invitrogen). The fragments of the target genes and ampicillin gene in the diluted cDNA were amplified in a PCR with AmpliTaq Gold by the primer sets: CA-RT-up, CA-RT-down, PC-RT-up, PC-RT-down and AMP-RT-up, AMP-RT-down (Table S1). The Ct value of the target gene was compared to that of the amp gene which contained in the pTrchisB plasmid to test the expression of the target genes on the transcriptional level.
To analyze the enzyme activities, cell extracts of the E. coli strains were prepared by washing the cell pellets with an appropriate buffer (50 mmol L−1 Tris-HCl (pH = 8.0), 1.0 mmol L−1 EDTA, 1.0 mmol L−1 EGTA, 0.05 % v/v NP40 and 0.1 mmol L−1 DTT) and disrupting the suspended cells by sonicating at 100 W for 10 min at 30 s intervals on ice. Cell debris was removed by centrifugation at 10,000g for 10 min at 4 °C. The supernatant was further centrifuged at 10,000g at 4 °C for 20 min, and the resulting supernatant was used for the enzyme activity assay. CA activity was expressed in Wilbur-Anderson (Wibur and Anderson 1948) units per milligram of protein and was calculated using the formula of CA activity = [(t 0/t − 1) × 10]/mg protein, where t 0 and t represent the time required for the pH to change from 8.0 to 7.0 in the buffer control and cell extracts, respectively. The PEP carboxylase (PEPC) activity was assayed by monitoring the decrease in absorbance of NADH at 340 nm using malate dehydrogenase as the coupling enzyme according to the Kodaki’s method (Kodaki et al. 1985). One milliliter reaction mixture for PEPC analysis is consisted of 50 mmol L−1 HEPES (pH = 7.3), 5.0 mmol L−1 PEP, 10 mmol L−1 MgCl2, 5.0 mmol L−1 NaHCO3, 4 units of malate dehydrogenase, 0.2 mmol L−1 NADH, and 25 μL cell extract. The extinction coefficient for NADH at 340 nm was 6.22 mmol L−1 cm−1. 1 U of PEPC activity was defined as the activity of oxidizing 1 μmol L−1 NADH min−1 at 30 °C.
Analytical methods
Glucose consumption and product (succinic acid, lactate, formate, acetate, and ethanol) formation during the fermentation were analyzed by using HPLC equipped with a Bio-Rad Aminex HPX-87H ion-exchange column (7.8 by 300 mm) and HP1200 chromatography working station system with UV absorbance and refractive index detectors. The samples of anaerobic culture were removed anoxically and centrifuged at 10,000 rpm for 10 min. The supernatants were then diluted with 10 volumes of 5 mM H2SO4 to be used as the HPLC samples. During each measurement, the column was eluted isocratically at a rate of 0.6 mL min−1 with 5 mM H2SO4 under 55 °C. The approximate retention time were glucose, 8.9 min; succinic acid, 11.5 min; lactic acid, 12.5 min; formic acid, 13.65 min; acetic acid, 14.86 min; and ethanol, 21.4 min.
Calculation of succinic acid yields
In this study, each measurement was repeated for three times, and the average values of the data were reported with the error bars of the standard deviation. The succinic acid yields were calculated from the maximum succinic acid concentration, the total amount of sugar consumed during the fermentation, and the final volume of the fermentation according to Eq. 1.
Results
Construction of parent plasmids containing the ecaA gene and artificial regulatory engineered E. coli library
Four parent plasmids (pTrc-ecaA1, pTrc-ecaA2, pTrc-ecaA3, pTrc-ecaA4), containing the ecaA gene from Anabaena sp. PCC 7120 under the control of the trc promoter, were constructed. Gel electrophoresis of the BamHI and HindIII digested samples was implemented to identify the positive clone. The approximately 900 bp DNA fragments digested by BamHI and HindIII were the same length of ecaA as expected. The four rbSSR spacer motifs were (A)16, (AC)8, (AT)8, and (C)16, respectively.
By taking advantage of the inherent instability of repeats during replication, especially in PCR, a plasmid library was generated by amplifying a region of each parent plasmid flanking the rbSSR sequence and reinserting the mutated fragments into a plasmid backbone. The resulting plasmids constituted the artificial regulatory pTrc-ecaA plasmid libraries.
The reinsertion fragments in the resulting plasmids were verified, and the results showed that the rbSSR sequences were mutated in two aspects: changed in length; mutated in base. For instance, the (A)16 was mutated into (A)13 or (A)15; (A)16 was also mutated into AAACAATAAAAGAAAA, AAGAACAATTAAAGAA in some clones. Similar mutations have been found in the other three parent plasmids, the rbSSR sequence of them are (AT)8, (AC)8, and (C)16, respectively.
Subsequently, the artificial regulatory plasmids were introduced into DC1515 to obtain the engineered E. coli library KCA1.
Effect of different sequences of rbSSR on the expression of ecaA gene on transcriptional level
A total of 320 colonies were selected from the engineered E. coli libraries KCA1. All the recombinant plasmids in the 320 colonies were sequenced at Shanghai Sangon Biological Engineering Technology & Service Co., Ltd. The effect of different sequences of rbSSR on the expression of ecaA gene on transcriptional level will be introduced in the following two aspects: different lengths and different bases of rbSSR on the expression of ecaA gene on transcriptional level.
Effect of rbSSR length on the expression of ecaA gene on transcriptional level
A previous study (Egbert and Klavins 2012) has reported that the gene expression level decreases with an increase in rbSSR length. In this study, four groups of colonies have been screened from the 320 colonies, in which the rbSSR sequences in the recombinant plasmids are (A) n , (AC) n , (AT) n , and (C) n , respectively (n: repeats of either a single or a pair of nucleotides). The ecaA gene expression on the transcriptional level was tested by the real-time quantitative PCR. As showed in Fig. 2a, the transcriptional level of ecaA gene in the four groups of colonies was not totally same as that deduced according to Egbert’s report. With motif (A) n , (AC) n , and (AT) n , in the approximately 6- to 8-bp range, the transcriptional level of ecaA gene increases with an increase in rbSSR length; while when the rbSSR length is out of the range, the transcriptional level of ecaA gene decreases with an increase in rbSSR length. Moreover, for the rbSSR sequences of (A) n and (AT) n , the best rbSSR length corresponding to the highest transcriptional level is 6; for the rbSSR sequence of (AC) n , the best rbSSR length is 8. However, for the rbSSR sequence of (C) n , the transcription of ecaA gene maintains at a low level with the change of the rbSSR length.

Effect of different rbSSR sequences on ecaA transcription (mRNA amplification). a Effect of different rbSSR sequence lengths on ecaA transcription (mRNA amplification); b effect of different base in rbSSR on ecaA transcription (mRNA amplification)
Effect of different base in rbSSR on the expression of ecaA gene on transcriptional level
Although the four groups of colonies screened from the 320 colonies revealed similar effects on the expression of ecaA gene on the transcriptional level, the best rbSSR length corresponding to the highest transcriptional level was different for different colonies. It is hypothesized that the compositions of base pairs in the rbSSR sequences of ecaA could affect the expression of ecaA gene. In order to testify the hypothesis, the other five groups of colonies were screened from the 320 colonies. Each group of colonies have the same rbSSR length of 4, 7, 10, 13, and 16, respectively, but their rbSSR bases are random. The rbSSR bases of the colonies were sequenced, and the coresponding ecaA gene expression on the transcriptional level was tested by the real-time quantitative PCR. The results in Fig. 2b showed that the difference of the rbSSR contents of A, T affects the expression of ecaA gene on the transcriptional level. For the rbSSR with a same length, a higher transcriptional level of ecaA gene can be achieved with a higher content of A, T bases.
Effect of the rbSSR libraries on the expression of pepc gene on the transcriptional level
The effect of artificial rbSSR library on the expression of pepc has also been tested. The same method was employed to construct the four parent plasmids (pTrc-pepc1, pTrc-pepc2, pTrc-pepc3, pTrc-pepc4) and to obtain the engineered E. coli library KPC1 containing 320 colonies.
Four groups of colonies were screened from the 320 colonies, the rbSSR spacer in the recombinant plasmids contained in the colonies is (A) n , (AC) n , (AT) n , (C) n , respectively. The pepc gene expression on the transcriptional level was tested by using the real-time quantitative PCR; as expected, similar results were obtained as the expression of gene ecaA (Fig. 3a). When the rbSSR sequence is (A) n , (AC) n , and (AT)n, respectively, the best rbSSR length attributed to the highest transcriptional level is 5, 6, and 5. For the rbSSR sequence of (C) n , a lower transcriptional level of pepc gene was obtained. The best rbSSR lengths for the four motifs corresponding to the highest transcriptional level of pepc are different, but they exhibit the same characteristic. When the rbSSR length is within the approximately 5- to 6-bp range, the transcriptional level of pepc genes increases with an increase in rbSSR length. While the rbSSR length is out of the range, the transcriptional level of pepc gene decreases with an increase in rbSSR length.

Effect of different rbSSR sequences on pepc transcription (mRNA amplification). a Effect of different rbSSR sequence lengths on pepc transcription (mRNA amplification); b effect of different base in rbSSR on pepc transcription (mRNA amplification)
In order to examine the effect of the A, T contents on the expression of pepc gene on the transcriptional level, two groups of colonies with the same rbSSR length of 12 and 15 were screened from the 320 colonies. The pepc gene expression on transcriptional level was tested by the real-time quantitative PCR, and the results were showed in Fig. 3b. Similar to the results of the ecaA gene, the transcriptional level of pepc gene increases with an increase in the content of A, T bases.
Effect of rbSSR sequence on the expression of the two genes on the translation level
In vivo, the gene expression was controlled at the two aspects: the transcriptional level and the translational level. Although the mRNA level in the cell can be tuned through the rbSSR library, the protein level may not be correspondingly regulated, i.e., the excessive mRNAs may not express excessive proteins. In order to verify whether the rbSSR library can regulate the gene expression simultaneously at both the transcriptional and translational level, eight colonies were randomly picked and grown in LB medium for enzyme assays. The rbSSR sequences in the eight colonies were sequenced, and the fold amplification of ecaA mRNA amplification was tested by using the real-time quantitative PCR, the gene expression strength on the translational level was tested by using enzyme assays and the results were showed in Table 1. With the variation of rbSSR sequences, the fold amplification of ecaA mRNA changes correspondingly, resulting in the different activities of the target enzyme. However, the activity of the target enzyme is almost proportional to the fold amplification of ecaA mRNA, a linear relationship could be built between the fold amplification of the enzyme activity as showed in Eq. 2 (for ecaA gene, R 2 = 0.9977) and Eq. 3 (for pepc gene, R 2 = 0.9964).
Constructing a fine-tuning system for the gene expression
By comparing the different rbSSR sequences with the homologous mRNA and the final gene expression level embodied by enzyme activities, a fine-tuning system for the gene expression could be established. The expression levels of sample gene can be controlled within the range of 0- to 50-folds by the rbSSR library predicted by the RBS calculator computer programs (Salis et al. 2009; http://salis.psu.edu/software). However, the aim of this study is to improve the succinic acid production, which only needs to regulate the two genes within a small range (0- to 7-folds). Eight groups of experimental dates within this range were compared with the computational predictions by the RBS calculator computer programs (Table 1), which verified that the experimental data match well with the computational results.
Optimization of the ecaA and pepc individual gene expression for improving the succinic acid production
The succinic acid production with individual ecaA, pepc gene expression under the control of rbSSR library was explored. The relationship of the expression level of ecaA gene, pepc gene, and the succinic acid production, productivity can be seen in Table 2. When the expression level of ecaA gene was 3.53-folds of the unmodified ecaA gene, the productivity of succinic acid was the highest (1.84 g L−1 h−1), and the yield of succinic acid reached to 0.82 g g−1. When the expression level of ecaA gene was 4.33-folds of the unmodified ecaA gene, the yield of succinic acid was 0.84 g g−1, but the productivity of succinic acid was relative lower. So, the best expression level of ecaA gene for succinic acid production by the engineered E. coli was 3.53-folds of the control. In Table 2, it was also found that the best expression level of pepc gene for succinic acid production by the engineered E. coli was 2.89-folds of the control. The yields of succinic acid and productivity were 0.86 g g−1 and 1.89 g L−1 h−1, respectively.
Optimization of the ecaA and pepc gene co-expression for improving the succinic acid production
Co-expression of the two genes by using the artificial regulatory rbSSR library was also studied to improve the succinic acid production. As shown in Table 3, when the expression level of ecaA gene is 3.53-fold of the unmodified ecaA gene and the expression level of pepc gene is 1.06-folds of the unmodified pepc gene, the yields of succinic acid and productivity reached the optimum values, which was 0.87 g g−1 and 2.01 g L−1 h−1, respectively.
Discussion
A preeminent engineered E. coli need some extrinsic key genes to be co-expressed in cells on an appropriate level, which can be fulfilled by using synthetic gene networks; however, the establishment of engineering reliable and predictable synthetic gene networks presents unique challenges because genetic parts such as promoters, RBS, and protein coding regions often behave unexpectedly when being used in novel designs (Cox et al. 2007; Mutalik et al. 2012; Sueel et al. 2007). The experiments described in this study suggested that it requires substantial tuning for a complex gene network to function as expected. It seems that the construction of the rbSSR library using sequence repeats is a reliable method for this purpose due to the instability of repeats during PCR replication. Accordingly, a very simple construct embedded in the spacer region between the RBS and initiation codon, which is called rbSSR, was used to tune gene networks for succinic acid production. The resulting rbSSR sequences demonstrated that our experimental scheme is feasible (Fig. 1). An rbSSR library and the engineered E. coli library LKCA1 and LKPC1 were obtained successfully.
In order to test whether the rbSSR library can regulate the target gene expression on the desired levels, the effect of the rbSSR sequences on the expression of ecaA gene was investigated (Fig. 2). The results indicated that an appropriate length of the rbSSR sequences is necessary for the ecaA gene transcription. Less efficient transcription, and then lower mRNA content, can be induced when the length is too long or too short. Moreover, a higher content of the A, T bases in the rbSSR sequences could result in a higher transcriptional level of ecaA gene. The same results were obtained for the pepc gene (Fig. 3), and this is a novel phenomenon that has not been reported in the literature. In the process of transcription, the initiation step is the coupling of the RNA polymerase on the DNA promoter to form the promoter complex then the double-stranded DNA becomes open, which is the rate-limiting step of transcription, taking much more time than the next step of RNA extension (Weisburg et al. 1991).
In the initial period of transcription, the σ factor in the RNA polymerase holoenzyme (RNAP) can recognize the DNA promoter, and thus, the RNAP is able to bind with the DNA promoter to the RNAP-promoter closed complex. Subsequently, the σ factor unwinds approximately one turn of the DNA promoter to form the RNAP-promoter open complex. After 8–9 bp mRNA is synthetized, the σ factor escapes the RNAP, while the RNAP escapes the promoter and enters into productive synthesis of RNA as RNAP-DNA elongation complex. More mRNA can be synthesized with more RNAPs binding to the DNA promoter (Weisburg et al. 1991). The rbSSR sequences can affect the ability of RNA polymerases to recognize the promoters. An appropriate length of the rbSSR sequences and more content of A, T bases in the rbSSR sequences can attract more RNA polymerase holoenzymes (RNAPs) to coupling on the promoter and finally lead to the increase of the target mRNA content in the host. It was reported that the sequences of TATAAT and TTGACA in E. coli can attract the σ factor (Pribnow 1975; Datsenko and Wanner 2000). These sequences have a common characteristic, i.e., they are basically made up of A, T bases. Therefore, the rbSSR sequences with a higher content of A, T bases could attract more σ factors, resulting in that more RNAPs gather around these sequences. Therefore, if the length of the rbSSR sequences is appropriate and the content of A, T bases in them is high, the RNAP concentration around the rbSSR sequences and the promoters would increase as the RNAP can couple on both the rbSSR sequences and the promoters simultaneously. However, if the length of the rbSSR sequences is too long, the RNAP coupling on the sequences cannot couple on the promoter, consequently, a higher RNAP concentration around the sequences would decrease the RNAP concentration around the promoters. If the length of the rbSSR sequences is too short, they cannot contain enough A, T bases to attract the RNAP. Therefore, an appropriate length of the rbSSR sequences is a prerequisite for increasing the RNAP concentration around the promoters.
Although previous studies (Egbert and Klavins 2012; Dong et al. 2012) showed that a same content of mRNAs may not be translated to a same content of proteins in a same host, the results in Table 1 demonstrated that a linear relationship exists between the fold amplification of target mRNA amplification and the target enzyme activity as showed in Eqs. 2 and 3. As the host cell does not express CA itself, the intercept of Eq. 2 is close to zero, while the PEPC can be expressed by the host cell, the intercept of Eq. 3 is 0.2386, indicating that if the pepc gene was not overexpressed in the host cell, the PEPC activity is about 0.2386 U mg−1.
Some studies (Egbert and Klavins 2012; Sakai et al. 2014; Orom et al. 2008) have reported that the 5′UTR can affect the gene expression at the translational level. One of the most recognized mechanism is the riboregulator theory (Sakai et al. 2014). Riboregulator is composed of two RNA molecules: cis-repressed mRNA (crRNA) and trans-activating noncoding sRNA (taRNA). The crRNA contains a sequence complementary to the RBS and forms a hairpin structure in its 5′UTR to prevent the ribosomal binding, thereby repressing expression. If the taRNA hybridizes with the crRNA through a linear-loop interaction that exposes the RBS by dissociating from the crRNA hairpin structure, posttranscriptional expression of the target gene could be activated. The results shown in Table 1 are not contradictory to this theory. Although the different sequences in 5′UTR may affect the bonding degree between crRNA and RBS, 5′UTR is divided into two parts by RBS: an upstream 5′UTR and a downstream 5′UTR. The crRNA lies in the upstream of 5′UTR, whereas the rbSSR sequences lies in the downstream of 5′UTR. So, the different rbSSR sequences might have little effect on gene expression on the translational level.
This study demonstrated that the gene expression regulated by the rbSSR sequences is mainly on the transcriptional level but not the translational level, which is an important theoretical basis to guide the construction of genetic tools for metabolic engineering. The mechanism of this phenomenon will be explored in our further study. Fine-tuning ecaA and pepc expression by rbSSR library in E. coli mutant was carried out with the aim to improve the succinic acid production. At appropriate gene co-expression levels, which is 3.53-folds for ecaA, 1.06-folds for pepc, the yields of succinic acid and productivity reach the optimum values, 0.87 g g−1 and 2.01 g L−1 h−1, respectively. The catalyzed carboxylation of phosphoenolpyruvate (PEP) to oxaloacetate (OAA) by PEP carboxylase (PEPC) is considered as the most important reaction in the succinic acid production by using E. coli, in which, 1 mol CO2 is assimilated in this step to form the first C4 metabolites OAA. This step can be enhanced by the better utilization of the PEP pool. In fact, the active substrate for PEPC is not CO2, but the chemically less reactive bicarbonate anion HCO3 − (Kai et al. 2003). Compared with CO2, a nonpolar micro-molecular, HCO3 − is hard to permeate the cell membrane. Furthermore, the hydration/dehydration reaction speed between CO2 and HCO3 − is relatively slow (Badger and Price 2003; Kozliak et al. 1995; Price and Badger 1989). So, there might not be sufficient HCO3 − spontaneously made in vivo to meet the biosynthetic needs, the supply of HCO3 − was improved in vivo for the succinic acid production by overexpressing a heterogeneous CA encoded by cyanobacterium Anabaena sp. 7120 ecaA gene. Increased PEPC activity by expressing pepc gene could enhance the carboxylation of PEP to oxaloacetate (OAA). OAA was sequentially reduced to malate and eventually converted to succinic acid by overexpressing pepc in wild E. coli (Millard et al. 1996; Wang et al. 2010). Furthermore, more energy and nutrients would be used if ecaA gene or pepc gene was expressed at a higher level, resulting in a metabolic burden that interfered with the cell need for basic maintenance and propagation and reduced the succinic acid production. Therefore, when the two genes were co-expressed, the optimization of the two gene expression levels is necessary for saving nutrition and energy cost while maintaining an appropriate enzymatic activity. Fine-tuning metabolic pathways by using the rbSSR library employed in this study could be extended to the production of other biochemicals.
References
Alper H, Fischer C, Nevoigt E, Setphanopoulos G (2005) Tuning genetic control through promoter engineering. Proc Natl Acad Sci U S A 102:12678–12683
Badger MR, Price GD (2003) CO2 concentrating mechanism in cyanobacteria: molecular components, their diversity and evolution. J Exp Bot 54:609–622
Bailey JE (1991) Toward a science of metabolic engineering. Science 252:1668–1675
Balzer GJ, Thakker C, Bennett GN, San KY (2013) Metabolic engineering of Escherichia coli to minimize byproduct formate and improving succinate productivity through increasing NADH availability by heterologous expression of NAD(+)-dependent formate dehydrogenase. Metab Eng 20:1–8
Barrick D, Villanueba K, Childs J, Kalil R, Schneider TD, Lawrence CE, Gold L, Stormo GD (1994) Quantitative-analysis of ribosome binding-sites in Escherichia coli. Nucleic Acids Res 22:1287–1295
Beato M (1989) Gene-regulation by steroid-hormones. Cell 56:335–344
Benner SA, Sismour AM (2005) Synthetic biology. Nat Rev Genet 6:533–543
Bi CH, Su P, Muller J, Yeh YC, Chhabra SR, Beller HR, Singer SW, Hillson NJ (2013) Development of a broad-host synthetic biology toolbox for Ralstonia eutropha and its application to engineering hydrocarbon biofuel production. Microb Cell Factories 12:107
Callura JM, Dwyer DJ, Isaacs FJ, Cantor CR, Collins JJ (2010) Tracking, tuning, and terminating microbial physiology using synthetic riboregulators. Proc Natl Acad Sci U S A 107:15898–15503
Chen H, Bijerknes M, Kumar R, Jay E (1994) Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res 22:4953–4957
Clark DP (1989) The fermentation pathways of Escherichia coli. FEMS Microbiol Rev 63:223–234
Cox RS, Surette MG, Elowitz MB (2007) Programming gene expression with combinatorial promoters. Mol Syst Biol 3:145
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad U S A 97(12):6640–6645
Dong HJ, Tao WW, Zhang YP, Li Y (2012) Development of an anhydrotetracycline-inducible gene expression system for solvent-producing Clostridium acetobutylicum: a useful tool for strain engineering. Metab Eng 14:59–67
Egbert RG, Klavins E (2012) Fine-tuning gene networks using simple sequence repeats. Proc Natl Acad Sci USA 109:16817–16822
Eiteman MA, Miller JH (1995) Effect of succinic acid on 2,3-butanediol production by Klebsiella oxytoca. Biotechnol Lett 17:1057–1062
Feist AM, Palsson BO (2008) The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol 26:659–667
Fischbach M, Voigt CA (2010) Prokaryotic gene clusters: a rich toolbox for synthetic biology. Biotechnol J 5:1277–1296
Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire MA, Benders GA, Montague MG, Ma L, Moodie MM, Merryman C, Vashee S, Krishnakumar R, Assas-Garcia N, Andrews-Pfannkoch C, Denisova EA, Young L, Qi ZQ, Segall-Shapiro TH, Calvey CH, Parmar PP, Hutchison CA, Smith HO, Venter JC (2010) Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329:52–56
Hawkins AS, McTernan PM, Lian H, Kelly RM, Adams MWW (2013) Biological conversion of carbon dioxide and hydrogen into liquid fuels and industrial chemicals. Curr Opin Biotechnol 24:376–384
Kai Y, Matsumura H, Lzui K (2003) Phosphoenolpyruvate carboxylase: three-dimensional structure and molecular mechanisms. Arch Biochem Biophys 414:170–179
Keasling JD (2010) Manufacturing molecules through metabolic engineering. Science 330:1355–1358
Kodaki T, Katagiri F, Asano M, Izui K, Katsuki H (1985) Cloning of phosphoenolpyruvate carboxylase gene from a cyanobacterium, Anacystis nidulans, in Escherichia coli. J Biochem 97:533–539
Kozliak EI, Fuchs JA, Guilloton MB, Anderson PM (1995) Role of bicarbonate/CO2 in the inhibition of Escherichia coli growth by cyanate. J Bacteriol 177:3213–3219
Laetitia M, Martin F (2006) Impact of RNA interference on gene networks. Metab Eng 8(6):672–683
Langer R, Tirrell DA (2004) Designing materials for biology and medicine. Nature 428:487–492
Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY (2012) Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol 8:536–546
Leitao AL, Engutia FJ (2014) Fungal extrolites as a new source for therapeutic compounds and as building blocks for applications in synthetic biology. Microbiol Res 169:652–665
Lescot M, Dehais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, Rouze P, Rombauts S (2002) PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res 30:325–327
Levin-Karp A, Barenholz U, Bareia T, Dayagi M, Zelcbuch L, Antonovsky N, Noor E, Milo R (2013) Quantifying translational coupling in E. coli synthetic operons using RBS modulation and fluorescent reporters. ACS Synth Biol 2:327–336
Liang LY, Liu RM, Ma JF, Chen KQ, Jiang M, Wei P (2011) Increased production of succinic acid in Escherichia coli by overexpression of malate dehydrogenase. Biotechnol Lett 33:2439–2444
Liu YP, Zheng P, Sun ZH, Ni Y, Dong JJ, Zhu LL (2008) Economical succinic acid production from cane molasses by Actinobacillus succinogenes. Bioresour Technol 99:1736–1742
Liu RM, Liang LY, Wu MK, Chen KQ, Jiang M, Ma JF, Wei P, Ouyang PK (2013) CO2 fixation for succinic acid production by engineered Escherichia coli co-expressing pyruvate carboxylase and nicotinic acid phosphoribosyltransferase. Biochem Eng J 79:77–83
Lu TK, Khalil AS, Collins JJ (2009) Next-generation synthetic gene networks. Nat Biotechnol 27:1139–1150
Mayfield S (2013) The green revolution 20: the potential of algae for the production of biofuels and bioproducts. Genome 56:551–555
McKinlay JB, Vieille C, Zeikus JG (2007) Prospects for a bio-based succinate industry. Appl Microbiol Biotechnol 76:727–740
Millard CS, Chao Y-P, Liao JC, Donnelly MI (1996) Enhanced production of succinic acid by overexpression of phosphoenolpyruvate carboxylase in Escherichia coli. Appl Environ Microbiol 62:1808–1810
Mossman BT, Borm PJ, Castranova V, Costa DL, Donaldson K, Kleeberger SR (2007) Mechanisms of action of inhaled fibers, particles and nanoparticles in lung and cardiovascular diseases. Part Fibre Toxicol 4:4
Mutalik VK, Qi L, Guimaraes JC, Lucks JB, Arkin AP (2012) Rationally designed families of orthogonal RNA regulators of translation. Nat Chem Biol 8:447–454
Oliver JWK, Machado IMP, Yoneda H, Atsumi S (2014) Combinatorial optimization of cyanobacterial 2,3-butanediol production. Metab Eng 22:76–82
Orom UA, Nielsen FC, Lund AH (2008) MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell 30:460–471
Paddon CJ, Keasling JD (2014) Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat Rev Microbiol 12:355–367
Pribnow D (1975) Nucleotide-sequence of an RNA-polymerase binding-site at an early T7 promoter. Proc Natl Acad U S A 72:784–788
Price GD, Badger MR (1989) Expression of human carbonic anhydrase in the cyanobacterium Synechococcus PCC7942 creates a high CO2-requiring phenotype. Evidence for a central role for carboxysomes in the CO2 concentrating mechanism. Plant Physiol 91:505–513
Rabinovitch-Deere CA, Oliver JWK, Rodriguez GM, Atsumi S (2013) Synthetic biology and metabolic engineering approaches to produce biofuels. Chem Rev 113:4611–4632
Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kridy J (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440:940–943
Sakai Y, Abe K, Nakashima S, Yoshida W, Ferri S, Sode K, Ikebukuro K (2014) Improving the gene-regulation ability of small RNAs by scaffold engineering in Escherichia coli. ACS Synth Biol 3:152–162
Salis HM, Mirsky EA, Voigt CA (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 27:946–950
Sueel GM, Kulkarni RP, Dworkin J, Garcia-Ojalvo J, Elowtiz MB (2007) Tunability and noise dependence in differentiation dynamics. Science 315:1716–1719
Thakker C, Zhu JF, San KY, Bennet G (2011) Heterologous pyc gene expression under various natural and engineered promoters in Escherichia coli for improved succinate production. J Biotechnol 155:236–243
Tong AHY, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CWV, Bussey H, Andrews B, Tyers M, Boone C (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294:2364–2368
Vellanoweth RL, Rabinowitz JC (1992) The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol Microbiol 6:1105–1114
Wang D, Li Q, Li WL, Xing JM, Su ZG (2009) Improvement of succinic acid production by overexpression of a cyanobacterial carbonic anhydrase in Escherichia coli. Enzyme Microbial Technol 45:491–497
Wang D, Li Q, Mao Y, Xing J, Su Z (2010) High-level succinic acid production and yield by lactose-induced expression of phosphoenolpyruvate carboxylase in ptsG mutant Escherichia coli. Appl Microbiol Biotechnol 87:2025–2035
Wang C, Zhang HL, Cai H, Zhou ZH, Chen YL, Ouyang PK (2014a) Succinic acid production from corn cob hydrolysates by genetically engineering Corynebacterium glutamicum. Appl Biochem Biotechnol 172:340–350
Wang J, Zhang BY, Zhang J, Wang HH, Zhao MH, Wang N, Dong LC, Zhou XH, Wang D (2014b) Enhanced succinic acid production and magnesium utilization by overexpression of magnesium transporter mgtA in Escherichia coli mutant. Bioresour Technol 170:125–131
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16s ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Werpy T, Petersen G (2004) Top value added chemicals from biomass. Department of energy. Washington, DC, pp 31–33
Wibur KM, Anderson NG (1948) Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 176:147–154
Yang DD, Francois JM, de Billerbeck GM (2012) Cloning, expression and characterization of an aryl-alcohol dehydrogenase from the white-rot fungus Phanerochaete chrysosporium strain BKM-F-1767. BMC Microbiol 12:126
Ye VM, Bhatia SK (2012) Metabolic engineering for the production of clinically important molecules: omega-3 fatty acids, artemisinin, and taxol. Biotechnol J 7:20–33
Yeh PJ, Hegreness MJ, Aiden AP, Kishony R (2009) Systems Microbiology. Opinion: drug interactions and the evolution of antibiotic resistance. Nat Rev Microbiol 7:460–466
Zelcbuch L, Antonovsky N, Bar-Even A, Levin-Karp A, Barenholz U, Dayagi M, Liebermeister W, Flamholz A, Noor E, Amram S, Brandis A, Bareia T, Yofe I, Jubran H, Milo R (2013) Spanning high-dimensional expression space using ribosome-binding site combinatorics. Nucleic Acids Res 41:e98
Zhang XL, Jantama K, Moore JC, Jarboe LR, Shanmugam KT, Ingram LO (2009) Metabolic evolution of energy-conserving pathways for succinate production in Escherichia coli. Proc Natl Acad U S A 106:20180–20185
Zhang F, Carothers JM, Keasling JD (2012) Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat Biotechnol 30:354–359
Zhu LW, Li XH, Zhang L, Li HM, Liu JH, Yuan ZP, Chen T, Tang YJ (2013) Activation of glyoxylate pathway without the activation of its related gene in succinate-producing engineered Escherichia coli. Metab Eng 20:9–19
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant 21106191, 21206175), the State Key Laboratory of Materials-Oriented Chemical Engineering (Grant KL14-11), and Fundamental Research Funds for the Central Universities (project no.CQDXWL-2013-019). The authors thank Professor Clark (Southern Illinois University) for strain DC1515.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
(DOC 58.5 kb)
Rights and permissions
About this article

Cite this article
Wang, J., Qin, D., Zhang, B. et al. Fine-tuning of ecaA and pepc gene expression increases succinic acid production in Escherichia coli . Appl Microbiol Biotechnol 99, 8575–8586 (2015). https://doi.org/10.1007/s00253-015-6734-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6734-7