
Abstract
Enzymatic production of biodiesel by transesterification of triglycerides and alcohol, catalyzed by lipases, offers an environmentally friendly and efficient alternative to the chemically catalyzed process while using low-grade feedstocks. Methanol is utilized frequently as the alcohol in the reaction due to its reactivity and low cost. However, one of the major drawbacks of the enzymatic system is the presence of high methanol concentrations which leads to methanol-induced unfolding and inactivation of the biocatalyst. Therefore, a methanol-stable lipase is of great interest for the biodiesel industry. In this study, protein engineering was applied to substitute charged surface residues with hydrophobic ones to enhance the stability in methanol of a lipase from Geobacillus stearothermophilus T6. We identified a methanol-stable variant, R374W, and combined it with a variant found previously, H86Y/A269T. The triple mutant, H86Y/A269T/R374W, had a half-life value at 70 % methanol of 324 min which reflects an 87-fold enhanced stability compared to the wild type together with elevated thermostability in buffer and in 50 % methanol. This variant also exhibited an improved biodiesel yield from waste chicken oil compared to commercial Lipolase 100L® and Novozyme® CALB. Crystal structures of the wild type and the methanol-stable variants provided insights regarding structure-stability correlations. The most prominent features were the extensive formation of new hydrogen bonds between surface residues directly or mediated by structural water molecules and the stabilization of Zn and Ca binding sites. Mutation sites were also characterized by lower B-factor values calculated from the X-ray structures indicating improved rigidity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
During the last decades, the use of biodiesel in compression-ignition engines has become popular as it offers an environment-friendly, biodegradable, nontoxic, and sustainable alternative to the traditional fossil diesel (Al-Zuhair 2007; Bajaj et al. 2010; Meher et al. 2006; Tan et al. 2010). Biodiesel comprises mono alkyl esters of long-chain fatty acids which is mainly produced by transesterification of triglycerides derived from renewable sources (e.g., plant oil or animal fat) and short-chain alcohol in a nonaqueous reaction system. Methanol is frequently used for biodiesel production due to its reactivity and low cost, and the reaction is termed as methanolysis (Al-Zuhair 2007; Lotti et al. 2014; Meher et al. 2006; Tan et al. 2010).
Most of the industrial biodiesel production is catalyzed by strong alkali catalysts resulting in high conversion yields and short reaction times of the process (Al-Zuhair 2007; Du et al. 2008; Tan et al. 2010). Biocatalytic processes based on lipases (EC 3.1.1.3) also exist. These enzymes are ubiquitous in all life domains where they play a key role in the biological turnover of lipids. Under natural aqueous conditions, they facilitate the hydrolysis of the carboxyl ester bonds present in triglycerides. However, in micro-aqueous environment, they possess the ability to catalyze esterification and transesterification reactions (Gupta et al. 2004; Jaeger et al. 1994, 1999; Lotti et al. 2014).
The enzymatic reaction system enables the use of low-grade and low-cost feedstock with high free fatty acids or water content (e.g., waste cooking oil or tallow). It reduces the amounts of alkali waste water that requires treatment and enables easy recovery of the product and the byproduct, glycerol. In addition, the biocatalyst is biodegradable and when immobilized can be reused for many production cycles and can lower the operational cost of the process. However, the major drawbacks of the enzymatic system are the biocatalyst cost and its limited stability in the presence of high methanol concentrations (Al-Zuhair 2007; Bajaj et al. 2010; Lotti et al. 2014; Tan et al. 2010). In order to achieve complete conversion of one oil molar equivalent to fatty acid methyl ester (FAME), three molar equivalents of methanol are necessary. However, in reaction systems with more than 1.5 methanol equivalents, the methanol is not completely dissolved and methanol droplets stay in the mixture (Shimada et al. 1999). The interaction of these methanol droplets with the lipase is most likely the reason for methanol-induced unfolding and inactivation.
Several explanations were offered for the destructive effect of polar solvents, such as methanol, on enzymes. Among them are the stripping off structurally important water molecules, penetration of the solvent into the protein, and disruption of the core hydrophobic interactions, as well as unbalanced electrostatic interactions between the enzyme surface and the organic solvent molecules (Arnold 1990; Chakravorty et al. 2012; Doukyu and Ogino 2010). Moreover, methanol has been shown by Pleiss et al. to be a competitive inhibitor of lipase B from Candida antarctica (CALB) (Kulschewski et al. 2013). Finally, methanol may influence lipases via its interactions with substrates (Smith and Canady 1992).
Different strategies were used to overcome the limited stability of lipases in methanol. The most popular strategy is the immobilization of the enzyme on various support matrices leading to structural rigidity and stability of the catalyst along with recycling ability (Dizge et al. 2009; Hsu et al. 2002; Noureddini et al. 2005; Yagiz et al. 2007). Process engineering was also used to lower the methanol concentration in the reaction by stepwise addition (Bélafi-Bakó et al. 2002; Nie et al. 2006; Soumanou and Bornscheuer 2003) or by the use of co-solvent systems. In addition, in recent years, few attempts were made to enhance lipases stability in methanol by applying protein engineering methods (Dror et al. 2014; Korman et al. 2013; Park et al. 2012). However, the protein engineering approach was hardly exploited due to the lack of structural information and profound understanding of methanol effect on the lipase structure and activity.
Different studies on lipase stability in polar organic solvents have highlighted the importance of the enzyme surface as a target for mutagenesis. Modification of surface charges by substitution with more hydrophobic residues was successfully used by Martinez and Arnold to enhance the stability of α-lytic protease in the presence of DMF (dimethylformamide) (Martinez and Arnold 1991). Furthermore, the introduction of new electrostatic interactions, hydrogen bonds, or salt bridges was reported to enhance enzyme stability (Dror et al. 2014; Kawata and Ogino 2009; Korman et al. 2013; Park et al. 2012; Reetz and Carballeira 2007; Reetz et al. 2010).
In this study, we employ a lipase isolated from the thermophilic bacterium, Geobacillus stearothermophilus T6. Lipase T6 is a highly active robust thermostable enzyme of the I.5 lipase subfamily (Meshulam-Simon 2001). These properties make it a potential biocatalyst for various industrial applications. Yet, its poor stability in the presence of short-chain alcohols, such as methanol or ethanol, prevents its application in reactions systems with high concentrations of these alcohols, like in the case of biodiesel production. In our previous work, we enhanced lipase T6 stability in methanol and ethanol by applying complimentary protein engineering approaches of random mutagenesis and structure guided consensus. We successfully evolved a methanol-stable variant, H86Y/A269T, which showed elevated thermostability together with improved methanolysis activity (Dror et al. 2014). In order to further improve the lipase stability in methanol, we substituted surface charged residues to hydrophobic ones. By combining several mutations, we obtained an additive effect and enhanced methanolysis activity of soybean oil and chicken oil. In addition, X-ray crystallography was used to determine the structures of wild-type lipase and its methanol-stable variants in order to gain insights on structure-stability correlations.
Materials and methods
Chemicals
Methanol, glycerol, and Triton X-100 were purchased from Bio-Labs (Jerusalem, Israel) and 2-propanol from J.T. Baker (Deventer, The Netherlands). Ethyl acetate, sulfuric acid 98 %, petroleum ether, and diethyl-ether were purchased from Gadot (Haifa, Israel), acetonitrile from Spectrum Chemical MFG (Gardena, CA, USA). Trizma-base, 4-nitrophenyl laurate (pNPL) and kanamycin were purchased from Sigma–Aldrich (Rehovot, Israel). p-Nitrophenol was purchased from Fluka (Switzerland). Methyl esters of palmitic acid, heptadecanoic acid, stearic acid, oleic acid, linoleic acid, linolenic acid, and polyethylene glycol (PEG 3015–3685) were purchased from Sigma–Aldrich (Rehovot, Israel). Refined soybean oil was purchased from a local grocery store, and waste chicken oil was obtained from Green City Ltd. (Ein Hamifratz, Israel). All materials used were of the highest purity available.
Bacterial strains, plasmids, and enzymes
Recombinant Geobacillus stearothermophilus T6 lip gene (EMBL, AF429311.1) (Meshulam-Simon 2001) with a histidine tag introduced to the gene C-terminus was cloned into vector pET9a (Novagen, Darmstadt, Germany) after double digestion with NdeI and BamHI (New England Biolabs, MA, USA) (Dror et al. 2014). The vector pET9a/lip T6His was transformed into Escherichia coli BL21 (DE3; Novagen, Darmstadt, Germany) for overexpression.
Novozymes® Lipolase 100L (Thermomyces lanuginosus lipase) and CALB (Candida antarctica lipase B) were purchased from Sigma–Aldrich (Rehovot, Israel).
Lipase activity assay
The lipase hydrolytic activity was determined using a simple colorimetric assay as described before by Dror et al. coworkers (Dror et al. 2014) with few modifications. Briefly, the assay was based on the hydrolysis of pNPL in 96-well plates format and periodic measurement of the liberated p-nitrophenol (pNP) absorbance at 405 nm using a multi-plate reader (Eon™ Microplate Spectrophotometer, BioTek Instruments, Inc., USA). A total of 10 μl of lipase solution (crude or purified) was added to 180 μl reaction mixture (50 mM Tris–HCl pH 8, 1.25 mM CaCl2, 4 % 2-propanol, and 1 % acetonitrile). The reaction was conducted at 40 °C and started with the addition of 10 μl 20 mM pNPL dissolved in 2-propanol. After incubation in methanol, the lipase solution was diluted with dilution buffer (0.5 mM CaCl2, 100 mM NaCl, 50 mM Tris–HCl pH 7.5 buffer) prior to the activity assay so that the methanol final concentration in the reaction mixture was <4 × 10−3 %. All experimental results represent triplicates unless pointed otherwise.
Surface charge modification
Positions (E23A, R104A, K139A, R215A, R231A, R336A, R374W, and R379A) were targeted for mutagenesis based on a 3D homology model with lipase from G. stearothermophilus P1 (PDB code 1JI3 (Tyndall et al. 2002)). Eight oligonucleotides were designed and obtained from Sigma–Aldrich (Rehovot, Israel) (see Table S1 in supplementary material). DNA library of lip T6His with randomization of the eight specific mutations was created by using the ISOR protocol (Incorporating Synthetic Oligonucleotides via Gene Reassembly (Herman and Tawfik 2007)) with modifications as previously described (Dror et al. 2014). After the random hybridization of the oligonucleotides, the assembly product was amplified using “nested” PCR reaction. 1.5 μl assembly product was amplified using Phusion DNA polymerase and 2 ng μl−1 of each primer (NdeI-F and BamHI-R, Table S2) in standard reaction conditions. After purification, the PCR product was incorporated into pET9a as described by Dror et al. (Dror et al. 2014). The resulting library was transformed into E. coli BL21 (DE3) cells via electroporation. Plasmids from 10 colonies were extracted using Mini Kit (Qiagen, CA, USA) and sequenced in order to test the reassembly success and hybridization rate. Ninety-six-well master plates were created with two positive control wells containing E. coli BL21 (DE3) expressing wild-type lipase T6 as previously described for the library screening (Dror et al. 2014).
Site-directed mutagenesis
The triple mutant H86Y/A269T/R374W was created by combining mutants R374W and H86Y/A269T using QuikChange® II Site-Directed Mutagenesis Kit (Stratagene, USA). Mutation R374W was added to pET9a/lip T6His-H86Y/A269T using primers R374W-F and R374W-R (Table S2). The plasmid pET9a/lip T6His-H86Y/A269T/R374W was transformed into E. coli BL21 (DE3) competent cells via electroporation.
Screening for methanol-stable mutants
In order to identify and isolate the methanol stable mutants, we used a high throughput screening in a 96-well plate format as described earlier (Dror et al. 2014). Briefly, colonies from the 96-well master plates were transferred into 96-deep-well plates for containing 1.2 ml of LB with 25 μg ml−1 kanamycin for overnight growth (at 37 °C with shaking at 200 rpm). After, the growth stage cells were harvested by centrifugation, and the cell pellets were broken by incubation in lysis buffer followed by freezing at −80 °C and thawing at 37 °C steps. The cell lysate was diluted by addition of 300 μl 50 mM sodium phosphate buffer pH 8. After additional centrifugation, the supernatant was used as the enzyme solution for activity assay before and after 30 min incubation in 60 % methanol. The relative residual activity of each variant was calculated by comparing its residual activity after incubation in 60 % methanol to the residual activity of the wild type (Dror et al. 2014).
Purification of lipase T6
His6-tag wild-type lipase T6 and its methanol stable mutants were purified in a one-step process using an Ni(II)-bound affinity column (HisTrap HP, Amersham Biosciences, Giles, UK) as described by Dror et al. (Dror et al. 2014) with few modifications. After overnight growth of single E. coli BL21 (DE3) cells harboring pET9a/lip T6His colonies in 0.5 l 0.5 mM CaCl2 TB medium with 25 μg ml−1 kanamycin, The cells were harvested by centrifugation and resuspended in a binding buffer (20 mM Tris–HCl buffer pH 7.5, 0.5 mM CaCl2, 0.1 mM Triton X-100, 500 mM NaCl, and 40 mM imidazole). The cells were broken using a homogenizer (EmulsiFlex-C3 High Pressure Homogenizer, AVESTIN, Ottawa, Canada) followed by centrifugation (16,000×g for 20 min at 15 °C) for the removal of cell debris. The supernatant was separated and exposed to heat treatment of 50 °C for 15 min followed by another centrifugation step. The supernatant was loaded on to Ni(II)-bound affinity column which was prior equilibrated with the binding buffer. The lipase was eluted from the column with a linear gradient of elution buffer (20 mM Tris–HCl buffer pH 7.5, 0.5 mM CaCl2, 0.1 mM Triton X-100, 500 mM NaCl, and 500 mM imidazole). The lipase fractions were collected and dialyzed against lipase buffer (2.5 % glycerol, 0.5 mM CaCl2, 100 mM NaCl 50 mM Tris–HCl pH 7.5 buffer) at room temperature for 24 h.
To prepare the protein for crystallization, the same purification method was used. However, the purification buffers were replaced in order to increase protein solubility and enable protein crystallization. Binding and elution buffers contained 20 mM Tris–HCl buffer, pH 8.5, 1 mM CaCl2, 0.1 mM Triton X-100, 500 mM NaCl, 10 mM 2-mercaptoethanol, 5 % 2-propanol, and 20 mM (binding) or 500 mM (elution) imidazole. The eluted lipase fractions were supplemented with 5 mM DTT and dialyzed against 20 mM Tris–HCl buffer pH 8.5, 0.5 mM CaCl2, 0.1 mM Triton X-100, 100 mM NaCl, 5 % 2-propanol, 0.5 mM ZnCl2, and 5 mM DTT.
Half-life determination in 70 % methanol
Half-life (t 1/2) values of lipase T6 and its mutants in 70 % methanol were determined by measuring their residual activity after different incubation periods (0–600 min) as previously described (Dror et al. 2014). Briefly, 300 μl of 0.2 mg ml−1 lipase solution were mixed with 700 μl methanol to a final volume of 1 ml. Twenty-microliter aliquots were periodically withdrawn from the incubation mixture and transferred into 180 μl lipase dilution buffer. This solution was used to determine the lipase residual activity as described above.
Thermal unfolding of lipase T6
The thermal induced unfolding temperatures of wild-type lipase T6 and the methanol-stable mutants R374W, H86Y/A269T, and R374W/H86Y/A269T were determined by differential scanning calorimetry (DSC) on a MicroCalVP-DSC (GE Healthcare Bio-Sciences AB, Sweden) as described previously (Dror et al. 2014) with a small modification in the lipase buffer. In addition, we tested the methanol effect on the Tm of wild type and R374W/H86Y/A269T by conducting the DSC measurements in 50 % methanol lipase buffer. The reaction cell contained 1 ml of 1.5 mg ml−1 lipase in lipase buffer (2.5 % glycerol, 0.5 mM CaCl2, 100 mM NaCl, 50 mM Tris–HCl, pH 7.5 buffer) or in 50 % methanol/lipase buffer, and the reference cell contained 1 ml of lipase buffer or 50 % methanol/ buffer without the lipase. The measurements were performed by scanning from 25 to 95 °C at 1 °C min−1 and analyzed using Origin software (MicroCal Inc.). Values represent an average of triplicates.
Fatty acid composition and water content of soybean and waste chicken oil
Chemical transesterification of soybean and waste chicken oil was conducted in order to determine the fatty acid composition of the oils. The chemical transesterification was catalyzed by sulfuric acid based on the reaction described by Liu (Liu 1994) with modifications. A total of 20 mg of oil were mixed with 20 ml 2.5 % H2SO4 methanol solution in an Erlenmeyer flask equipped with a condenser. The mixture was stirred on a hot plate at 90 °C using a magnetic stirrer for 2.5 h. Subsequently, the flask was cooled down to room temperature for 15 min followed by addition of 5 ml of 1:1 petroleum ether:diethyl–ether solution. The mixture was transferred into 100 ml measuring bottle followed by addition of distilled water until the organic phase was clearly seen at the bottle neck. After 30 min, the organic phase containing the fatty acid methyl esters (FAME) was transferred into a glass vial, the solvent was evaporated, and FAME mixture was suspended in 1 ml ethyl acetate. A total of 10 μl of the FAME mixture was mixed with 490 μl ethyl acetate and taken for gas chromatographic analysis. The water content of the oils was determined by the Karl Fisher method (Fischer 1935) using 787 KF Titrino (Metrohm, Switzerland).
Enzymatic methanolysis of soybean oil and waste chicken oil
Enzymatic methanolysis of soybean oil catalyzed by lipase T6 and its methanol stable mutants was carried out in order to evaluate the mutations effect on the transesterification activity. In addition, we evaluated the methanolysis of soybean oil and waste chicken oil by wild-type lipase T6, variant H86Y/A269T/R374W and two commercial lipases, T. lanuginosus lipase (Novozymes®, Lipolase 100 L, TLL) and Candida antarctica lipase B (Novozymes® CALB), which were often used in biodiesel production studies (Maceiras et al. 2011; Raita et al. 2011; Rodrigues et al. 2008; Tan et al. 2010). The protein concentration of the lipase solutions was determined by the method of Bradford (Bradford 1976), and samples were analyzed on SDS-PAGE (Sambrook and Russel 2001) to ensure their protein purity. The commercial lipases were diluted with lipase buffer to 2 mg ml−1 lipase. The methanolysis reactions were carried out in 14-ml closed glass vials filled with 2 g oil. In the methanolysis of soybean oil by wild type and methanol stable variants, we used 1.5:1, methanol to soybean oil molar ratio. One hundred microliters of 2 mg ml−1 lipase solution (0.5 mM CaCl2, 2.5 % glycerol, 100 mM NaCl, 50 mM Tris–HCl, pH 7.5) was added to the reaction mixture resulting in 0.01 % lipase and 5 % water content based on oil weight.
For evaluating lipase T6 performance in methanolysis of soybean oil and waste chicken oil in comparison with commercial enzymes, experiments were conducted in high methanol concentrations typically used in industry (methanol to oil ratio of 4.5:1). For both substrates, we added to the reaction mixture 200 μl of 2 mg ml−1 lipase solution resulting in 0.02 % lipase and 10 % water content based on oil weight. Prior to lipase addition, the methanol and oil were mixed to avoid lipase contact with high methanol concentrations. The glass vials with the reaction mixture were maintained at 45 °C in a Vibramax 100 orbital shaker (Heidolph Instruments, Germany) at 1350 rpm. A total of 50 μl samples were taken from the reaction mixture periodically and centrifuged for 3 min at 20,000×g. A total of 10 μl aliquots of the upper layer were weighted and mixed with 490 μl of ethyl acetate with 1 mg ml−1 heptadecanoic acid methyl ester (internal standard) for gas chromatographic analysis.
Gas chromatography analysis of FAME
The FAME content of the chemical and enzymatic transesterification samples was analyzed and quantified using a 6890N GC instrument (Agilent Technologies, CA, USA) equipped with a capillary DB-23 column (60 m × 250 μm × 0.25 μm, Agilent Technologies) and a flame ionization detector as previously described (Dror et al. 2014). The identification and quantification of FAME in the enzymatic methanolysis were done based on FAME standards (Sigma–Aldrich, Rehovot, Israel) retention times and calibration curves described by Dror et al. (Dror et al. 2014). FAME yields were defined as percentage of the sample weight. The FAME percentage in the chemical transesterification was calculated based on the peak area of each FAME in the sample.
Crystallization and data collection
Crystallization trials were performed in 96-well hanging drop plates using MOSQUITO robot with a PEGs Suite (www.qiagen.com) at 20 °C. The crystals appeared after a week at a wide range of crystallization conditions, which were further optimized. The optimal conditions contained 0.2 M sodium citrate and 25 % PEG3350. The hanging drop contained 2 μl protein solution (4 mg ml−1) and 2 μl crystallization condition.
X-ray diffraction data of lipase T6 wild type, H86Y/A269T, and H86Y/A269T/R374W were collected in-house X-ray beam R-Axis HTC (Rigaku), while the X-ray diffraction data of A269T variant were collected at the European Synchrotron Radiation Facility (ESRF), Grenoble, France, at beamline BM14 at 9.8 keV corresponding to the anomalous signal of zinc. Diffraction data was indexed, integrated, and reduced with Moslfm and Scala (Leslie and Joint 1992) or with HKL2000 (Otwinowski and Minor 2001).
The structure of A269T variant was solved by molecular replacement using Phaser (McCoy 2007) and the coordinates of a thermostable lipase from G. stearothermophilus P1 (PDB code 1JI3), which has a 95 % sequence identity to lipase T6. Refinement was performed using PHENIX (Adams et al. 2010). Manual model building, real-space refinement, and structure validations were performed using Coot (Emsley and Cowtan 2004). The structure of wild-type lipase T6 and variants H86Y/A269T and H86Y/A269T/R374W were solved using coordinates of the A269T variant. Crystal parameters and data statistics are summarized in Table S3 in the supplementary material.
Protein data bank accession numbers
The coordinates and structure factors of wild-type lipase T6, A269T, H86Y/A269T, and H86Y/A269T/R374W have been deposited in the RCSB PDB under accession codes 4X6U, 4X71, 4X7B, and 4X85, respectively.
Results
Mutagenesis at surface residues
A semi-rational protein engineering approach of surface charge modification was used in order to enhance the stability of lipase T6 in the presence of high methanol concentrations. This approach suggests that substitution of charged residues on the enzyme surface by more hydrophobic residues may stabilize the protein structure in the presence of polar organic solvents (Arnold 1990). In the absence of a crystal structure of lipase T6 at this point of the research, a 3D homology model of wild-type lipase T6 was generated based on the crystal structure of lipase from G. stearothermophilus P1 (PDB code 1JI3 (Tyndall et al. 2002)) and charged residues on the lipase surface were located. In order to avoid activity loss, we excluded residues placed on the α-helix lid. In addition, in order to prevent destabilization, no residues which were likely to participate in stabilizing interaction (e.g., salt bridges or hydrogen bonds) or placed on the calcium- or zinc-binding domains were chosen for substitution. Considering these criteria, we located eight surface charged residues for substitution (Glu23, Arg104, Lys139, Arg215, Arg231, Arg336, Arg374, and Arg379). All of the residues, besides Arg374, were substituted to alanine. Based on the lipase model, position of Arg374 on the surface was relatively close to Trp41. Therefore, we substituted it to tryptophan in order to generate π interactions between these two residues. A combinatorial library based on these eight residues was generated and screened (Herman and Tawfik 2007).
Over 1000 colonies were screened for enhanced stability in methanol by measuring their residual activity before and after 30 min incubation in 60 % methanol. Only one variant, R374W, showed 2.3-fold improved stability in methanol compared to the wild type. This variant was purified for further characterization. The mutation R374W was added to the double variant H86Y/A269T which was previously found as the most stable variant in methanol (Dror et al. 2014) to create the triple variant H86Y/A269T/R374W.
Half-life determination in 70 % methanol
Half-life values of lipase T6 variants R374W, H86Y/A269T, and H86Y/A269T/R374W were determined in 70 % methanol by periodically measuring their residual activity (E/E 0) and calculating their half-life values from their deactivation profiles (Fig. 1). E 0 and E were defined as the lipase activity in the standard activity assay conditions before and after incubation in 70 % methanol. The half-life value (t 1/2) of the lipase was defined as the time required for the enzyme to lose half of its original activity in 70 % methanol. The half-life values were calculated using the exponential model E/E 0 = exp(−K d t). The correlation coefficient of the model was R 2 > 0.93 for all mutants. This model was previously found to have good agreement with the deactivation profiles of lipase T6 in methanol and ethanol (Dror et al. 2014) and to other enzymes in polar organic solvents (Fishman et al. 2002). In the model, (t) represents the incubation time while (K d) represents the enzyme inactivation constant.

Deactivation profiles of wild-type lipase T6 and methanol-stable variants in 70 % methanol. The activity of purified lipase T6 variants in the standard activity assay conditions (0.002 μg enzyme in 200 μl of 50 mM Tris–HCl pH 8 buffer containing 1 mM pNPL as substrate) was measured after different incubation times in 70 % methanol. The residual activity (E/E 0) was calculated by comparing the activity of before (E) and after (E 0 ) incubation in 70 % methanol. The wild-type data was adapted from Dror et al. (2014). The results represent triplicates
The initial specific hydrolytic activity in buffer was similar for all enzymes. Values of 6.2 ± 0.1, 6.0 ± 0.3, and 6.7 ± 0.2 μmole pNP mg−1 min−1 were measured for wild-type, R374W, and H86Y/A269T/R374W, respectively. In our previous work, we found that 70 % methanol had a strong destructive effect on the wild type as evidenced from a low half-life value of 4 min (Dror et al. 2014). The single mutant, R374W, and the double mutant, H86Y/A269T, showed half-life values of 96 and 224 min, respectively. The combination of these mutations to the triple variant, H86Y/A269T/R374W resulted in an additive stabilizing effect of 324 min half-life value which reflects an 87-fold enhanced stability compared to the wild type (Fig. 1, Table 1).
Thermal unfolding of lipase T6
The thermal induced unfolding (Tm) of lipase T6 and the methanol-stable variants was measured by DCS. In our previous work, we have found a positive correlation between the lipase thermostability and its stability in methanol (Dror et al. 2014). This correlation was found for other enzymes in organic solvents as well (Reetz et al. 2010; Vazquez-Figueroa et al. 2008). The wild-type lipase showed thermal-induced unfolding Tm of 66.8 ± 0.2 °C and H86Y/A269T showed significant elevation of Tm to 70.9 ± 0.1. However, the addition of mutation R374W did not affect the unfolding temperature (P < 0.05). R374W and H86Y/A269T/R374W showed Tm values of 66.2 ± 0.2 and 70.6 ± 0.1 °C, respectively.
In order to evaluate the influence of methanol on Tm, we conducted DSC measurements for wild type and the triple mutant, H86Y/A269T/R374W, in the presence of 50 % methanol. The DSC results in the presence of 50 % methanol showed a significant decrease in the Tm of 23.6 and 21.2 °C for the wild type and R374W/H86Y/A269T, respectively, compared to their Tm in buffer (Table 2 and Fig. 2). In addition, the delta Tm between the wild type and the triple variant was increased from 3.8 °C in buffer to 6.2 °C in 50 % methanol (Table 2). A decrease in thermostability in the presence of methanol was previously reported for Pseudomonas cepacia lipase (Tanaka 1998) and Bacillus subtilis lipase (Kamal et al. 2013). The effect of the “catalyst” factor (wild type or mutant) and the “medium” factor (buffer or 50 % methanol) on the lipase Tm was found significant for each factor separately (P < 0.001). In addition, post hoc ANOVA analysis revealed that the interaction between the “catalyst” and the “medium” factors was significant (P < 0.001). These results emphasize the mutations’ contribution to the lipase conformational stability since the mutant thermal stability was significantly elevated compared to the wild type in the presence of methanol (Table 2 and Fig. 2).

Typical DSC thermograms of wild-type lipase T6 and variant H86Y/A269T/R374W in lipase buffer or 50 % methanol. The unfolding temperatures of purified wild-type lipase T6 and variant H86Y/A269T/R374W were measured by DSC in buffer (0.5 mM CaCl2, 2.5 %glycerol, 100 mM NaCl, 50 mM Tris–HCl, pH 7.5) or in 50 % methanol (% V/V). The thermal-induced unfolding of lipase T6 occurs as a single transition. The thermograms were generated by Origin software (MicroCal Inc.)
Fatty acid composition and water content of soybean and waste chicken oil
The fatty acid (FA) composition of soybean oil and waste chicken oil was determined by chemical transesterification. In the soybean oil, the main fatty acid was linoleic acid (18:2, 52.1 %) and five fatty acids (16:0, 18:0, 18:1, 18:2, and 18:3) composed 99.8 % of the total FA (Table S4). In the waste chicken oil, the main fatty acid was oleic acid (18:1, 38 %) and the main six fatty acids (16:0, 16:1, 18:0, 18:1, 18:2, and 18:3) composed 94.5 % of the total FA. In addition, there was a significant amount of myristic acid (14:0) and unsaturated long-chain FA which were absent in the soybean oil (16:1, 20:1, 20:2, 20:4 and 24:1) (Table S4). Furthermore, the percentage of saturated FA in the soybean oil and in the waste chicken oil was 15.2 and 26.1 %, respectively. The FA composition and the percentage of saturated FA of the waste chicken oil were similar to previous reports (Kaewmeesri et al. 2015; Nizar et al. 2013). The water content of the soybean oil was very low, 0.047 ± 0.002 %, as expected for refined edible oil. In contrast, the water content of the waste chicken oil was 5-folds higher (0.246 ± 0.004 %). This water content was in the typical range for waste chicken oil according to literature (Kaewmeesri et al. 2015).
Enzymatic methanolysis of soybean oil and waste chicken oil
Enzymatic methanolysis of oil for biodiesel production holds many advantages over the chemically catalyzed process. However, the limited stability of the lipase in the presence of high methanol concentrations present in the reaction mixture is a major obstacle for industrial implementation of the enzyme catalyzed process (Al-Zuhair 2007; Lotti et al. 2014).
In this work, we carried out methanolysis reactions using soluble forms of the enzymes. For the evaluation of the mutation effect on the lipase performance in methanolysis, we used soybean oil and a 1.5:1 methanol to oil molar ratio which enables maximal theoretical FAME yield of 50 %. The results indicated a positive correlation between lipase T6 variants’ stability in methanol and their methanolysis activity (Figure S1). The highest conversion rate for all variants was measured during the first 2 h of the reaction and decreased with time. The methanol stable variant, H86Y/A269T/R374W showed a slow decrease in activity after 2 h and after 24 h reached 46 % FAME yield, which is close to the theoretical maximal yield. The decrease in rate can be attributed to lower availability of the substrate (methanol) or to methanol-induced unfolding. On the other hand, wild-type lipase T6 showed poor methanolysis activity, and its activity rate was steeply decreased after 2 h reaction and after 24 h reached 8.6 % FAME yield (Figure S1). These results suggest that lipase T6 poor methanolysis activity was a result of methanol-induced unfolding which led to inactivation.
Wild-type lipase T6, variant H86Y/A269T/R374W, and the commercial lipases were evaluated in methanolysis of soybean oil and waste chicken oil in the presence of a high methanol concentration (4.5:1 methanol:oil). These conditions represent the industrial reaction system in which excess of methanol is used in order to obtain full conversion of oil to FAME. In the soybean oil reaction system, the best activity was observed for TLL which had a very high conversion rate (55 % after 2 h) and reached 95 % FAME yield after 24 h (Fig. 3a). Wild-type and variant H86Y/A269T/R374W showed lower conversion rates (16 and 26 % after 2 h, respectively) with a steep decline in the activity after the first 2 h (Fig. 3a). On the other hand, in the waste chicken oil reaction system, the best activity was observed for H86Y/A269T/R374W (44 % after 2 h) which reached 64 % FAME yield after 24 h (Fig. 3b). In this reaction system, the wild type and TLL showed a much lower activity and reached only 19 and 16 % FAME yield after 24 h (Fig. 3b). The commercial lipase, CALB, showed a very poor methanolysis activity in both reaction systems, and it was almost inactivate after 2 h (Figures 3a, b).

Methanolysis of soybean oil (a) and chicken waste oil (b) by wild-type lipase T6, variant H86Y/A269T/R374W, T. lanuginosus lipase (Novozymes® Lipolase 100 L, TLL), and Candida antarctica lipase B (Novozymes® CALB). Reaction conditions: 2 g oil, 10 % water, 4.5:1 methanol to oil molar ratio, and 0.02 % lipase content based on oil weight, 1350 rpm, 45 °C. The results represent triplicates
Overall structure of lipase T6
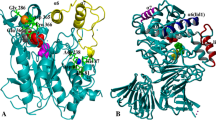
Crystals of wild type and variants diffracted to a 2.19–2.4 Å and belonged to the P212121 space group with the same cell parameters and molecular packing of one molecule in the asymmetric unit (Table S3). The overall structure of lipase T6 is similar to lipase P1 from G. stearothermophilus (PDB code 1JI3, rms 0.225) (Tyndall et al. 2002) and consists of 10 β-strands and 18 α-helices. The active site of lipase T6 is located in a hydrophobic pocket and composed of a catalytic triad Ser114, His359, and Asp318, while a hydrophobic helical lid (residues 176–192) in closed conformation isolates the active site from the solvent. Two metal ions Zn2+ and Ca2+ were found in the structures of lipase T6. These metal ions were also reported for other lipases belonging to I.5 subfamily and contribute to the thermostability of the enzymes (Guncheva and Zhiryakova 2011). The Ca2+ is coordinated by Asp366, Glu361, Pro367, and Gly287 and two water molecules, which were visualized only in variant A269T. The Zn2+ is coordinated by His82, His88, Asp62, and Asp239 (Figs. 4 and 5). The Zn2+ presence was confirmed using zinc anomalous scattering (Figure S2).

Crystal structure of wild-type lipase T6. The α-helix lid, α9, (Phe177-Ala192) is marked in red. The catalytic triad residues (Ser114, Asp318, and His359) are shown in green sticks. The calcium-binding residues (Glu361,Gly287, Pro367, and Asp366) are shown in magenta sticks, and zinc-binding residues (Asp62, His88, Asp239, and His82) are shown in cyan sticks. Calcium and zinc metal ions are shown as yellow and gray spheres, respectively. Residues subjected to mutagenesis (H86, A269, and R374) are shown in orange sticks. All figures presented in this work were generated using PyMOL (http://www.pymol.org/)

Close-up of structural mutation sites in wild-type lipase T6 and variant H86Y/A269T/R374W. Wild-type A269 (a) and A269T (b). Wild-type H86 (c) and H86Y (d). Wild-type R374 (e) and R374W (f). The residues subjected to mutagenesis are in orange sticks. Zinc and calcium metal ions are shown as gray and yellow spheres, respectively. Water molecules are shown as red spheres. Conserved water molecules discussed in the text are marked by yellow asterisks. Possible hydrogen bonds are marked in black dashed lines; α18
Effect of mutations on methanol-stability
Structures of the methanol-stable variants were solved and compared to the structure of wild-type enzyme. The overall structures are very similar (rms 0.144–0.212), suggesting that the stabilizing effect in methanol did not occur as a result of significant conformational changes but due to local ones. It was observed that the replacement of Ala to Thr at position 269, enabled the formation of hydrogen bonds with two structurally conserved water molecules. These water molecules connect between β8 and α16 thus stabilizing this entire region (Figure 5a, b). The replacement of His to Tyr at position 86 provoked the movement of a conserved water molecule, this movement widened a hydrogen bond network and enabled more direct contact with His82 which coordinates Zn2+. In addition Tyr86 also formed a hydrogen bond with Lys85 which stabilizes the region around the Zn2+ binding site (Figure 5c, d).
The most interesting observation was at position 374, in which we substituted Arg with Trp to presumably form π-interactions with Trp41. However, in contrast to our expectation, Trp41 made a flip of 100° and formed a hydrogen bond with the back bone of Trp374. This hydrogen bond connects helices α1 and α18 and thus provides structural stability (Figure 5e, f). In addition, Trp374 formed a hydrogen bond with a conserved water molecule which in turn formed a hydrogen bond with Arg35. Arg35 plays a key role in coordination and stabilization of the Ca2+ binding site residues through hydrogen bond interactions with the backbone of Phe371, Asn368, and a structural water molecule. This water molecule generates hydrogen bonds with the backbone of Asp366 and Glu361 which ligate Ca2+ in the binding site. Moreover, a larger network of hydrogen bonds between water molecules, Arg35 and the Ca2+ binding site loop was formed (Figure 5e, f).
Another way to evaluate the stabilizing effect of the mutations on the lipase structure was to calculate the change in the average B factor values surrounding the mutation positions. The B-factor or the temperature factor in X-ray data reflects the flexibility of an atom position. Therefore, we calculated the changes in B-factor values between the wild type and variant H86Y/A269T/R374W (Table 3). A decrease of 2.4–4.0 Å2 in the B-factors corresponds well with the improved structural stability of the triple variant H86Y/A269T/R374W.
Discussion
Enzymatic esterification and transesterification reactions, catalyzed by lipases, hold a great promise for environmentally friendly, sustainable, and economical industrial production of biodiesel. However, the biocatalyst cost and its poor stability in methanol hinder the exploitation of the process on a large scale. Consequently, methanol stable lipases are of great interest for the biodiesel and chemical industries. Protein engineering by a semi-rational approach was applied to modify surface charged residues. These residues did not participate in electrostatic interactions, and it was generally expected that their charge modification may eliminate a potential force for structural unfolding in the presence of methanol. Eight different charged residues were substituted by hydrophobic ones and screened for improved stability. However, only one mutation, R374W, was found as beneficial to enhance the lipase stability in methanol (Fig. 1, Table 1). Structural visualization of this mutation revealed the formation of new hydrogen bonds between helices and structural water molecules which stabilized the Ca2+ binding site loop (Fig. 5e, f). These observations suggest that the stabilizing effect was more likely a result of new hydrogen bonds formation on the lipase surface rather than charge modification or π interactions as was presumed. In our study, we have chosen alanine as the alternative amino acid for seven charged surface residues. Although these substitutions eliminated the surface charge, they were not effective in stabilizing the lipase in methanol presumably since new stabilizing interactions were not introduced (e.g., hydrogen bonds, salt bridges). Therefore, we assume that by using saturation mutagenesis at the predetermined positions rather than the introduction of specific alternations, we could have found more beneficial substitutions which could have created new stabilizing interactions. Alternatively, a polar residue such as serine could have been superior. Martinez and Arnold (1991) have successfully used saturation mutagenesis to modify the surface charge of α-lytic protease to enhance its stability in DMF. Their results have shown that for each position different substitutions contributed to the enzyme stability, and their stabilizing effect was not solely attributed to the charge modification (Martinez and Arnold 1991).
The combination of variants R374W and H86Y/A269T to create the triple variant, H86Y/A269T/R374W, resulted in an additive stabilizing effect in methanol and improved performance in methanolysis of soybean oil and waste chicken oil (Figs. 1, S1, 3, and Table 1) while the hydrolytic activity remained unchanged. In addition, all three mutations are located on the enzyme surface and have no direct contact with structural regions directly associated with the lipase activity (e.g., lid helices, active site) (Fig. 4). Based on these observations, we suggest that the improved methanolysis activity of the triple mutant was a result of its enhanced stability in methanol rather than a change in the catalytic activity. A positive correlation between lipase-enhanced stability in methanol, and improved methanolysis activity was also reported by Korman et al. who applied directed evolution to Proteus mirabilis lipase (Korman et al. 2013). Furthermore, the mutation stabilizing effect was confirmed by the DSC analysis in which the ∆Tm between the triple mutant, and the wild type in 50 % methanol was nearly twice larger than in buffer (Fig. 2 and Table 2).
The overall goal of our study was to enhance lipase T6 stability in methanol in order to improve its performance in biodiesel synthesis. Therefore, we compared the methanolysis activity of wild type and variant H86Y/A269T/R374W with two commercial lipases (Fig. 3). These lipases in their immobilized form, Lipozyme®TL IM (TLL) and Novozym®435 (CALB) were considered as the leading biocatalysts for biodiesel synthesis (Fernandez-Lafuente 2010; Hernández-Martín and Otero 2008). However, methanol-induced inactivation in reaction systems with more than 1.5 methanol molar equivalent to oil was previously reported for TLL (Xu et al. 2004), CALB (Shimada et al. 1999), and lipase T6 (Dror et al. 2014). For the methanolysis reactions, we used soybean and waste chicken oil. Waste chicken oil is a slaughterhouse byproduct, and its low cost and low grade make it a perfect feedstock for enzymatic biodiesel production. The percentage of saturated FA in the waste chicken oil was significantly higher than in soybean oil (Table S4), making it a more suitable feedstock for biodiesel production. A higher percentage of saturated FA may increase the cetane number and increase the cloud point of the FAME (Karmakar et al. 2010). The methanolysis results showed that for both oil feedstocks, variant H86Y/A269T/R374W was better than the wild type as a result of its enhanced stability in methanol. In the soybean oil system, TLL showed the best activity and closely reached full conversion of oil to FAME while H86Y/A269T/R374W was the second best (Fig. 3a). However, in the methanolysis of waste chicken oil, H86Y/A269T/R374W was much better than TLL and greatly surpassed CALB (Fig. 3b). The different activities of H86Y/A269T/R374W and TLL on different oil feedstocks are most likely a result of their composition. Refined soybean oil is composed mainly of triglycerides while the waste chicken oil was composed of 89.5 triglycerides and 10.5 % of diglycerides, monoglycerides, and FFA (according to the supplier). TLL was reported to have high stability in methanol (Fernandez-Lafuente 2010), and this explains the high conversion of the soybean oil. However, its poorer activity in the waste chicken oil can be attributed to inhibition by detergent-like molecules such as FFA, monoglycerides, or phospholipids present in the feedstock. It was previously reported by Mogensen et al. that TLL activity was inhibited by detergents (Mogensen et al. 2005). It was suggested that the inhibition may occur when the detergent molecules bind in the active site pocket, and as a result, the substrate access into the active site is prevented (Mogensen et al. 2005). Methanolysis inhibition by phospholipids was also reported for Aspergillus niger lipase (Li et al. 2014) and CALB (Watanabe et al. 2002). On the other hand, H86Y/A269T/R374W methanolysis of soybean oil stopped after 2 h reaction time probably due to direct contact of the lipase with methanol droplets which led to methanol-induced unfolding. We suggest that the high content of polar “detergent-like” substances in the waste chicken oil improved the methanol solubility and decreased the lipase T6 unfolding resulting in improved methanolysis (Fig. 3b).
The results presented in this work show that by introducing three mutations, lipase T6 poor methanolysis activity can be elevated to the level of the commercial lipase TLL (Fig. 3). In addition, the results of the waste chicken oil methanolysis showed the great potential of lipase T6 as a biocatalyst for biodiesel production from low-grade feedstocks. Nevertheless, further improvement of lipase T6 stability in methanol will enable its application in reaction systems with higher methanol content which will improve the FAME yield and shorten reaction times.
In our previous work, we used homology modeling to study the contribution of A269T and H86Y to the stability in methanol. We suggested that these mutations enable the formation of new hydrogen bonds with other residues (Dror et al. 2014). In this study, the crystal structures of the lipase variants have confirmed our assumptions and provided a detailed view of the stabilizing interactions which could not have been seen in the model. The most prominent feature discovered from the structures was the extensive formation of new hydrogen bonds between surface residues directly or mediated by structural water molecules (Figs. 4 and 5). Mutations on the protein surface were also found to have a great influence on the stability in polar organic solvents of Pseudomonas aeruginosa LST-03 lipase (Kawata and Ogino 2009), Salinivibrio proteolyticus metalloprotease (Badoei-Dalfard et al. 2010), CALB (Park et al. 2012) and Bacillus subtilis lipase (Reetz et al. 2010). It was noted that polar solvents, such as methanol, can destabilize the protein structure by penetrating into the hydrophobic core, thus leading to unfolding and inactivation (Doukyu and Ogino 2010). We suggest that lipase T6-enhanced stability in methanol resulted from the extension of the hydrogen bonds network on the lipase surface which contributed to its rigidity and prevented the methanol penetration into the enzyme core and thus assisted in maintaining its active conformation. Similar observations were reported by Reetz et al. (2010) who concluded that the enhanced stability of B. subtilis lipase mutants in three polar organic solvents was a result of a new amino acid cooperative network which was formed by salt bridges and hydrogen bonds on the lipase surface.
One of the mechanisms which was proposed to trigger enzymes unfolding in the presence of polar organic solvents was stripping of structurally essential water molecules from the protein surface which may lead to a decrease in the protein conformational stability and lower the protein Tm (Doukyu and Ogino 2010; Kamal et al. 2013; Klibanov 1997). Our results support this theory as all three mutations enabled the formation of hydrogen bonds to structural water molecules and elevated the lipase thermostability in 50 % methanol (Figs. 2, 5, and Table 2). The substitution of alanine with threonine at position 269 enabled the formation of two new hydrogen bonds to structural water molecules. In addition, this mutation was also reported to elevate the unfolding temperature of the lipase by 3.4 °C (Dror et al. 2014). The stabilizing effect in methanol of mutations H86Y and R374W can be attributed not only to hydrogen bond formation but also to the stabilization of the Zn2+ and Ca2+ binding sites, respectively (Fig. 5). It was previously suggested that the Zn2+ and Ca2+ binding sites of the homologues G. stearothermophilus P1 and L1 lipases contribute to the enzymes stability (Kim et al. 2000; Tyndall et al. 2002). Mutation H86Y enlarged the hydrogen bond network around the Zn2+ binding site thus providing further stabilization and increased unfolding temperature as well as protein stability in methanol. Similarly, mutation R374W stabilized the Ca2+ binding site loop and greatly increased the stability in methanol (Fig. 1). Similar results were observed for P. mirabilis lipase, where the reorganization of a loop around the Ca2+ binding site, improved protein stability in methanol environment (Korman et al. 2013).
The mutation stabilizing effect on the lipase structure was also reflected by the decrease in the B-factor values (Table 3). It was recently proposed by Reetz and Carballeira (2007) that improved thermostability of an enzyme could be achieved by targeting residues with high B-factors for mutagenesis (B-FIT method). This strategy was successfully applied to enhance the thermostability of B. subtilis lipase which was shown subsequently to express enhanced stability in polar organic solvents (Reetz and Carballeira 2007; Reetz et al. 2010). However, the most stabilizing single mutation, A269T, which showed the highest improvement in thermostability together with enhanced stability in methanol, would not have been found by applying the B-FIT method on lipase T6 since the position itself is stable. Therefore, we suggest that a more effective tactic for stabilization of enzymes in polar organic solvents is the use of complementary approaches, namely the B-FIT approach and the structure-guided consensus approach.
In conclusion, we have successfully applied complimentary protein engineering approaches to create a robust variant of lipase T6 which showed enhanced stability in methanol, elevated thermostability, and improved performance in biodiesel synthesis. Crystal structures revealed that the enhanced stability in methanol and elevated thermostability were a result of surface mutations which enabled the formation of new hydrogen bonds and stabilization of metal ion binding sites which further contributed to the overall stability of the enzyme. Furthermore, these results provide a structural support for the proposed protein unfolding mechanism by stripping off essential water molecules. Our study demonstrates the potential of protein engineering to evolve robust and effective biocatalysts for biodiesel production which has hardly been exploited for this purpose. We believe that further improvement of lipase T6 for biodiesel production can be achieved by combining rational design, B-FIT, and structure-guided consensus approaches on the lipase surface. The evolved lipase will be immobilized for its reuse for multiple biodiesel production cycles.
References
Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D 66(2):213–221
Al-Zuhair S (2007) Production of biodiesel: possibilities and challenges. Biofuels Bioprod Bioref 1(1):57–66
Arnold FH (1990) Engineering enzymes for non-aqueous solvents. Trends Biotechnol 8:244–249
Badoei-Dalfard A, Khajeh K, Asghari SM, Ranjbar B, Karbalaei-Heidari HR (2010) Enhanced activity and stability in the presence of organic solvents by increased active site polarity and stabilization of a surface loop in a metalloprotease. J Biochem 148(2):231–238
Bajaj A, Lohan P, Jha PN, Mehrotra R (2010) Biodiesel production through lipase catalyzed transesterification: an overview. J Mol Catal B Enzym 62(1):9–14
Bélafi-Bakó K, Kovács F, Gubicza L, Hancsók J (2002) Enzymatic biodiesel production from sunflower oil by Candida antarctica lipase in a solvent-free system. Biocatal Biotransform 20(6):437–439
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(1–2):248–254
Chakravorty D, Parameswaran S, Dubey V, Patra S (2012) Unraveling the rationale behind organic solvent stability of lipases. Appl Biochem Biotechnol 167(3):439–461
Dizge N, Aydiner C, Imer DY, Bayramoglu M, Tanriseven A, Keskinler B (2009) Biodiesel production from sunflower, soybean, and waste cooking oils by transesterification using lipase immobilized onto a novel microporous polymer. Bioresour Technol 100(6):1983–1991
Doukyu N, Ogino H (2010) Organic solvent-tolerant enzymes. Biochem Eng J 48(3):270–282
Dror A, Shemesh E, Dayan N, Fishman A (2014) Protein engineering by random mutagenesis and structure-guided consensus of Geobacillus stearothermophilus lipase T6 for enhanced stability in methanol. Appl Environ Microbiol 80(4):1515–1527
Du W, Li W, Sun T, Chen X, Liu D (2008) Perspectives for biotechnological production of biodiesel and impacts. Appl Microbiol Biotechnol 79(3):331–337
Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60(12 Part 1):2126–2132
Fernandez-Lafuente R (2010) Lipase from Thermomyces lanuginosus: uses and prospects as an industrial biocatalyst. J Mol Catal B Enzym 62(3–4):197–212
Fischer K (1935) Neues Verfahren zur maßanalytischen Bestimmung des Wassergehaltes von Flüssigkeiten und festen Körpern. Angew Chem 48(26):394–396
Fishman A, Levy I, Cogan U, Shoseyov O (2002) Stabilization of horseradish peroxidase in aqueous-organic media by immobilization onto cellulose using a cellulose-binding-domain. J Mol Catal B Enzym 18(1–3):121–131
Guncheva M, Zhiryakova D (2011) Catalytic properties and potential applications of Bacillus lipases. J Mol Catal B Enzym 68(1):1–21
Gupta R, Gupta N, Rathi P (2004) Bacterial lipases: an overview of production, purification and biochemical properties. Appl Microbiol Biotechnol 64(6):763–781
Herman A, Tawfik DS (2007) Incorporating synthetic oligonucleotides via gene reassembly (ISOR): a versatile tool for generating targeted libraries. Protein Eng Des Sel 20(5):219–226
Hernández-Martín E, Otero C (2008) Different enzyme requirements for the synthesis of biodiesel: Novozym® 435 and Lipozyme® TL IM. Bioresour Technol 99(2):277–286
Hsu A-F, Jones K, Foglia TA, Marmer WN (2002) Immobilized lipase-catalysed production of alkyl esters of restaurant grease as biodiesel. Biotechnol Appl Biochem 36(3):181–186
Jaeger KE, Dijkstra BW, Reetz MT (1999) Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu Rev Microbiol 53(1):315–351
Jaeger KE, Ransac S, Dijkstra BW, Colson C, van Heuvel M, Misset O (1994) Bacterial lipases. FEMS Microbiol Rev 15(v15i0001):29–63
Kaewmeesri R, Srifa A, Itthibenchapong V, Faungnawakij K (2015) Deoxygenation of waste chicken fats to green diesel over Ni/Al2O3: effect of water and free fatty acid content. Energy Fuel 29(2):833–840
Kamal MZ, Yedavalli P, Deshmukh MV, Rao NM (2013) Lipase in aqueous-polar organic solvents: activity, structure, and stability. Protein Sci 22(7):904–915
Karmakar A, Karmakar S, Mukherjee S (2010) Properties of various plants and animals feedstocks for biodiesel production. Bioresour Technol 101(19):7201–7210
Kawata T, Ogino H (2009) Enhancement of the organic solvent-stability of the LST-03 lipase by directed evolution. Biotechnol Prog 25(6):1605–1611
Kim M-H, Kim H-K, Lee J-K, Park S-Y, Oh T-K (2000) Thermostable lipase of Bacillus stearothermophilus: high-level production, purification, and calcium-dependent thermostability. Biosci Biotechnol Biochem 64(2):280–286
Klibanov AM (1997) Why are enzymes less active in organic solvents than in water? Trends Biotechnol 15(3):97–101
Korman T, Sahachartsiri B, Charbonneau D, Huang G, Beauregard M, Bowie J (2013) Dieselzymes: development of a stable and methanol tolerant lipase for biodiesel production by directed evolution. Biotechnol Biofuels 6(1):70
Kulschewski T, Sasso F, Secundo F, Lotti M, Pleiss J (2013) Molecular mechanism of deactivation of C. antarctica lipase B by methanol. J Biotechnol 168(4):462–469
Leslie A, Joint C (1992) ESF-EACMB newsletter on protein crystallography No. 26. Daresbury Laboratory, Warrington
Li Y, Du W, Liu D (2014) Exploration on the effect of phospholipids on free lipase-mediated biodiesel production. J Mol Catal B Enzym 102:88–93
Liu K-S (1994) Preparation of fatty acid methyl esters for gas-chromatographic analysis of lipids in biological materials. J Am Oil Chem Soc 71(11):1179–1187
Lotti M, Pleiss J, Valero F, Ferrer P (2014) Effects of methanol on lipases: molecular, kinetic and process issues in the production of biodiesel. Biotechnol J:1–9
Maceiras R, Vega M, Costa C, Ramos P, Márquez MC (2011) Enzyme deactivation during biodiesel production. Chem Eng J 166(1):358–361
Martinez P, Arnold FH (1991) Surface charge substitutions increase the stability of α-lytic protease in organic solvents. J Am Chem Soc 113(16):6336–6337
McCoy A (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D 63(1):32–41
Meher LC, Vidya Sagar D, Naik SN (2006) Technical aspects of biodiesel production by transesterification—a review. Renew Sust Energ Rev 10(3):248–268
Meshulam-Simon G (2001) Isolation and characterization of lipases from thermophilic bacteria for the preparation of optically active compounds. Ph.D. thesis, Technion—Israel Institute of technology. Haifa. Israel
Mogensen JE, Sehgal P, Otzen DE (2005) Activation, inhibition, and destabilization of Thermomyces lanuginosus lipase by detergents. Biochemistry (Mosc) 44(5):1719–1730
Nie K, Xie F, Wang F, Tan T (2006) Lipase catalyzed methanolysis to produce biodiesel: optimization of the biodiesel production. J Mol Catal B Enzym 43(1–4):142–147
Nizar NNA, NMJ M, Hashim DM (2013) Differentiation of lard, chicken fat, beef fat and mutton fat by GCMS and EA-IRMS techniques. J Oleo Sci 62(7):459–464
Noureddini H, Gao X, Philkana RS (2005) Immobilized Pseudomonas cepacia lipase for biodiesel fuel production from soybean oil. Bioresour Technol 96(7):769–777
Otwinowski Z, Minor W (2001) DENZO and SCALEPACK. In: Rossmann MG, Arnold E (eds) International tables for crystallography volume F: crystallography of biological macromolecules. International tables for crystallography, vol F. Springer, Netherlands, pp 226–235
Park H, Joo J, Park K, Yoo Y (2012) Stabilization of Candida antarctica lipase B in hydrophilic organic solvent by rational design of hydrogen bond. Biotechnol Bioprocess Eng 17(4):722–728
Raita M, Laothanachareon T, Champreda V, Laosiripojana N (2011) Biocatalytic esterification of palm oil fatty acids for biodiesel production using glycine-based cross-linked protein coated microcrystalline lipase. J Mol Catal B Enzym 73(1–4):74–79
Reetz MT, Carballeira JD (2007) Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc 2(4):891–903
Reetz MT, Soni P, Fernandez L, Gumulya Y, Carballeira JD (2010) Increasing the stability of an enzyme toward hostile organic solvents by directed evolution based on iterative saturation mutagenesis using the B-FIT method. Chem Commun 46(45):8657–8658
Rodrigues RC, Volpato G, Ayub MAZ, Wada K (2008) Lipase-catalyzed ethanolysis of soybean oil in a solvent-free system using central composite design and response surface methodology. J Chem Technol Biotechnol 83(6):849–854
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Shimada Y, Watanabe Y, Samukawa T, Sugihara A, Noda H, Fukuda H, Tominaga Y (1999) Conversion of vegetable oil to biodiesel using immobilized Candida antarctica lipase. J Am Oil Chem Soc 76(7):789–793
Smith RR, Canady WJ (1992) Solvation effects upon the thermodynamic substrate activity; correlation with the kinetics of enzyme catalyzed reactions. I. Effects of added reagents such as methanol upon alpha-chymotrypsin. Biophys Chem 43(2):173–187
Soumanou MM, Bornscheuer UT (2003) Improvement in lipase-catalyzed synthesis of fatty acid methyl esters from sunflower oil. Enzym Microb Technol 33(1):97–103
Tan T, Lu J, Nie K, Deng L, Wang F (2010) Biodiesel production with immobilized lipase: a review. Biotechnol Adv 28(5):628–634
Tanaka A (1998) Differential scanning calorimetric studies on the thermal unfolding of Pseudomonas cepacia lipase in the absence and presence of alcohols. J Biochem 123(2):289–293
Tyndall JDA, Sinchaikul S, Fothergill-Gilmore LA, Taylor P, Walkinshaw MD (2002) Crystal structure of a thermostable lipase from Bacillus stearothermophilus P1. J Mol Biol 323(5):859–869
Vazquez-Figueroa E, Yeh V, Broering JM, Chaparro-Riggers JF, Bommarius AS (2008) Thermostable variants constructed via the structure-guided consensus method also show increased stability in salts solutions and homogeneous aqueous-organic media. Protein Eng Des Sel 21(11):673–680
Watanabe Y, Shimada Y, Sugihara A, Tominaga Y (2002) Conversion of degummed soybean oil to biodiesel fuel with immobilized Candida antarctica lipase. J Mol Catal B Enzym 17(3–5):151–155
Xu Y, Du W, Zeng J, Liu D (2004) Conversion of soybean oil to biodiesel fuel using lipozyme TL IM in a solvent-free medium. Biocatal Biotransform 22(1):45–48
Yagiz F, Kazan D, Akin AN (2007) Biodiesel production from waste oils by using lipase immobilized on hydrotalcite and zeolites. Chem Eng J 134(1–3):262–267
Acknowledgments
This project was funded in part by the Israel Ministry of Environmental Protection, grant number 132-2-2. This research benefited from use of the Technion Center of Structural Biology facility of the Lorry I. Lokey Center for Life Sciences and Engineering and the Russell Berrie Nanotechnology Institute. We thank Dr. Hay Dvir from the Technion Center of Structural Biology facility for assistance in diffraction data collection. We also thank the staff of the European Synchrotron Radiation Facility in France, beamline BM14, for the provision of synchrotron radiation facilities and assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 397 kb)
Rights and permissions
About this article

Cite this article
Dror, A., Kanteev, M., Kagan, I. et al. Structural insights into methanol-stable variants of lipase T6 from Geobacillus stearothermophilus . Appl Microbiol Biotechnol 99, 9449–9461 (2015). https://doi.org/10.1007/s00253-015-6700-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6700-4