
Abstract
Thermoanaerobacterium aotearoense P8G3#4 produced β-glucosidase (BGL) intracellularly when grown in liquid culture on cellobiose. The gene bgl, encoding β-glucosidase, was cloned and sequenced. Analysis revealed that the bgl contained an open reading frame of 1314 bp encoding a protein of 446 amino acid residues, and the product belonged to the glycoside hydrolase family 1 with the canonical glycoside hydrolase family 1 (GH1) (β/α)8 TIM barrel fold. Expression of pET-bgl together with a chaperone gene cloned in vector pGro7 in Escherichia coli dramatically enhanced the crude enzyme activity to a specific activity of 256.3 U/mg wet cells, which resulted in a 9.2-fold increase of that obtained from the expression without any chaperones. The purified BGL exhibited relatively high thermostability and pH stability with its highest activity at 60 °C and pH 6.0. In addition, the activities of BGL were remarkably stimulated by the addition of 5 mM Na+ or K+. The enzyme showed strong ability to hydrolyze cellobiose with a K m and V max of 25.45 mM and 740.5 U/mg, respectively. The BGL was activated by glucose at concentration varying from 50 to 250 mM and tolerant to glucose inhibition with a K i of 800 mM glucose. The supplement of the purified BGL to the sugarcane bagasse hydrolysis mixture containing a commercial cellulase resulted in about 20 % enhancement of the released reducing sugars. These properties of the purified BGL should have important practical implication in its potential applications for better industrial production of glucose or bioethanol started from lignocellulosic biomass.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Global energy consumption and fossil fuel depletion have promoted the production of biofuels and bioproducts from lignocellulosic biomass since lignocellulosic material is the most abundantly renewable source of energy on the Earth (Sørensen et al. 2013). Enzymatic hydrolysis of plant biomass converting cellulose to glucose is recognized as a critical step in the production of biofuels and platform molecules in the synthesis of chemicals from lignocellulosic biomass (Gilbert et al. 2008). Design of better cellulase cocktails with increased performance is one of the most important tasks in obtaining high yields of fermentable sugars from lignocellulosic biomass.
Enzymatic release of monosaccharides from cellulose is a multistep complex process synergistically catalyzed by a mixture of different cellulolytic enzymes: endo-1,4-β-glucanases (EC 3.2.1.4), cellobiohydrolases (or exo-1,4-β-glucanases) (EC 3.2.1.91), and β-glucosidases (BGL, EC 3.2.1.21) (Gilbert et al. 2008; Lynd et al. 2002). The general consensus of enzymatic cellulose hydrolysis is as follows: endoglucanases randomly hydrolyze the internal glycosidic linkages, resulting in a rapid decrease in polymer length and a gradual increase in the number of released reducing ends. Cellobiohydrolases hydrolyze the cellulose polymer from either the reducing or nonreducing ends, liberating cellobiose as the main product, and finally, BGLs hydrolyze oligosaccharides and cellobiose to produce glucose. However, cellobiose acts as a strong inhibitor of both endoglucanases and cellobiohydrolases (Singhania et al. 2013). It is also to be noted that BGL is often itself inhibited by the high concentration of its product glucose (Krogh et al. 2010; Lu et al. 2013), making BGL the rate-limiting enzyme. Therefore, producing highly active and glucose-tolerant BGLs with a weak product inhibition has become important for relieving the product inhibition and increasing the hydrolysis rate of cellulose (Borges et al. 2014).
Trichoderma reesei is today the paradigm for industrial scale production of cellulase. However, the amount of BGL generated by the filamentous fungi represents a very low percentage of the total secreted enzymes, which leads to inefficient and incomplete industrial cellulose hydrolysis (Borges et al. 2014; Herpoël-Gimbert et al. 2008). Many species of fungi, especially Aspergillus, have been widely studied to produce BGLs (Decker et al. 2001; Gunata and Vallier 1999; Yan and Lin 1997). Some other microbial β-glucosidases from Penicillium (Krogh et al. 2010), Pyrococcus (Kengen et al. 1993), and Thermotoga (Park et al. 2005) were also identified and characterized. However, most of these BGLs lack high glucose tolerance or high specific activity for cellobiose conversion. Therefore, overexpression of BGLs with advantages discussed above has become important.
Thermoanaerobacterium aotearoense is a strict anaerobe isolated from a hot spring in China (Li et al. 2010). Several studies have described that this strain can grow on a variety of carbohydrates including glucose, xylose, cellobiose, mannose, and trehalose at 55 °C, which have attracted considerable interest with regards to ethanol (Cai et al. 2011), hydrogen (Lai et al. 2014; Li et al. 2010), and lactic acid (Yang et al. 2013) production from lignocellulosic biomass. And, our previous studies have shown that it was a good potential enzyme producer, especially of carbohydrate digestive enzymes (Lai et al. 2014).
In this paper, we report the sequence analysis, cloning, overexpression in Escherichia coli, and detailed biochemical characterization of β-glucosidase from T. aotearoense P8G3#4. The hydrolytic capability of the BGL was evaluated by doping in combination with cellulase and the purified BGL on sugarcane bagasse.
Materials and methods
Distribution of β-glucosidase in T. aotearoense P8G3#4
The MTC medium composition and culture conditions for T. aotearoense P8G3#4 (CGMCC No. 9000) were reported in Li et al. (2010) and Yang et al. (2013). The overnight culture of T. aotearoense P8G3#4 was inoculated into fresh MTC medium, in which cellobiose was used as the sole carbon source instead of glucose or xylose. After anaerobic fermentation at 55 °C for 24 h, 10 OD600 of cells (about 3 mL) were collected by centrifugation at 6000×g at 4 °C for 10 min. The supernatant was assayed for extracellular β-glucosidase activity. The cell pellets were suspended in 1-mL lysis buffer (50 mM Tris-Cl, pH 7.2, 5 % glycerol, 50 mM NaCl), then frozen in liquid nitrogen for 1 min, and thawed in a 25 °C water bath for 5 min. The freezing and defrosting process was repeated three times. Then, the cell suspension was sonicated for 30 pulses in an ice-water bath (400 W, 3 s each with a 3-s interval), and the sonicants were centrifuged at 14,000×g at 4 °C for 2 min. The supernatant was used for intracellular enzyme activity, and the cell pellets were washed with water twice and then assayed as the enzymatic fraction bound at the mycelium surface.
Nucleic acid manipulations
Genome DNA from T. aotearoense P8G3#4 was prepared using a genome extraction kit (Sangon, Shanghai, China) as described as manufacturer’s handbook. The β-glucosidase gene (bgl) was amplified by PCR reaction using PrimeSTAR HS DNA Polymerase (TaKaRa, Dalian, China) with the genomic DNA of T. aotearoense P8G3#4 as template. The forward primer used was 5′-TAGCCCCATATGGCTAATTTTCCAAAAGGT-3′ (where the underlines indicate the NdeI site), and the reverse primer was 5′-TCCGTCTCGAGAAAAACAATTGAAGCTCTATTTAT-3′ (the underlines indicating the XhoI site). The bgl DNA fragment was amplified under defined PCR conditions of initial denaturation at 98 °C for 2 min followed by 30 cycles of 98 °C for 10 s, 51 °C for 10 s, and 72 °C for 1 min 20 s in 50-μL reaction buffer with a final 10-min extension at 72 °C. Then, the bgl gene was digested with NdeI and XhoI (Fermentas of Thermo Scientific, Pittsburgh, PA), then ligated with the double-digested pET30a vector (Novagen, Wisconsin, USA) yielding the plasmid pET-bgl. The E. coli DH5α (Invitrogen, CA, USA) chemically competent cells were used as the host for cloning. Transformed cells were spread on Luria–Bertani (LB) agar plates containing 50 ng/μL of kanamycin incubated at 37 °C overnight. The positive clones, screened by colony PCR and confirmed by DNA sequence analysis, were selected.
The sequence similarity search was performed with the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/).
Molecular modeling
Modeling templates were identified through the on-line Position-Specific Iterated Basic Local Alignment Search Tool (PSI-BLAST, http://www.ebi.ac.uk/Tools/sss/psiblast/) with default parameters under the Protein Data Bank (PDB) database. To refine loop and discrete optimized protein energy (DOPE) values, three thermostable β-glucosidases from Halothermothrix orenii (PDB 4PTV (Hassan et al. 2015)), Clostridium cellulovorans (PDB 3AHX (Jeng et al. 2011)), and a soil metagenome protein (PDB 4HZ6A (Nam et al. 2010)) were selected as the structural models, to build the 3D structure of T. aotearoense BGL by advanced modeling techniques with MODELLER9v14 (Sali et al. 1995). The structural model was evaluated using PROCHECK (Laskowski et al. 1993) and visualized with PyMol (The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC., New York).
Expression of the recombinant BGL
Plasmid pET-bgl was transformed into E. coli BL21(DE3) (Novagen, Madison, WI, USA) and induced to express recombinant BGL by adding isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM at OD600 approximately 0.8 and incubated further at 30 °C for 12 h. Cells were then collected and lysed for soluble protein extraction. The supernatant fractions (soluble protein) and cell pellets (insoluble protein) were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a 12 % acrylamide gel.
In order to achieve high yields of soluble BGL, competent E. coli BL21(DE3) cells were first transformed with appropriate chaperone combinations from the Molecular Chaperone Plasmid Sets (Table 1) obtained from TaKaRa (Dalian, China), and a respective transformant was made competent. Chaperone-overexpressing cells were finally transformed with the plasmid pET-bgl. Recombinant cells from saturated overnight cultures were diluted 100-fold into LB medium containing 20 ng/μL chloramphenicol and 50 ng/μL kanamycin and grown at 37 °C, 250 rpm to reach an OD600 of 0.3–0.4. Then, the expression of chaperones was initiated with 1 mg/mL l-arabinose and/or 5 mg/mL tetracycline for about 2 h. When the OD600 was about 0.6, cell cultures were supplemented with 1 mM IPTG to induce the BGL gene expression at 25 °C for 6 h.
Purification of the recombinant BGL
Cell cultures (1 L) were separated by centrifugation at 5000×g for 20 min. The cell pellets were resuspended in 35-mL binding buffer (20 mM phosphate buffer, 500 mM NaCl, 20 mM imidazole, pH 7.4); then, 450 pulses of sonication (400 W, 3 s each with a 3 s interval) in an ice water bath were applied. After centrifugation (11,000×g at 4 °C for 30 min), the supernatant was passed through a 0.22-μm filter and applied to a HiTrap™ Chelating HP column (GE Healthcare, Piscataway, NJ, USA), and the purification followed standard nickel affinity chromatography procedures. Aliquots containing BGL activity were pooled and loaded onto a HiPrep™ 26/10 desalting column (GE Healthcare). Fractions of the elute were stored in 100 mM Bis-Tris-HCl buffer pH 6.5 containing 20 % (v/v) glycerol at −20 °C.
Enzyme activity and protein assays
SDS-PAGE was used in determination of the purity of BGL. The protein concentration was determined by a BCA Protein Assay Kit (Sangon, Shanghai, China).
β-Glucosidase activity was routinely assayed by using a 0.2-mL reaction mixture containing 2.5 mM p-nitrophenyl-β-d-glucopyranoside (pNPGlu) (Sigma, St. Louis, MO., USA), 100 mM citric acid-sodium phosphate buffer (pH 6.0), and an appropriate dilution of β-glucosidase preparation. After 5 min of incubation at 60 °C, the reaction was stopped by adding 0.6 mL of 1 M Na2CO3. The p-nitrophenol was determined by monitoring the absorbance at 405 nm (Harnpicharnchai et al. 2009). One unit of β-glucosidase activity is equivalent to 1 μmol of p-nitrophenol released from the pNPGlu in 1 min under these conditions. β-Galactosidase and β-xylosidase activities were assayed under the same conditions, except that p-nitrophenyl-β-d-galactopyranoside (pNPGal) and p-nitrophenyl-β-d-xylopyranoside (pNPXyl), all purchased from Sigma, were the respective substrates. Cellobiase activity was measured as described above using 100 mg/mL of substrate, and the glucose released was determined by high-performance liquid chromatography (HPLC) (Waters 2695, Milford, MA) using an Aminex HPX-87P column (Bio-Rad, Hercules, CA) with a refractive index detector and distilled water as the eluent (Li et al. 2010). Activity against the esculin (6,7-dihydroxycoumarin 6-glucoside) and CM-cellulose (CMC) was assayed by monitoring the A 540 by the method of Miller (1959), using glucose as the standard. One enzyme unit (U) was defined as the amount of enzyme that releases 1 μmol of product per minute. Specific activity was expressed as U/mg protein.
The Michaelis-Menten constant (K m) and maximum activity (V max) values were determined by measuring the initial rates at various pNPGlu concentrations (0.25, 0.5, 0.75, 1.0, 2.0, 3.0, 4.0, and 5.0 mM) or various cellobiose concentration (10, 20, 30, 40, 50, 60, 70, 80, and 90 mM) under optimal reaction conditions and calculated by the double-reciprocal plot method of Lineweaver and Burk (1934) using the GraphPad software program (GraphPad Software, Inc. CA, USA).
Characterization of the recombinant BGL
In order to determine the pH and temperature profiles, the enzymatic reaction was carried out at different pH values in 100 mM citric acid-sodium phosphate buffer (pH 4.0 to 8.0) and various temperatures (40 to 80 °C). The enzyme activity obtained at the optimum condition was used to calculate the relative percentage of enzyme activity at other conditions. To estimate the thermostability, the purified BGL (0.15 μg) was incubated at 50 and 55 °C for 120 min. Samples were taken at different times, and the catalytic activity was measured. The pH stability of BGL was assessed by incubating enzyme in buffers with different pH values between 4.0 and 8.0 with increments of 0.4 for 24 h. The residual activities were determined under optimum temperature and pH conditions using the method as described above. In the pH and temperature stability determination, the initial activity was assumed to be 100 % and used to calculate the enzyme activities as percentage of the initial activity during the incubation period. The assays were performed in three independent experiments.
The effect of various metal ions on the BGL activity was determined in the presence of 5 mM of K+, Na+, Li+, Mg2+, Ca2+, Fe3+, Fe2+, Mn2+, Co2+, Ni2+, Cu2+, Ag+, and Zn2+. The initial concentration of the metal ions was prepared by dissolving them in deionized water. Purified enzyme (20 μL) was preincubated with 20 μL of the metal ion at 50 °C and pH 6.0 for 10 min in a water bath. Then, the enzyme-metal ion mixtures were incubated with 200 μL of 2.5 mM of pNPGlu as the substrate in 100 mM citric acid-sodium phosphate buffer (pH 6.0) to initiate the enzyme reaction. In addition, the impact of enzyme inhibitors on the enzyme activity was also investigated using 5 mM EDTA and 1 % SDS. Activity was determined as the standard β-glucosidase assay and expressed as a percentage of the activity obtained in the absence of the chemical reagents and metal cations.
Sugarcane bagasse hydrolysis
Sugarcane bagasse was kindly provided by the Guangzhou Sugarcane Industry Research Institute (Guangzhou, China). The natural sugarcane bagasse was air-dried, milled, and screened through a 0.3-mm sieve and stored at 4 °C. Batch enzymatic hydrolysis of sugarcane bagasse was carried out at 2 % (w/v) consistency in a citric acid-sodium phosphate buffer (100 mM, pH 6.0). The cellulase (Cellic® Ctec2, Novozymes, Bagsvaerd, Denmark) load was 2 filter paper unit (FPU) per gram of sugarcane bagasse in all the experiments, whereas the purified BGL load was 1 U. The flasks with the reaction mixture were carried out in a rotary shaker at 50 °C and 150 rpm, and the hydrolysis carried out during 120 h. Samples withdrawn at different intervals were filtered (0.45 μm), and the supernatant was analyzed for glucose released. All the experiments were performed in duplicate.
Nucleotide sequence accession number
The nucleotide sequence of bgl has been deposited in the GenBank database under the accession number KP772230.
Results
Cellular distribution of β-glucosidase in T. aotearoense P8G3#4
About 85.8 % of the β-glucosidase activity (21.8 ± 0.8 U/OD) in T. aotearoense P8G3#4 induced by cellobiose was intracellular, which was detected by assaying the supernatant of cell lysates using pNPGlu as substrate. And, the remaining enzyme activity (3.6 ± 0.1 U/OD) was detected by the measurement of cell pellets from lysates, indicating that some of the BGL was surface-bounded to the debris of cell disruption. No enzyme activity was detected in the supernatant of the fermentation culture, implying that the extracellular enzyme activity was negligible. Cellular distribution analysis of β-glucosidase indicated that T. aotearoense P8G3#4 can produce some kind of enzyme showing β-glucosidase activity.
Cloning and sequence analysis of BGL
In the reference genome sequence of Thermoanaerobacterium saccharolyticum JW/SL-YS485 (GenBank: CP003184), there is only one obvious gene for a β-glucosidase. A putative open reading frame (ORF) encoding T. aotearoense P8G3#4 BGL was amplified by PCR from the genomic DNA with primers designed from the annotated β-glucosidase ORF deposited in GenBank (CP003184). The sequenced PCR product was 1341 bp and encoded an ORF of 446 amino acid residues. The protein product has a calculated pI of 6.16 and a molecular weight of 49.1 kDa. A protein-protein BLAST search showed that the encoded BGL shares the highest amino acid sequence similarity of 87 % with the glycoside hydrolase family 1 (GH 1) protein from Thermoanaerobacterium xylanolyticum LX-11 (GenBank: AEF18219.1), and 81 % with the enzymes from Thermoanaerobacter siderophilus (EIV99244.1), Thermoanaerobacter thermohydrosulfuricus (WP_004399779.1), and Thermoanaerobacter thermocopriae (WP_028991588), which are all annotated as β-glucosidase in the whole-genome sequencing data. However, the characterization of the enzymes has not been done so far.
Molecular modelling
Multiple templates were selected for homology modeling for loop refinement and DOPE minimum evaluation. Three thermostable β-glucosidase from H. orenii (PDB code 4PTV (Hassan et al. 2015)), C. cellulovorans (PDB code 3AHX (Jeng et al. 2011)), and a soil metagenome protein (PDB code 4HZ6A (Nam et al. 2010)), sharing 50–55 % identity with the cloned BGL, were used as the structural models. Three-dimensional modeling of the BGL was performed by the advanced modeling technique with MODELLER9v14 (Sali et al. 1995). The generated structure showed a typical (β/α)8-TIM barrel scaffold (Fig. 1a).

Three-dimensional structure of BGL from T. aotearoense P8G3#4 generated by molecular modelling. a Overall structure of (β/α)8-TIM barrels scaffold. b Closeup view of the putative catalytic active pocket. The residues involved in enzymatic hydrolysis are shown as sticks. Two catalytic residues, Glu163 and Glu351 (red sticks with dots for electron cloud), play likely roles as proton donor and nucleophile in the hydrolytic mechanism, respectively. The other six amino acid residues (yellow sticks) play likely essential roles in catalysis, including Asn162 and Glu405 that provide polar interaction with −1 glucose, Gln18, His118, and Trp406 form hydrogen bonds to the glycosyl moiety, and Tyr295 likely reduces the energy barrier in the deglycosylation step (Badieyan et al. 2012)
BGL expression and purification in E. coli
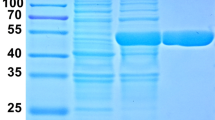
The bgl gene was expressed as an N-terminal 6 × His tag fusion protein using pET30a expression vector in E. coli BL21(DE3). SDS-PAGE analysis of the BGL showed a protein band corresponding to about 46 kDa (Fig. 2), which was discrepant with the calculated molecular weight by the amino acid sequence (52.6 kDa). The anomalous mobility of proteins on SDS-PAGE appears to be common and possibly originate from altered SDS/protein aggregate stoichiometry (Rath et al. 2009).

Expression and purification of BGL in E. coli BL21(DE3) cells. SDS-PAGE analysis of BGL with (a) or without (b) molecular chaperone sets. “no IPTG” indicates that the cells were induced by the chaperone inducer but without IPTG addition; “1 mM IPTG” means that the recombinant BGL were induced through 1 mM IPTG supplement. Supernant fractions for the cell extracts are denoted as “S,” and for the insoluble pellets of the lysates are denoted as “I.” Corresponding BGL positions are marked by arrows. Different molecular chaperones band are indicated by the protein name. M, protein marker. c Specific activity (U/mg wet cells) of the soluble fractions from the recombinant cells without chaperones (column 1), with chaperone vector pGro7 (column 2), pKJE7 (column 3), pTf16 (column 4), pG-KJE8 (column 5), and pG-tf2 (column 6), respectively. Measurements were performed using pNPGlu as the substrate in three independant experiments. d Purification of recombinant BGL from E. coli BL21(DE3) harboring pET-bgl and pGro7. Lane M, protein marker; lane 1, without any inducer; 2, induced by l-arabinose; 3, cell lysates induced by l-arabinose and IPTG; 4, purified BGL
There was substantive expression of BGL enzyme in E. coli with a yield of about 22 % of total protein. Disappointingly, most of the overexpressed BGL was found in the insoluble fraction after the centrifugation following the cell lysis. No obvious soluble fraction of BGL was observed. The hydrolysis activity against pNPGlu was about 27.7 U/mg wet cells. We also tried several other expression parameters, including incubation temperature, time, and the inducer concentration, to increase the soluble fraction level, but did not obtain sufficient amounts of soluble enzyme suitable for further study (data not shown).
In order to obtain more of the overexpressed BGL in soluble fraction, we simultaneously overexpressed the protein with a set of prokaryotic chaperone plasmids (Table 1). Experiments were carried out with E. coli BL21(DE3) cells harboring a pair of expression plasmids, i.e., pET-bgl combined with a chaperone vector. Results with different chaperone vectors differed from each other. As shown in Fig. 2a, b, the BGL protein, most part of which was insoluble in the cells without chaperone proteins, was partially converted to the soluble form when coexpressed with the chaperone team involving GroEL-GroES. The β-glucosidase activity of the crude cell extracts was dramatically increased to 256.3 U/mg wet cells with the chaperone plasmid pGro7 (encoding GroEL-GroES) (Fig. 2c). The β-glucosidase activities for the coexpression of pET-bgl with pG-KJE8 or pG-Tf2 were only 78.1 and 79.5 U/mg wet cells, respectively (Fig. 2c).
Enzyme BGL was isolated from the crude extract of E. coli BL21(DE3) containing pGro7 and pET-bgl by two-step-purification. The yield of active β-glucosidase activity was 3355 U/L and 11.6 U/mg protein. The final BGL was purified about 9.14-fold, and the enzyme was obtained with a recovery yield of 40.4 % (Table 2). The BGL purity after desalting reached 98 % with trace of GroEL chaperone detected on SDS-PAGE gel, and the molecular mass of the enzyme was estimated to be 46 kDa by gel mobility consistent with the result above (Fig. 2d).
Biochemical characterization of recombinant BGL
The effect of pH and temperature on the purified BGL activity was determined using the standard assay described previously. BGL exhibited relatively high activities in a pH range (pH 5.5–6.5) with an optimum activity at pH 6.0 in the citric acid-sodium phosphate buffer (Fig. 3a). It was stable over a slightly acidic pH range, retaining more than 90 % of the initial activity after incubation at pH 5.2–7.2 for 24 h (Fig. 3b). However, the enzyme activity reduced nearly to 40 and 30 % at pH 8.0 and pH 4.0, respectively. The thermal profile of the enzyme was evaluated at various temperatures from 40 to 80 °C (Fig. 3c). The activity increased linearly with increase in temperature up to 60 °C; thereafter, it declined to 50.1 and 43.9 % of the maximum at 70 and 75 °C, respectively. The thermostability of the enzyme was determined in citric acid-sodium phosphate buffer (pH 6.0) at 50 and 55 °C (Fig. 3d). The purified BGL was found to be stable at 50 °C with 65.9 % of its initial activity after incubation for 2 h. However, the inactivation of BGL proceeded a slightly faster rate at 55 °C than that at 50 °C.

Effect of pH and temperature on activity and stability of BGL from T. aotearoense P8G3#4. a Effect of pH on the β-glucosidase activity; b stability of β-glucosidase incubated at buffers with different pH value for 24 h; c effect of temperature on the activity. Enzyme activity was assayed at various temperatures of 40–80 °C in 100 mM citric acid-sodium phosphate buffer (pH 6.0); d stability of β-glucosidase at 50 °C (black circle) and 55 °C (black square). The relative activity represents average of mean ± SD of triplicate
The effects of metal ions and some chemicals at concentrations of 5 mM on the recombinant BGL activity were investigated (Table 3). The β-glucosidase activity was remarkably increased by K+ or Na+, and completely inactivated by Ag+, Cu2+, Zn2+, and 1 % SDS. The enzyme activity was strongly inhibited by Co2+, Ni2+, Mn2+, Fe2+, and Fe3+. No obvious effect was detected with Mg2+ (100 ± 5 %). The enzyme activity was moderately affected by Li+, Ca2+, and EDTA (100 ± 20 %).
Among the different substrates tested (Table 4), the recombinant BGL exhibited the best hydrolyzing capacity against cellobiose (740.39 U/mg) followed by p-nitrophenyl-β-d-glucopyranoside (pNPGlu) and esculin with 103.8 and 76.63 U/mg, respectively. pNPGal was hydrolyzed at 30.5 % of that of pNPGlu, while little enzyme activity was detected using pNPXyl as the substrate. And, there was no observable activity on CMC or sucrose. Kinetic constant determination under optimal conditions showed that the recombinant BGL displayed standard Michaelis-Menten kinetics (Fig. 4a, c). The K m values were calculated from a Lineweaver-Burk double reciprocal plots (Fig. 4b, d) as 0.66 mM for pNPGlu and 25.45 mM for cellobiose, respectively. The overall V max values for pNPGlu and cellobiose were 180.6 and 740.5 U/mg, while the catalytic efficiency (K cat/K m) was 226.33 and 314.38 mM−1 s−1, respectively.

Nonlinear Michaelis-Menten plots (a, c) and Lineweaver-Burk plots (b, d) of the purified BGL from T. aotearoense P8G3#4. a, b Using pNPGlu as the substrate; c, d using cellobiose as the substrate. The BGL had a K m value of 0.66 mM and a V max value of 180.0.2 U/mg for pNPGlu, and a K m value of 25.45 mM and a V max value of 740.5 U/mg for cellobiose, respectively
Different concentrations of glucose were added to the reaction system, and the β-glucosidase activity was measured (Fig. 5). The concentrations of glucose below 250 mM stimulated the pNPGlu hydrolysis. It should be noted that the enzyme activity was dramatically enhanced by 141.9 % when 100 mM glucose was supplemented in the reaction mixture. However, when the glucose concentration was further increased over 250 mM, the BGL activity was gradually decreased with a K i of 800 mM glucose. This suggested that the recombinant BGL from T. aotearoense P8G3#4 was highly tolerant to glucose inhibition, which is desirable for practical applications in cellulose degradation.

Effects of glucose on BGL activity using 1 M pNPGlu as the substrate. The β-glucosidase activity without glucose supplement was defined as 100 %. The dash line indicates the 100 % of the initial activity. Error bars represent standard deviation from a duplicate analysis
Cellobiose and sugarcane bagasse hydrolysis
Glucose production from 100 g/L cellobiose (290 mM) catalyzed by the purified BGL was analyzed by HPLC (Fig. 6a). The glucose was released quickly with a theoretical yield of 25.3 % at the beginning 30 min of the reaction. After 2-h incubation, more than 50 % of cellobiose were degraded to glucose with a concentration of 58.2 g/L. The final concentration of glucose in this reaction reached about 105 g/L (maximum theoretical value) after incubating at 50 °C for 7 h. At the initial stage of reaction, the catalysis rate was up to 44 g/L/h. The fast reaction lasted only for a short period of time. An hour later, the catalysis speed was found to slow down with the prolonging of reaction time, which was partly attributed to loss of enzymatic activity.

Substrate hydrolysis by the recombinant BGL from T. aotearoense P8G3#4. a Cellobiose hydrolysis. The substrate of cellobiose (100 g/L) was incubated with BGL (5 U) at 50 °C for 9 h. Reaction mixture was taken at regular time interval for the glucose analysis by HPLC. b Sugarcane bagasse hydrolysis by the purified BGL and Cellic® Ctec2 (Novozymes, Denmark). 0.2 g of sugarcane bagasse was hydrolyzed in 20 mL of 0.1 M acid-sodium phosphate buffer buffer (pH 6.0). Black square 1 U of BGL (based on the activity toward pNPGlu) with 0.4 filter paper unit (FPU) of Cellic® Ctec2; black circle, 0.4 FPU of Cellic® Ctec2; black up-pointing triangle, 1 U of BGL; black down-pointing triangle no enzyme addition. The experiments were performed in triplicate
Efficient hydrolysis of lignocellulosic biomass into fermentable sugars is one of the key steps in the production of biobased chemicals. The time profiles of sugarcane bagasse hydrolysis by the purified BGL with or without Cellic® Ctec2 are shown in Fig. 6b. Reducing sugars were released in the reaction system containing the commercial cellulase preparation (Cellic® Ctec2, 0.4 FPU). Addition of the purified BGL into the reaction mixture led to about 20 % increase of production of reducing sugars, achieving 24.6 mg per gram of sugarcane bagasse. However, when the recombinant BGL was used as the single hydrolyzing enzyme, no reducing sugars were detected, indicating that this purified enzyme could not hydrolyze lignocellulosic biomass by itself. In industrial scale biomass hydrolysis, the enzymatic catalytic activity was usually low when the product concentration was high, which significantly reduced the overall productivity (Hodge et al. 2008). This study shows that the recombinant BGL can play a key role for continued reduced sugar production at higher product concentration, indicating that the effect of glucose inhibition on β-glucosidase from Cellic® Ctec2 was higher than that on the recombinant BGL from T. aotearoense P8G3#4.
Discussion
β-Glucosidase is one of the essential enzymes in efficient hydrolysis of cellulosic biomass, as it catalyzes the reaction converting cellubiose to glucose and relieves the inhibition of cellobiose to cellobiohydrolases and endoglucanases. A number of thermostable enzymes, including β-glucosidase, have been isolated and characterized from the members of the genus Thermoanaerobacterium (Pei et al. 2012; Sansenya et al. 2015; Zhao et al. 2013a). In this study, we cloned the bgl gene, encoding β-glucosidase in T. aotearoense P8G3#4. Analysis based on the amino acid sequence indicated that its product belongs to the superfamily glycoside hydrolase family 1 (GH1). Three-dimensional modeling of the overexpressed BGL implied that its structure has a (β/α)8 barrel scaffold (Fig. 1a), which is one of the characteristics of the GH1 enzymes. Based on the X-ray structures of β-glucosidase from H. orenii and C. cellulovorans, the active pocket of the studied BGL is predicted to be formed by Gln18, His118, Asn162, Glu163, Tyr295, Glu351, Glu405, and Trp406 (Fig. 1b). Two active site amino acid residues (Glu163 and Glu351), respectively, functioning as a nucleophile and a proton donor, play key roles in the hydrolysis reaction (Vuong and Wilson 2010). Recently, the catalytic mechanism of a GH1 β-glucosidase from Oryza sativa was systematically studied by Badieyan et al. (2012). Amino acid sequence alignment (Fig. S1 in the Supplementary Material) indicated that residues Asn162 and Glu405 from the cloned BGL, conserved in clan A of glycoside hydrolases (except GH26), provide polar interaction with −1 glucose (Gloster and Davies 2010). The absolutely conserved Tyr295 forms a hydrogen bond to both the pyranoside ring and the nucleophile Glu351 and is critical for keeping the substrate and nucleophile in appropriate positions for nucleophilic attack, by lowering the energy barrier in the deglycosylation step (Badieyan et al. 2012). In contrast, Gln18, His118, and Trp406, which also form hydrogen bonds to the glycosyl moiety, showed relatively less effect on the catalysis.
Recombinant proteins overexpressed in E. coli often form insoluble aggregates of non-native proteins, known as inclusion bodies, because of their inability to reach a correct tertiary conformation due to anomalies in protein folding (Baneyx and Mujacic 2004). In some cases, coexpression of molecular chaperones could facilitate target protein folding and enhance production of active proteins (Baneyx and Mujacic 2004; Hartl 2011). In the E. coli cytoplasm, de novo folding involves three chaperone systems: trigger factor (TF), DnaK-DnaJ-GrpE, and GroEL-GroES teams (Baneyx and Mujacic 2004; Hartl and Hayer-Hartl 2002). TF and the dnaK-dnaJ-grpE chaperone team maintain nascent or other preexisting proteins in unfolded states (Hartl and Hayer-Hartl 2002; Nishihara et al. 2000). TF is ideally positioned to interact with short nascent chains. Longer nascent chains or newly synthesized proteins may alternatively be captured by DnaK, a chaperone whose substrate pool overlaps with that of TF. GroEL and GroES interact with partially folded polypeptides and assist in the additional folding (Hartl 2011). In the attempt to obtain functional BGL in E. coli, five prokaryotic chaperone combinations were used in this study for coexpression with pET-bgl, respectively. The GroEL-GroES chaperone remarkablely enhanced β-glucosidase activity in the crude cell extracts. However, no synergistic effect in formation of soluble BGL was observed when coexpressed with pG-KJE8 and pG-Tf2, respectively. Conversely, the existence of TF or dnaK-dnaJ-grpE chaperones in the GroEL-GroES team had negative impact on the BGL expression. These results do not completely agree with the observation for some other proteins reported by Nishihara et al. (1998, 2000). In addition, the chaperones of pKJE7 and pTf16 could not increase the solubility of the overexpressed BGL. This was confirmed by the fact that there was no significant variation in the β-glucosidase activities for the supernatant fraction between the BGL expression without any chaperones and the BGL coexpression with pKJE7 or pTf16 (Fig. 2c). These results further demonstrated that despite many proven success as folding modulators in recombinant protein production, molecular chaperones also show unsatisfactory effects related to their activities in promoting proteolysis of target proteins (Martínez-Alonso et al. 2010).
The recombinant BGL from T. aotearoense P8G3#4 exhibited the highest activity at 60 °C (Fig. 3c), and its half-life time was 199.2 and 96.9 min at 50 and 55 °C, respectively (Fig. 3d). According to the van’t Hoff rule (van’t Hoff and Lehfeldt 1898), an enzyme catalyzed reaction rate is roughly double with every 10 °C increase of temperature under the assumption that the reaction enthalpy ΔH is constant. Thus, the lignocellulosic biomass hydrolysis at higher temperatures necessitates the search for thermophilic β-glucosidases. The optimal temperature and thermostability of the purified BGL obtained in this study were relatively higher than those of the β-glucosidases discovered from Aspergillus oryzae (Riou et al. 1998), Streptomyces sp. (Mai et al. 2013), Candida peltata (Saha and Bothast 1996), Scytalidium thermophilum (Zanoelo et al. 2004), and Prunus domestica (Chen et al. 2012) (Table 5). This indicates that the recombinant BGL from T. aotearoense P8G3#4 could be a suitable candidate in various industrial processes, especially in the production of fermentable sugars from biomass.
Some metal ions and reagents are reported to affect β-glucosidase activity. As a common trend, many of the β-glucosidases are inhibited by heavy metals, such as Ag+, Cu2+, Hg2+, Zn2+, Mn2+ (Pei et al. 2012; Yan and Lin 1997; Zhao et al. 2013b). In this study, the purified BGL activity was also completely inhibited by Ag+, Cu2+, and Zn2+, and moderately inhibited by Mn2+. However, a dramatic enhancement of β-glucosidase by Na+ and K+ was observed in this study. Similar effects were not reported for the BGLs from Thermotoga thermarum (Zhao et al. 2013b), T. thermosaccharolyticum (Pei et al. 2012), and A. niger (Yan and Lin 1997). In addition, EDTA (5 mM) addition to the reaction buffer stimulated the activity of BGL to 115.6 % of the enzyme activity measured without EDTA (Table 3), suggesting that T. aotearoense P8G3#4 BGL may not require metal ions for enzyme activity.
Enzymatic hydrolysis of cellulose is a complex process, in which activities of cellobiohydrolases and endoglucanases are often inhibited by cellobiose. Therefore, the cellobiose conversion by BGL is the main bottleneck in the efficient biomass degradation by cellulase. In this study, though the recombinant BGL has a lower affinity toward cellobiose than pNPGlu, it can hydrolyze the former substrate about 4 times faster than the latter with a V max of 740.5 U/mg. To our best knowledge, this is the fastest rate in the cellobiose break down catalyzed by β-glucosidase (Table 5). Substrate specificity of BGL varies markedly from different sources (Table 5). According to their substrate specificity, β-glucosidase may be divided into three groups: acryl-β-glucosidases, cellobiases, and broad-specificity β-glucosidases. The first group exhibits an extreme preference toward hydrolysis of acryl-β-glucosidases, whereas cellobiases hydrolyze cello-oligosaccharides only (including cellobiose). Members of the broad-specificity β-glucosidases show significant activity on both substrate types and represent the most commonly observed group in cellulolytic microbes (Bhatia et al. 2002). Substrate specificity of the recombinant BGL from T. aotearoense P8G3#4 indicated that it belonged to the third group.
Moreover, BGL is often itself inhibited by its product glucose (Krogh et al. 2010; Lu et al. 2013), making BGL the rate-limiting enzyme. Some β-glucosidases with high glucose tolerance from species of Aspergilli have been cloned and characterized (Decker et al. 2001; Gunata and Vallier 1999; Riou et al. 1998; Yan and Lin 1997). Some other microbial β-glucosidases from C. peltata (Saha and Bothast 1996), P. domestica seeds (Chen et al. 2012), T. thermarum (Zhao et al. 2013b), and T. thermosaccharolyticum (Pei et al. 2012) were also identified. A β-glucosidase gene isolated by metagenomic library screening and expressed in E. coli showed the highest glucose tolerance with a K i of 1500 mM (Lu et al. 2013) (Table 5). However, all of the reported glucose-tolerant BGLs showed a relatively lower specific activity for cellobiose than the T. aotearoense BGL. The recombinant BGL from T. aotearoense P8G3#4 was activated by glucose at concentrations varying from 50 to 250 mM, and it is shown to be highly tolerant to glucose inhibition, with a K i of 800 mM. The recombinant BGL from T. aotearoense P8G3#4 was the only β-glucosidase that has been reported not only to be resistant to high concentration of glucose, but it has in addition a fast catalytic rate for cellobiose.
In this study, we successfully overexpressed soluble and functional BGL from T. aotearoense P8G3#4 with the help of the GroEL-GroES chaperone system in E. coli. The purified BGL showed the fastest rate in cellobiose breakdown (based on the literature data), and higher tolerance to glucose and better thermostability than most of the reported β-glucosidases. In relation to industrial biomass conversion, cellobiose hydrolysis rate, inhibitors, and stability are often restrictive for maintaining high conversion rates throughout the hydrolysis. It is obvious that the recombinant BGL has the potential to increase the rate and extent of lignocellulose deconstruction to fermentable sugars.
References
Badieyan S, Bevan DR, Zhang C (2012) Probing the active site chemistry of β-glucosidases along the hydrolysis reaction pathway. Biochemistry 51:8907–8918
Baneyx F, Mujacic M (2004) Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol 22:1399–1408
Bhatia Y, Mishra S, Bisaria VS (2002) Microbial β-glucosidases: cloning, properties, and applications. Crit Rev Biotechnol 22:375–407
Borges DG, Baraldo A, Farinas CS, Giordano RDC, Tardioli PW (2014) Enhanced saccharification of sugarcane bagasse using soluble cellulase supplemented with immobilized β-glucosidase. Bioresour Technol 167:206–213
Cai Y, Lai C, Li S, Liang Z, Zhu M, Liang S, Wang J (2011) Disruption of lactate dehydrogenase through homologous recombination to improve bioethanol production in Thermoanaerobacterium aotearoense. Enzym Microb Technol 48:155–161
Chen L, Li N, Zong M-H (2012) A glucose-tolerant β-glucosidase from Prunus domestica seeds: purification and characterization. Process Biochem 47:127–132
Decker CH, Visser J, Schreier P (2001) β-Glucosidase multiplicity from Aspergillus tubingensis CBS 643.92: purification and characterization of four β-glucosidases and their differentiation with respect to substrate specificity, glucose inhibition and acid tolerance. Appl Microbiol Biotechnol 55:157–163
Gilbert HJ, Stålbrand H, Brumer H (2008) How the walls come crumbling down: recent structural biochemistry of plant polysaccharide degradation. Curr Opin Plant Biol 11:338–348
Gloster TM, Davies GJ (2010) Glycosidase inhibition: assessing mimicry of the transition state. Org Biomol Chem 8:305–320
Gunata Z, Vallier MJ (1999) Production of a highly glucose-tolerant extracellular β-glucosidase by three Aspergillus strains. Biotechnol Lett 21:219–223
Harnpicharnchai P, Champreda V, Sornlake W, Eurwilaichitr L (2009) A thermotolerant β-glucosidase isolated from an endophytic fungi, Periconia sp., with a possible use for biomass conversion to sugars. Protein Expr Purif 67:61–69
Hartl FU (2011) Chaperone-assisted protein folding: the path to discovery from a personal perspective. Nat Med 17:1206–1210
Hartl FU, Hayer-Hartl M (2002) Protein folding - molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295:1852–1858
Hassan N, Nguyen T-H, Intanon M, Kori L, Patel BC, Haltrich D, Divne C, Tan T (2015) Biochemical and structural characterization of a thermostable β-glucosidase from Halothermothrix orenii for galacto-oligosaccharide synthesis. Appl Microbiol Biotechnol 99:1731–1744
Herpoël-Gimbert I, Margeot A, Dolla A, Jan G, Mollé D, Lignon S, Mathis H, Sigoillot J-C, Monot F, Asther M (2008) Comparative secretome analyses of two Trichoderma reesei RUT-C30 and CL847 hypersecretory strains. Biotechnol Biofuels 1:18
Hodge DB, Karim MN, Schell DJ, McMillan JD (2008) Soluble and insoluble solids contributions to high-solids enzymatic hydrolysis of lignocellulose. Bioresour Technol 99:8940–8948
Hoff JH v’t, Lehfeldt RA (1898) Lectures on theoretical and physical chemistry. E. Arnold, London
Jabbour D, Klippel B, Antranikian G (2012) A novel thermostable and glucose-tolerant β-glucosidase from Fervidobacterium islandicum. Appl Microbiol Biotechnol 93:1947–1956
Jeng W-Y, Wang N-C, Lin M-H, Lin C-T, Liaw Y-C, Chang W-J, Liu C-I, Liang P-H, Wang AHJ (2011) Structural and functional analysis of three β-glucosidases from bacterium Clostridium cellulovorans, fungus Trichoderma reesei and termite Neotermes koshunensis. J Struct Biol 173:46–56
Kengen SW, Luesink EJ, Stams AJ, Zehnder AJ (1993) Purification and characterization of an extremely thermostable β-glucosidase from the hyperthermophilic archaeon Pyrococcus furiosus. Eur J Biochem 213:305–312
Krogh KBRM, Harris PV, Olsen CL, Johansen KS, Hojer-Pedersen J, Borjesson J, Olsson L (2010) Characterization and kinetic analysis of a thermostable GH3 β-glucosidase from Penicillium brasilianum. Appl Microbiol Biotechnol 86:143–154
Lai Z, Zhu M, Yang X, Wang J, Li S (2014) Optimization of key factors affecting hydrogen production from sugarcane bagasse by a thermophilic anaerobic pure culture. Biotechnol Biofuels 7:119
Laskowski RA, Macarthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291
Li S, Lai C, Cai Y, Yang X, Yang S, Zhu M, Wang J, Wang X (2010) High efficiency hydrogen production from glucose/xylose by the ldh-deleted Thermoanaerobacterium strain. Bioresour Technol 101:8718–8724
Lineweaver H, Burk D (1934) The determination of enzyme dissociation constants. J Am Chem Soc 56:658–666
Lu J, Du L, Wei Y, Hu Y, Huang R (2013) Expression and characterization of a novel highly glucose-tolerant β-glucosidase from a soil metagenome. Acta Biochim Biophys Sin 45:664–673
Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS (2002) Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev 66:506–577
Mai Z, Yang J, Tian X, Li J, Zhang S (2013) Gene cloning and characterization of a novel salt-tolerant and glucose-enhanced β-glucosidase from a marine Streptomycete. Appl Biochem Biotechnol 169:1512–1522
Martínez-Alonso M, García-Fruitós E, Ferrer-Miralles N, Rinas U, Villaverde A (2010) Side effects of chaperone gene co-expression in recombinant protein production. Microb Cell Fact 9:64
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of ruducing sugar. Anal Chem 31:426–428
Nam KH, Sung MW, Hwang KY (2010) Structural insights into the substrate recognition properties of β-glucosidase. Biochem Biophys Res Commun 391:1131–1135
Nishihara K, Kanemori M, Kitagawa M, Yanagi H, Yura T (1998) Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl Environ Microbiol 64:1694–1699
Nishihara K, Kanemori M, Yanagi H, Yura T (2000) Overexpression of trigger factor prevents aggregation of recombinant proteins in Escherichia coli. Appl Environ Microbiol 66:884–889
Park TH, Choi KW, Park CS, Lee SB, Kang HY, Shon KJ, Park JS, Cha J (2005) Substrate specificity and transglycosylation catalyzed by a thermostable β-glucosidase from marine hyperthermophile Thermotoga neapolitana. Appl Microbiol Biotechnol 69:411–422
Pei J, Pang Q, Zhao L, Fan S, Shi H (2012) Thermoanaerobacterium thermosaccharolyticum β-glucosidase: a glucose-tolerant enzyme with high specific activity for cellobiose. Biotechnol Biofuels 5:31
Rajasree KP, Mathew GM, Pandey A, Sukumaran RK (2013) Highly glucose tolerant β-glucosidase from Aspergillus unguis: NII 08123 for enhanced hydrolysis of biomass. J Ind Microbiol Biotechnol 40:967–975
Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM (2009) Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Natl Acad Sci U S A 106:1760–1765
Riou C, Salmon JM, Vallier MJ, Gunata Z, Barre P (1998) Purification, characterization, and substrate specificity of a novel highly glucose-tolerant β-glucosidase from Aspergillus oryzae. Appl Environ Microbiol 64:3607–3614
Saha BC, Bothast RJ (1996) Production, purification, and characterization of a highly glucose-tolerant novel β-glucosidase from Candida peltata. Appl Environ Microbiol 62:3165–3170
Sali A, Potterton L, Yuan F, Vanvlijmen H, Karplus M (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23:318–326
Sansenya S, Mutoh R, Charoenwattanasatien R, Kurisu G, Ketudat Cairns JR (2015) Expression and crystallization of a bacterial glycoside hydrolase family 116 β-glucosidase from Thermoanaerobacterium xylanolyticum. Acta Crystallogr F 71:41–44
Singhania RR, Patel AK, Sukumaran RK, Larroche C, Pandey A (2013) Role and significance of β-glucosidases in the hydrolysis of cellulose for bioethanol production. Bioresour Technol 127:500–507
Sørensen A, Lübeck M, Lübeck PS, Ahring BK (2013) Fungal β-glucosidases: a bottleneck in industrial use of lignocellulosic materials. Biomolecules 3:612–631
Uchima CA, Tokuda G, Watanabe H, Kitamoto K, Arioka M (2012) Heterologous expression in Pichia pastoris and characterization of an endogenous thermostable and high-glucose-tolerant β-glucosidase from the termite Nasutitermes takasagoensis. Appl Environ Microbiol 78:4288–4293
Vuong TV, Wilson DB (2010) Glycoside hydrolases: catalytic base/nucleophile diversity. Biotechnol Bioeng 107:195–205
Yan TR, Lin CL (1997) Purification and characterization of a glucose-tolerant β-glucosidase from Aspergillus niger CCRC 31494. Biosci Biotechnol Biochem 61:965–970
Yang X, Lai Z, Lai C, Zhu M, Li S, Wang J, Wang X (2013) Efficient production of L-lactic acid by an engineered Thermoanaerobacterium aotearoense with broad substrate specificity. Biotechnol Biofuels 6:124
Zanoelo FF, Polizeli M, Terenzi HF, Jorge JA (2004) β-Glucosidase activity from the thermophilic fungus Scytalidium thermophilum is stimulated by glucose and xylose. FEMS Microbiol Lett 240:137–143
Zhao L, Pang Q, Xie J, Pei J, Wang F, Fan S (2013a) Enzymatic properties of Thermoanaerobacterium thermosaccharolyticum β-glucosidase fused to Clostridium cellulovorans cellulose binding domain and its application in hydrolysis of microcrystalline cellulose. BMC Biotechnol 13:101
Zhao L, Xie J, Zhang X, Cao F, Pei J (2013b) Overexpression and characterization of a glucose-tolerant β-glucosidase from Thermotoga thermarum DSM 5069T with high catalytic efficiency of ginsenoside Rb1 to Rd. J Mol Catal B-Enzym 95:62–69
Acknowledgments
The authors gratefully appreciate the financial support by the National Natural Science Foundation of China (NSFC 21276096), and the Open Project Program of Guangdong Key Laboratory of Fermentation and Enzyme Engineering, SCUT (FJ2013006). And Dr. Shuang Li was funded by the Pearl River New-Star of Science & Technology supported by Guangzhou City (2012 J2200012).
Conflict of interest
No conflict of interest exits in the submission of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 506 kb)
Rights and permissions
About this article

Cite this article
Yang, F., Yang, X., Li, Z. et al. Overexpression and characterization of a glucose-tolerant β-glucosidase from T. aotearoense with high specific activity for cellobiose. Appl Microbiol Biotechnol 99, 8903–8915 (2015). https://doi.org/10.1007/s00253-015-6619-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6619-9