
Abstract
Enzyme fusion was investigated as a strategy to improve productivity of a two-step whole-cell biocatalysis. The biotransformation of long-chain sec-alcohols into esters by an alcohol dehydrogenase (ADH) and Baeyer–Villiger monooxygenases (BVMOs) was used as the model reaction. The recombinant Escherichia coli, expressing the fusion enzymes between the ADH of Micrococcus luteus NCTC2665 and the BVMO of Pseudomonas putida KT2440 or Rhodococcus jostii RHA1, showed significantly greater bioconversion activity with long-chain sec-alcohols (e.g., 12-hydroxyoctadec-9-enoic acid (1a), 13-hydroxyoctadec-9-enoic acid (2a), 14-hydroxyicos-11-enoic acid (4a)) when compared to the recombinant E. coli expressing the ADH and BVMOs independently. For instance, activity of the recombinant E. coli expressing the ADH-Gly-BVMO, in which glycine-rich peptide was used as the linker, with 1a was increased up to 22 μmol g dry cells−1 min−1. This value is over 40 % greater than the recombinant E. coli expressing the ADH and BVMO independently. The substantial improvement appeared to be driven by an increase in the functional expression of the BVMOs and/or an increase in mass transport efficiency by localizing two active sites in close proximity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biocatalysis is widely used to synthesize a variety of molecules covering natural and unnatural compounds (Balke et al. 2012; Bornscheuer et al. 2012; Holtmann et al. 2014; Ladkau et al. 2014; Lee et al. 2012; Lin et al. 2014; Zhao et al. 2013). In particular, multistep enzyme biotransformations allow not only synthesis of structurally complex molecules but also simplification and intensification of the biocatalytic processes avoiding the expensive isolation of intermediates (Ladkau et al. 2014; Lopez-Gallego and Schmidt-Dannert 2010). However, performance of multistep biocatalysis, especially multistep whole-cell biocatalysis, may suffer from difficulty in functional expression and/or low stability of some of the enzymes involved resulting in undesired accumulation of reaction intermediates and thereby reduced productivity and yield of the final products.
Herein, we investigated an approach to attenuate or overcome the imbalance problems in activity of the catalytic enzymes via a two-step biocatalysis. We used molecular fusion of the enzymes via covalent linkage, which are present in serial order for the two-step biocatalysis. Fusion with the soluble peptides and/or proteins may enhance functional expression of the counterpart enzymes (Chen et al. 2013; Zhang et al. 2009). Furthermore, enzyme fusion is expected to improve protein–protein interactions, which may increase cascade biotransformation rates of the rather large and insoluble substrates either by channeling intermediates between enzymes or by localizing two active sites in close proximity.
Baeyer–Villiger monooxygenases (BVMOs) are oxidative enzymes that catalyze the Baeyer–Villiger oxidations and sulfoxidations with high chemoselectivity, regioselectivity, and enantioselectivity (Balke et al. 2012; de Gonzalo et al. 2010; Orru et al. 2011). Since a variety of aldehydes and (a)cyclic ketones could be converted into their corresponding esters and lactones, BVMOs are interesting candidates for various synthetic applications (de Gonzalo et al. 2010; Holtmann et al. 2014; Pazmino et al. 2009). However, a number of BVMO-encoding genes cloned were difficult to be overexpressed in a functional form in conventional microbial biocatalysts (e.g., Escherichia coli, Saccharomyces cerevisiae) (Cheesman et al. 2001; Kirschner et al. 2007; Lee et al. 2004; Rehdorf et al. 2007; van Beek et al. 2014).
Alcohol dehydrogenases (ADHs) are a group of dehydrogenase enzymes that catalyze the interconversion between alcohols and aldehydes or ketones. Since the enzymes are able to catalyze formation of a variety of ketones, which are the substrates of the BVMOs, the ADH reactions can be coupled with the BVMO to generate the industrially relevant carboxyl synthons (Bisogno et al. 2010; Mallin et al. 2013; Oberleitner et al. 2013; Rioz-Martinez et al. 2010; Song et al. 2013; Staudt et al. 2013). For instance, serial reaction of hydroxy fatty acids (e.g., ricinoleic acid, lesquerolic acid) with the ADH from Micrococcus luteus NCTC2665 and the BVMO from Pseudomonas putida KT2440 led to formation of esters, which can be converted into ω-hydroxycarboxylic acids and n-alkanoic acids (Song et al. 2013). ε-Caprolactone could be produced from cyclohexanol through the coupled reaction of an ADH and a BVMO with efficient internal cofactor recycling (Mallin et al. 2013; Staudt et al. 2013).
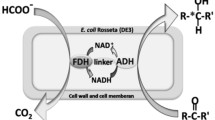
The ADHs are usually easier to be overexpressed in a functional form in microbial cells when compared to the BVMOs. For example, a secondary ADH from M. luteus NCTC2665 was overproduced in functional form in E. coli cells and stable under reaction conditions (Figs. S1 and S2 in the Supporting Information). Thereby, a protein fusion between the BVMOs from P. putida KT2440 (Rehdorf et al. 2007) or Rhodococcus jostii RHA1 (Szolkowy et al. 2009) and the ADH of M. luteus was investigated with an aim to increase functional expression and activity of the BVMOs under reaction conditions. The model reaction was the biotransformation of sec-alcohols (1a–7a) into the esters (1c–7c), which can be hydrolyzed to ω-hydroxycarboxylic acids and n-alkanoic acids (Song et al. 2013) (Fig. 1).

The biotransformation pathway. Secondary alcohols (i.e., hydroxy fatty acids) (a) are converted into ester c via the corresponding ketones (b). The products can be hydrolyzed to ω-hydroxycarboxylic acids and n-alkanoic acids
Materials and methods
Microbial strains and culture media
Recombinant E. coli BL21(DE3), expressing the ADH and BVMO independently or the ADH-BVMO fusion enzymes, was cultivated in Riesenberg medium, which was supplemented with 10 g L−1 glucose and the appropriate antibiotics for plasmid maintenance (Table S1). The Riesenberg medium consisted of 4 g L−1 (NH4)2HPO4, 13.5 g L−1 KH2PO4, 1.7 g L−1 citric acid, 1.4 g L−1 MgSO4, and 10 mL L−1 trace metal solution (10 g L−1 FeSO4, 2.25 g L−1 ZnSO4, 1.0 g L−1 CuSO4, 0.5 g L−1 MnSO4, 0.23 g L−1 Na2B4O7, 2.0 g L−1 CaCl2, and 0.1 g L−1 (NH4)6Mo7O24. Recombinant E. coli BL21(DE3) pACYC-ADH, expressing the ADH only, was cultivated in lysogeny broth (LB) medium. Expression of the ADH was induced by adding 0.1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) into the culture broth at the early exponential phase.
Reagents
Ricinoleic acid, linoleic acid, 5-hydroxydecanoic acid, n-heptanoic acid, and palmitic acid were purchased from Sigma-Aldrich. Lesquerolic acid methyl ester was purchased from Santa Cruz Biotechnology. 12-Hydroxystearic acid, 10-hydroxystearic acid, and other carboxyl products were purified in our lab as described previously (Song et al. 2013).
Gene cloning
The pACYC-ADH/BVMO used for the independent expression of ADH and BVMO was constructed via PCR of the BVMO gene of P. putida KT2440 from pJOE-KT2440 (Rehdorf et al. 2007). The BVMO_nde1_F and BVMO_pvu1_R (Table S2 in the Supporting Information) were used as the forward and reverse primers, respectively. The resulting fragments were inserted into the NdeI-PvuI site of pACYC-ADH (Song et al. 2013). The fusion gene encoding the ADH-BVMO fusion enzyme (Table 1) was synthesized via PCR of the ADH part with ADH_001_F and ADH_001_R as the forward and reverse primers, respectively (Table S2 in the Supporting Information). The BVMO part was amplified with BVMO_001_F and BVMO_001_R as the forward and reverse primers. Afterward, the two fragments were fused via isothermal assembly PCR (Gibson et al. 2009) and inserted into the EcoRI–HindIII site of pACYC-duet vector. The fusion genes encoding the ADH-FOM-BVMO and ADH-Gly-BVMO (Table 1) were synthesized via two-step PCR. The first-step PCR for construction of the ADH-FOM-BVMO was carried out with ADH_001_F and ADH_101_R1 as the forward and reverse primers using pACYC-ADH as template. The resulting fragment was used as the template of the second-step PCR with the primers ADH_001_F and ADH_101_R2. The BVMO part was also synthesized via two-step PCR, but using the different primers (i.e., BVMO_101_F1 and BVMO_001_R for the first step and BVMO_101_F2 and BVMO_001_R for the second step). Afterward, the two fragments were fused via isothermal assembly PCR and inserted into the EcoRI–HindIII site of pACYC-duet vector. The first-step PCR for construction of the ADH-Gly-BVMO was carried out with the primers ADH_001_F and ADH_201_R1 using pACYC-ADH as template. The resulting fragment was used as the template of the second-step PCR with the primers ADH_001_F and ADH_201_R2. The BVMO part was also synthesized via two-step PCR, but using the different primers (i.e., BVMO_201_F1 and BVMO_001_R for the first step and BVMO_201_F2 and BVMO_001_R for the second step). Afterward, the two fragments were fused via isothermal assembly PCR and inserted into the EcoRI–HindIII site of pACYC-duet vector.
The pACYC-ADH/MO16 used for the independent expression of ADH and MO16 was synthesized via PCR of the MO16 gene of R. jostii RHA1 from pET-MO16 (Szolkowy et al. 2009). The MO16_nde1_F and MO16_nde1_R (Table S2) were used as the forward and reverse primers, respectively. The resulting fragments were inserted into the NdeI site of pACYC-ADH. The fusion gene encoding the ADH-MO16 fusion enzyme was constructed via PCR of the ADH part with ADH_300_F and ADH_300_R as the forward and reverse primers, respectively (Table S2). The MO16 part was amplified with the forward and reverse primers MO16_001_F and MO16_001_R. Afterward, the two fragments were fused via isothermal assembly PCR and inserted into the HindIII site of pACYC-duet vector. The fusion gene encoding ADH-FOM-MO16 was constructed via PCR of the ADH part with the forward and reverse primers ADH_300_F and ADH_301_R using the pACYC-ADH-FOM-BVMO as template (Table S2). The MO16 part was amplified with the forward and reverse primers MO16_101_F and MO16_001_R using pET-MO16 as template. Afterward, the two fragments were fused via isothermal assembly PCR and inserted into the HindIII site of pACYC-duet vector. The fusion gene encoding ADH-Gly-MO16 was synthesized via PCR of the ADH part with the forward and reverse primers ADH_300_F and ADH_302_R using the pACYC-ADH-Gly-BVMO as template (Table S2). The MO16 part was amplified with the forward and reverse primers MO16_201_F and MO16_001_R using pET-MO16 as template. Afterward, the two fragments were fused via isothermal assembly PCR and inserted into the HindIII site of pACYC-duet vector.
Purification of ADH
The enzymes were purified via affinity chromatography on a Ni-NTA gel matrix (Qiagen, Crawley, UK), after cultivation of the recombinant E. coli BL21(DE3) pACYC-ADH in the LB medium and cell lysis by sonication. A column filled with 3 mL of Ni-NTA resin was equilibrated with 15 volumes of buffer containing 20 mM Tris, 500 mM NaCl, and 5 mM imidazole, and the supernatant was loaded onto the column. The column was washed with 10 volumes of wash buffer containing 20 mM Tris, pH 8.0, 500 mM NaCl, and 20 mM imidazole. The proteins were then eluted by increasing the imidazole concentration to 0.3 M. Fractions containing the recombinant proteins were pooled and dialyzed to remove imidazole.
Activity assay of ADH
The ADH activity was evaluated as described previously (Niehaus et al. 1978). In brief, the activity of the ADH was measured on a basis of reduction rate of NAD+, thereby increase of observance at 340 nm at 30 °C in the presence of 10-hydroxystearic acid as the substrate. The reaction was initiated by adding 0.4 μM ADH into 50 mM sodium pyrophosphate buffer (pH 9.0) containing 0.2 mM 10-hydroxystearic acid and 0.25 mM NAD+.
Protein electrophoresis
Cell lysates were prepared using a bacteria cell lysis buffer supplemented with protease inhibitor cocktail (Roche, Zurich, Switzerland). Whole-cell lysates prepared from cell broth (OD 5) were run on 9 % sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE), and the proteins were stained with Coomassie Brilliant Blue R-250.
Whole-cell biotransformation
Biotransformation with recombinant E. coli cells was conducted on the basis of our previous work (Jang et al. 2014). In brief, the recombinant E. coli cells were cultivated in Riesenberg medium at 30 °C. Expression of the recombinant genes was induced at 16 °C by adding 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After cell growth reached at the stationary growth phase (ca. 3 g dry cells L−1), the culture pH was adjusted to 8.0 with 1 N NaOH and the reaction substrate was added to 5 or 10 mM in the medium. Tween 80 was also added to 0.5 g L−1. The reaction was incubated at 35 °C and 200 rpm in shaking incubator.
Product analysis by GC/MS
Concentrations of remaining hydroxy fatty acids and accumulating fatty acid products in the medium (i.e., ketone b and ester c or d) were determined as described previously (Song et al. 2013). The reaction medium was mixed with an equal volume of ethyl acetate containing methyl palmitate as an internal standard. The organic phase was harvested after vigorous vortexing and then subjected to derivatization with N-methyl-N-(trimethylsilyl)trifluoroacetamide (TMS). The TMS derivatives were analyzed using a Thermo Ultra Trace GC system connected to an ion trap mass detector (Thermo ITQ1100 GC-ion Trap MS, Thermo Scientific). The derivatives were separated on a nonpolar capillary column (30-m length, 0.25-μm film thickness, HP-5MS, Agilent). A linear temperature gradient was programmed as follows: 90 °C, 5 °C min−1 to 280 °C. The injection port temperature was 230 °C. Mass spectra were obtained by electron impact ionization at 70 eV. Scan spectra were obtained within the range of 100–600 m/z. Selected ion monitoring (SIM) was used for the detection and fragmentation analysis of the reaction products. Composition of the esters c and d was confirmed by analyzing ester hydrolysis products (e.g., alkanoic acids, ω-hydroxycarboxylic acids, alkanols, and α,ω-dicarboxylic acids), which were prepared with esterase of Pseudomonas fluorescens WI SIK as described in an earlier study (Song et al. 2013). Concentration of the reaction substrates and products was determined on a basis of calibration curves, which were determined using commercially available products or products isolated in our lab.
Results
Design and construction of the fusion enzymes
Molecular fusion of the ADH and the BVMOs was initiated by mimicking the fatty acid β-oxidation multienzyme (FOM) complex of Pseudomonas fragi, which is involved in the β-oxidation pathway of fatty acids (Ishikawa et al. 2004). There, the ADH component is connected to a hydratase through a rather rigid α-helix linker, which consists of 30 amino acids (Table 1). Thereby, the first fusion enzymes were constructed by connecting the C-terminal of ADH to the α-helix linker of the FOM and then to the N-terminal of BVMOs (Table 1). Other fusion enzymes were constructed by using a glycine-rich peptide linker, consisting of 12 amino acids, which was reported to be flexible in structure and would hardly inhibit natural movement of the enzymes (Pazmino et al. 2009).
When the ADH and the BVMO from P. putida KT2440 were expressed independently, the ADH showed a thick band in the soluble fraction of the extracts, but the BVMO band in the soluble fraction was very weak (Fig. S3). On the other hand, the fusion enzymes exhibited significantly denser bands in the soluble fractions compared to the BVMO expressed alone. In particular, the ADH-BVMO fusion enzyme, which was connected by the glycine-rich linker (i.e., ADH-Gly-BVMO), showed the highest expression level in soluble form. This result indicated that expression of the BVMO gene was facilitated by fusion with the ADH gene.
Catalytic activity of the fusion enzymes
The ADH-BVMO fusion enzymes were isolated under various conditions based on a previous study (Rehdorf et al. 2007). The fusion enzymes showed significantly higher NADPH oxidation activity in the presence of 12-ketooleic acid (1b) compared to the BVMO expressed independently (Fig. S4). However, the fusion enzymes were also unstable; they lost their activity rather fast after isolation in the reaction buffer, as shown for the cyclohexanone monooxygenase from Acinetobacter sp. NCIMB9871 (van Beek et al. 2014). Thereby, we have examined the catalytic activity of the enzymes via whole-cell reaction with ricinoleic acid (1a) as the substrate (Fig. 2). The recombinant E. coli BL21(DE3) pACYC-ADH/BVMO, expressing the ADH and BVMO independently, converted ricinoleic acid into the ester (1c) at a rate of 9.3 μmol g dry cells−1 min−1 at t < 3 h (see the “Materials and methods” section for details). However, the product formation was soon ceased and the intermediate (1b) remained accumulated in the reaction medium (Fig. 2a). This indicated that the BVMO lost its catalytic activity during biotransformation and the oxygenation reaction would be a rate-limiting step. On the other hand, the recombinant E. coli BL21(DE3) cells, expressing the ADH and BVMO fusion enzymes, led to higher ester formation rate while the reaction intermediate accumulated less in the medium. Especially, the ADH-Gly-BVMO construct appeared to be the most active. The recombinant cells expressing the ADH-Gly-BVMO construct showed the greatest biotransformation performance, which was estimated from the final concentration of the ester product (Fig. 2d). Notably, the ADH reaction rates, which were estimated based on the concentrations of the intermediate (1b) and ester (1c) in the medium, were not markedly dependent upon the enzymes used. This indicated that the BVMO activity was mainly improved by protein fusion (Table 1). The low concentration of the intermediate (1b) with the recombinant E. coli expressing the ADH-Gly-BVMO construct also indicated that the substrate was transported efficiently from the active site of the ADH to the BVMO active site in the fusion system. Based on the whole-cell biotransformation data, we assumed that molecular fusion of the enzymes and type of the linkers are important to performance of the cascade biotransformation of the rather large and hydrophobic substrates. The flexible linker (i.e., glycine-rich linker) led to greater performance of the fusion enzymes compared to the rigid linker (i.e., α-helix FOM linker).

Biotransformation of ricinoleic acid (1a) into ester (1c) by recombinant E. coli BL21(DE3) pACYC-ADH/BVMO expressing the ADH and BVMO independently (a), E. coli BL21(DE3) pACYC-ADH-BVMO expressing the ADH-BVMO fusion enzyme (b), E. coli BL21(DE3) pACYC-ADH-FOM-BVMO expressing the ADH-FOM-BVMO fusion enzyme (c), and E. coli BL21(DE3) pACYC-ADH-Gly-BVMO expressing the ADH-Gly-BVMO fusion enzyme (d). The biotransformation was initiated at the stationary growth phase (cell density, 3 g dry cells L−1) in Riesenberg medium by adding 10 mM ricinoleic acid and 0.5 g L−1 Tween 80 to the culture broth at 8 h after inducing gene expression with 0.1 mM IPTG at 16 °C. Symbols indicate concentrations of ricinoleic acid (1a) (solid circle), 10-ketooleic acid (1b) (solid triangle), and ester 1c (solid square)
Effect of induction temperature on biocatalytic activity
Activity of the BVMO of P. putida KT2440 expressed in E. coli was dependent on the culture temperature. The optimal temperature was around 16 °C (Rehdorf et al. 2007). At higher temperatures, the enzymes did not appear to fold in an active form. Here, we examined folding efficiency of the fusion enzymes by investigating the effect of gene expression temperature on their soluble expression level and whole-cell biotransformation activity with ricinoleic acid (1a). Increase in induction temperature from 16 to 30 °C led to increased expression of the fusion enzyme in an insoluble form (Fig. S5). However, soluble expression level of the fusion enzyme remained rather unchanged even at 30 °C, and thereby, the whole-cell biotransformation activity indicating the in vivo enzyme activity was not markedly reduced at the temperature (Table 2). This result suggests that the fusion enzyme is relatively better expressed in a soluble form at higher temperatures as compared to the wild-type BVMO.
Biotransformation activity of the fusion enzymes
Biotransformation of 13-hydroxyoctadec-9-enoic acid (2a), 10-hydroxyoctadec-12-enoic acid (3a), 14-hydroxyicos-11-enoic acid (4a), 12-hydroxyoctadecanoic acid (5a), 10-hydroxyoctadecanoic acid (6a), and 5-hydroxydecanoic acid (7a) (Fig. 1) was also carried out with the recombinant E. coli BL21(DE3) cells, expressing the ADH and BVMO independently or the ADH-BVMO fusion enzymes (Table 3). As compared to the recombinant E. coli expressing ADH and BVMO independently, the recombinant E. coli expressing ADH-Gly-BVMO fusion enzyme showed significantly higher bioconversion with 2a, 3a, 4a, and 7a as substrates, which have the three-dimensional conformation similar to 1a. The alcohols 2a, 3a, and 4a possess a double bond in the carbon skeleton. 7a is a medium-chain sec-alcohol (i.e., 5-hydroxydecanoic acid). In contrast, the whole-cell biocatalysts did not show significant difference in bioconversion with long-chain aliphatic ester fatty acids without double bonds (i.e., 5a, 6a). Overall, the relative catalytic activity of the fusion enzymes was dependent upon the structure of the substrates (e.g., position of a double bond, carbon chain length). Another interesting point was the formation of the “abnormal” regioisomeric esters 2d or 3d in the biotransformation of 2a and 3a. The ratio of ester d to ester c appeared to be influenced by the enzymes used. The ADH-Gly-BVMO fusion enzyme resulted in smaller ratio of ester d to c than the other enzymes (Table 3). The reason for the difference in regioselectivity remains to be investigated.
Fusion of R. jostii BVMO and M. luteus ADH
The BVMO from R. jostii RHA1 (i.e., MO16) (Szolkowy et al. 2009) is much more vulnerable to inclusion body formation compared to the P. putida BVMO, when expressed in E. coli (see Fig. S6). Therefore, we have examined fusion of the R. jostii BVMO with the ADH from M. luteus. Fusion between the both enzymes also led to a significant increase of soluble expression of the BVMO, MO16 (Fig. S6). The greater expression level in the soluble form allowed the recombinant E. coli expressing the ADH-MO16 fusion enzymes to reach higher biotransformation rates for 1a and 2a when compared to the enzymes expressed independently (Fig. 3) (Table 4). Activity of the recombinant E. coli expressing the ADH-Gly-MO16 with 1a was about twofold greater than that of the recombinant E. coli expressing the ADH and MO16 independently. This result indicates that fusion of insoluble enzymes with soluble enzymes could be useful to enhance functional expression of the insoluble enzymes and improve the overall performance of whole-cell biocatalysis.

Biotransformation of ricinoleic acid (1a) into ester (1c) by recombinant E. coli BL21(DE3) pACYC-ADH/MO16 expressing the ADH and MO16 independently (a), E. coli BL21(DE3) pACYC-ADH-MO16 expressing the ADH-MO16 fusion enzyme (b), E. coli BL21(DE3) pACYC-ADH-FOM-MO16 expressing the ADH-FOM-MO16 fusion enzyme (c), and E. coli BL21(DE3) pACYC-ADH-Gly-MO16 expressing the ADH-Gly-MO16 fusion enzyme (d). The biotransformation was initiated at the stationary growth phase (cell density, 3 g dry cells L−1) in Riesenberg medium by adding 10 mM ricinoleic acid and 0.5 g L−1 Tween 80 to the culture broth at 8 h after inducing gene expression with 0.1 mM IPTG at 16 °C. Symbols indicate concentrations of ricinoleic acid (1a) (solid circle), 10-ketooleic acid (1b) (solid triangle), and ester 1c (solid square)
Discussion
During the last decade, protein fusion technologies were widely used to enhance soluble expression of the proteins (e.g., the use of soluble tags) as well as to facilitate enzyme purification (e.g., the use of glutathione S-transferase (GST) tags) (Chen et al. 2013; Zhang et al. 2009). This concept was hardly used in the context of synthetic applications. Only a few studies have reported on enzyme fusion to achieve self-sufficient cofactor regeneration (e.g., fusion of BVMOs to NADPH-regenerating phosphite dehydrogenase) (de Gonzalo et al. 2011; Pazmino et al. 2008, 2009).
We demonstrated here that enzyme fusion is a promising technology to improve performance of two-step whole-cell biocatalysis. Oxygenation activity of the recombinant E. coli cells with respect to long-chain unsaturated sec-alcohols (e.g., 12-hydroxyoctadec-9-enoic acid (1a), 13-hydroxyoctadec-9-enoic acid (2a), 14-hydroxyicos-11-enoic acid (4a)) and medium-chain saturated sec-alcohols (e.g., 5-hydroxydecanoic acid) into the corresponding esters was significantly improved by using the ADH-BVMO fusion enzymes (Tables 3 and 4). The protein fusion appeared to contribute to functional expression of the catalytic enzymes (i.e., BVMOs) in recombinant E. coli cells. In addition, the substantial improvement in biotransformation activity of the recombinant E. coli cells might be also ascribed to an increase in mass transport efficiency of the reaction substrate. Enzyme fusion was reported to improve protein–protein interactions, which may lead to channeling intermediates between enzymes or localizing two active sites in close proximity (Chen et al. 2013). However, why the relative catalytic activity of the fusion enzymes was dependent upon the structure of the reaction substrates (e.g., position of a double bond, carbon chain length) remains to be investigated.
The biotransformation activity of the recombinant cells was markedly influenced by type of the fusion linkers (Tables 3 and 4). The flexible linker (i.e., glycine-rich linker)-based fusion enzymes such as ADH-Gly-BVMO and ADH-Gly-MO16 allowed the whole cells to reach higher productivity than the cells expressing the rather rigid α-helix linker-based fusion enzymes (i.e., ADH-FOM-BVMO, ADH-FOM-MO16). Thereby, optimization of the fusion linkers may allow further increase in functional expression and activity of the biotransformation enzymes.
All the results suggest that fusion of the catalytic enzymes could be used to improve productivity and product yields in the cascade reactions of substrates with low water solubility (e.g., amination of primary alcohols (Sattler et al. 2012; Song et al. 2014), conversion of flavanones into dihydrochalcones (Gall et al. 2013), and halogenation of substituted tryptophan derivatives (Frese et al. 2014)). Therefore, we assumed that protein fusion technologies are promising tools to improve performance of multistep biocatalysis.
References
Balke K, Kadow M, Mallin H, Sass S, Bornscheuer UT (2012) Discovery, application and protein engineering of Baeyer-Villiger monooxygenases for organic synthesis. Org Biomol Chem 10:6249–6265. doi:10.1039/c2ob25704a
Bisogno FR, Rioz-Martinez A, Rodriguez C, Lavandera I, de Gonzalo G, Pazmino DET, Fraaije MW, Gotor V (2010) Oxidoreductases working together: concurrent obtaining of valuable derivatives by employing the PIKAT method. ChemCatChem 2:946–949. doi:10.1002/cctc.201000115
Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K (2012) Engineering the third wave of biocatalysis. Nature 485:185–194. doi:10.1038/nature11117
Cheesman MJ, Kneller MB, Kelly EJ, Thompson SJ, Yeung CK, Eaton DL, Rettie AE (2001) Purification and characterization of hexahistidine-tagged cyclohexanone monooxygenase expressed in Saccharomyces cerevisiae and Escherichia coli. Protein Expr Purif 21:81–86. doi:10.1006/prep.2000.1340
Chen X, Zaro JL, Shen W-C (2013) Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 64:1357–1369. doi:10.1016/j.addr.2012.09.039
de Gonzalo G, Mihovilovic MD, Fraaije MW (2010) Recent developments in the application of Baeyer-Villiger monooxygenases as biocatalysts. ChemBioChem 11:2208–2231. doi:10.1002/cbic.201000395
de Gonzalo G, Smit C, Jin J, Minnaard AJ, Fraaije MW (2011) Turning a riboflavin-binding protein into a self-sufficient monooxygenase by cofactor redesign. Chem Commun 47:11050–11052. doi:10.1039/c1cc14039f
Frese M, Guzowska PH, Voss H, Sewald N (2014) Regioselective enzymatic halogenation of substituted tryptophan derivatives using the FAD-dependent halogenase RebH. ChemCatChem 6:1270–1276. doi:10.1002/cctc.201301090
Gall M, Thomsen M, Peters C, Pavlidis IV, Jonczyk P, Gruenert PP, Beutel S, Scheper T, Gross E, Backes M, Geissler T, Ley JP, Hilmer J-M, Krammer G, Palm GJ, Hinrichs W, Bornscheuer UT (2013) Enzymatic conversion of flavonoids using bacterial chalcone isomerase and enoate reductase. Angew Chem Int Ed 53:1439–1442. doi:10.1002/anie.201306952
Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA III, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi:10.1038/nmeth.1318
Holtmann D, Fraaije MW, Arends IWCE, Opperman DJ, Hollmann F (2014) The taming of oxygen: biocatalytic oxyfunctionalisations. Chem Commun. doi:10.1039/c3cc49747j
Ishikawa M, Tsuchiya D, Oyama T, Tsunaka Y, Morikawa K (2004) Structural basis for channelling mechanism of a fatty acid beta-oxidation multienzyme complex. EMBO J 23:2745–2754. doi:10.1038/sj.emboj.7600298
Jang H-Y, Jeon E-Y, Baek AH, Lee S-M, Park J-B (2014) Production of ω-hydroxyundec-9-enoic acid and n-heptanoic acid from ricinoleic acid by recombinant Escherichia coli-based biocatalyst. Process Biochem 49:617–622. doi:10.1016/j.procbio.2014.01.025
Kirschner A, Altenbuchner J, Bornscheuer UT (2007) Cloning, expression, and characterization of a Baeyer-Villiger monooxygenase from Pseudomonas fluorescens DSM 50106 in E. coli. Appl Microbiol Biotechnol 73:1065–1072. doi:10.1007/s00253-006-0556-6
Ladkau N, Schmid A, Buehler B (2014) The microbial cell—functional unit for energy dependent multistep biocatalysis. Curr Opin Biotechnol 30:178–189. doi:10.1016/j.copbio.2014.06.003
Lee DH, Kim MD, Lee WH, Kweon DH, Seo JH (2004) Consortium of fold-catalyzing proteins increases soluble expression of cyclohexanone monooxygenase in recombinant Escherichia coli. Appl Microbiol Biotechnol 63:549–552. doi:10.1007/s00253-003-1370-z
Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY (2012) Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol 8:536–546. doi:10.1038/nchembio.970
Lin Y, Jain R, Yan Y (2014) Microbial production of antioxidant food ingredients via metabolic engineering. Curr Opin Biotechnol 26:71–78. doi:10.1016/j.copbio.2013.10.004
Lopez-Gallego F, Schmidt-Dannert C (2010) Multi-enzymatic synthesis. Curr Opin Chem Biol 14:174–183. doi:10.1016/j.cbpa.2009.11.023
Mallin H, Wulf H, Bornscheuer UT (2013) A self-sufficient Baeyer-Villiger biocatalysis system for the synthesis of ε-caprolactone from cyclohexanol. Enzym Microb Technol 53:283–287. doi:10.1016/j.enzmictec.2013.01.007
Niehaus JRWG, Frielle T, Kingsley JREA (1978) Purification and characterization of a secondary alcohol dehydrogenase from a pseudomonad. J Bacteriol 134:177–183
Oberleitner N, Peters C, Muschiol J, Kadow M, Sass S, Bayer T, Schaaf P, Iqbal N, Rudroff F, Mihovilovic MD, Bornscheuer UT (2013) An enzymatic toolbox for cascade reactions: a showcase for an in vivo redox sequence in asymmetric synthesis. ChemCatChem 5:3524–3528. doi:10.1002/cctc.201300604
Orru R, Dudek HM, Martinoli C, Pazmino DET, Royant A, Weik M, Fraaije MW, Mattevi A (2011) Snapshots of enzymatic Baeyer-Villiger catalysis: oxygen activation and intermediate stabilization. J Biol Chem 286:29284–29291. doi:10.1074/jbc.M111.255075
Pazmino DET, Snajdrova R, Baas B-J, Ghobrial M, Mihovilovic MD, Fraaije MW (2008) Self-sufficient Baeyer-Villiger monooxygenases: effective coenzyme regeneration for biooxygenation by fusion engineering. Angew Chem Int Ed 47:2275–2278. doi:10.1002/anie.200704630
Pazmino DET, Riebel A, de Lange J, Rudroff F, Mihovilovic MD, Fraaije MW (2009) Efficient biooxidations catalyzed by a new generation of self-sufficient Baeyer-Villiger monooxygenases. ChemBioChem 10:2595–2598. doi:10.1002/cbic.200900480
Rehdorf J, Kirschner A, Bornscheuer UT (2007) Cloning, expression and characterization of a Baeyer-Villiger monooxygenase from Pseudomonas putida KT2440. Biotechnol Lett 29:1393–1398. doi:10.1007/s10529-007-9401-y
Rioz-Martinez A, Bisogno FR, Rodriguez C, de Gonzalo G, Lavandera I, Pazmino DET, Fraaije MW, Gotor V (2010) Biocatalysed concurrent production of enantioenriched compounds through parallel interconnected kinetic asymmetric transformations. Org Biomol Chem 8:1431–1437. doi:10.1039/b925377g
Sattler JH, Fuchs M, Tauber K, Mutti FG, Faber K, Pfeffer J, Haas T, Kroutil W (2012) Redox self-sufficient biocatalyst network for the amination of primary alcohols. Angew Chem Int Ed 51:9156–9159. doi:10.1002/anie.201204683
Song JW, Jeon EY, Song DH, Jang HY, Bornscheuer UT, Oh DK, Park JB (2013) Multistep enzymatic synthesis of long-chain alpha, ω-dicarboxylic and ω-hydroxycarboxylic acids from renewable fatty acids and plant oils. Angew Chem Int Ed 52:2534–2537. doi:10.1002/anie.201209187
Song JW, Lee J-H, Bornscheuer UT, Park JB (2014) Microbial synthesis of medium chain α, ω-dicarboxylic acids and ω-aminocarboxylic acids from renewable long chain fatty acids. Adv Synth Catal 356:1782–1788. doi:10.1002/adsc.201300784
Staudt S, Bornscheuer UT, Menyes U, Hummel W, Groeger H (2013) Direct biocatalytic one-pot-transformation of cyclohexanol with molecular oxygen into ε-caprolactone. Enzym Microb Technol 53:288–292. doi:10.1016/j.enzmictec.2013.03.011
Szolkowy C, Eltis LD, Bruce NC, Grogan G (2009) Insights into sequence-activity relationships amongst Baeyer-Villiger monooxygenases as revealed by the intragenomic complement of enzymes from Rhodococcus jostii RHA1. ChemBioChem 10:1208–1217. doi:10.1002/cbic.200900011
van Beek HL, Wijma HJ, Fromont L, Janssen DB, Fraaije MW (2014) Stabilization of cyclohexanone monooxygenase by a computationally designed disulfide bond spanning only one residue. FEBS Open Bio 4:168–174. doi:10.1016/j.fob.2014.01.009
Zhang J, Yun J, Shang Z, Zhang X, Pan B (2009) Design and optimization of a linker for fusion protein construction. Prog Nat Sci 19:1197–1200. doi:10.1016/j.pnsc.2008.12.007
Zhao S, Kumar R, Sakai A, Vetting MW, Wood BM, Brown S, Bonanno JB, Hillerich BS, Seidel RD, Babbitt PC, Almo SC, Sweedler JV, Gerlt JA, Cronan JE, Jacobson MP (2013) Discovery of new enzymes and metabolic pathways by using structure and genome context. Nature 502(7473):698–702. doi:10.1038/nature12576
Acknowledgments
This study was supported by the Marine Biomaterials Research Center grant from Marine Biotechnology Program funded by the Ministry of Oceans and Fisheries, Korea.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 662 kb)
Rights and permissions
About this article

Cite this article
Jeon, EY., Baek, AH., Bornscheuer, U.T. et al. Enzyme fusion for whole-cell biotransformation of long-chain sec-alcohols into esters. Appl Microbiol Biotechnol 99, 6267–6275 (2015). https://doi.org/10.1007/s00253-015-6392-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6392-9