
Abstract
Historically used in textile and paper industry, hemp fibres have started to find new applications in composite materials with important economic and ecological advantages. However, their applications are limited since manufacturers have some difficulties to standardise fabrication processes. This study is a first step before selection and isolation of strains that could later be used to optimise microbial retting efficiency and hence fibre quality. We studied six samples harvested on different ground types, at different dates and with different retting durations on field to obtain an exhaustive representation of the process. After DNA extraction, total bacteria and fungi associated with stems during retting were specifically quantified using real-time PCR. Then, using sequence analysis of randomly cloned 16S and 18S ribosomal RNA (rRNA) genes, a phylogenetic characterisation of the dominant microorganisms was carried out. Quantitatively, we showed that there were 8.1–9.5 log10 16S rRNA gene copies per gram of hemp straw for bacteria and 8.6–9.6 log10 18S rRNA gene copies per gram for fungi. Qualitatively, we noticed a higher bacterial diversity in comparison to fungi. This work showed that in the different samples, the same species were present but in significantly different proportions according to ground type, harvest dates and retting durations on field. The most frequent bacterial sequences were affiliated to species Escherichia coli, Pantoea agglomerans, Pseudomonas rhizosphaerae, Rhodobacter sp., Pseudomonas fulva, Rhizobium huautlense and Massilia timonae, whereas fungal sequences were principally related to the genera Cladosporium and Cryptococcus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bast fibre plants such as flax, hemp, and nettle produced valuable sclerenchymatous fibres which can be used in various products like textiles and high-quality papers (McGovern 1983). Hemp fibre plant (Cannabis sativa L.) has been recently used in bio-based composite materials for the automobile, the building, the household electrical appliances and the packaging (Pickering et al. 2007). Glass fibres have been the most used fibres to reinforce moulds due to their low production cost. Nevertheless, the incorporation of plant fibres in composite materials instead of glass fibres is an already marketed concept (Pickering et al. 2007).
The substitution of the plant fibres in mineral or synthetic fibres presents numerous advantages, such as the renewable character of the resource, the biodegradability and the reduction in the environmental impact and costs (Tahir et al. 2011). Nevertheless, certain drawbacks can put a brake on the industrial development, in particular the heterogeneity of the quantity and quality of the plant fibres. In plant stem, elementary cellulose fibres are glued together to form bundles located between the epidermis and the xylem core and parallel to the longitudinal axis of the stem. In these bundles, fibres are embedded in a pectic polysaccharidic network (Cronier et al. 2005; Vignon and Garcia-Jaldon 1996). The variability of hemp fibres is due to the preliminary retting, indispensable stage to the later use of fibres, which depends on environmental factors such as temperature, moisture, duration, etc. Stem retting is a process which facilitates the separation of fibres from other stem tissues. The main idea is to degrade the pectins (backbone of galacturonic acid residues with side chains of l-rhamnose, arabinose, galactose and xylose) and other cementing compounds that bind the bast fibres and fibre bundles to other tissues and thereby separate fibres and nonfibre materials ((Akin 2013) for a review). Enzymes hydrolysing pectins are broadly known as pectinases and include polygalacturonases, pectin esterases, pectin lyases and pectate lyases (Alkorta et al. 1998). A wide variety of microorganisms produce pectinases. Retting is thus a fermentation process involving several bacteria (e.g., Bacillus, Clostridium) and fungi (e.g., Aspergillus, Penicillium) which degrade the bark and release fibres (Sharma and Robinson 1983). To achieve anaerobic retting, straw sheaves are submerged in water pits, in concrete tanks or in running fresh water. The first microorganisms able to grow belong to the genus Bacillus in the aerobic phase; at that point, tank or other stagnant retts become depleted of dissolved oxygen encouraging the development of a different microbiota, notably Clostridium acetobutylicum and Clostridium felsineum for hemp water retting (Tamburini et al. 2004). Achromobacter parvulus, several Clostridia and Pseudomonas aeruginosa were identified as bacterial retting agents implicated in the water retting of Brazilian flax (Rosemberg 1965). Pollution due to this fermentation process as well as important costs linked to labour and drying lead this anaerobic retting to be replaced by dew retting (an aerobic process) (Henriksson et al. 1997). In dew (or field) retting, plant straw is thinly spread on the ground and exposed to the action of fungi and aerobic bacteria for 2–10 weeks. Species of Cladosporium, Penicillium, Aspergillus and Rhodotorula have been isolated from dew-retted plants (Fogarty and Ward 1972). Fungi such as Cladosporium herbarum, Epicoccum nigrum, Alternaria alternata, Fusarium sp., Aureobasidium pullulans, Phoma sp., Mucor spp., Rhizomucor pusillus and Rhizopus sp. were also identified during flax dew retting (Akin et al. 1998; Henriksson et al. 1997; Sharma 1986a). Since hemp is more lignified than flax, different enzymes should be needed. A mechanical step is then used to clean the separated fibres. The main disadvantage of the whole microbial process is the risk that cellulolytic enzymes secreted by the microbiota damage fibres. The duration is thus extremely important.
For the industries, it is necessary to master the retting to obtain value-added fibres with a constant quality and a high yield. Furthermore, there are only few data in the literature concerning the dew retting of hemp. The objective of this study was to gather information on the dynamics of the retting, especially on the identification of microorganisms involved during this primary process. On the one hand, quantitative information will permit the estimation of the colonisation level of the various microorganisms present using real-time PCR. On the other hand, qualitative information will characterise the retting profile by identifying the microbial species involved. The phylogenetic analysis of bacterial 16S and fungal 18S ribosomal RNA (rRNA) genes, amplified directly from complex communities, provides an efficient strategy for exploring the biodiversity and had been applied to many ecosystems (Borneman and Hartin 2000; Renault et al. 2012; Song et al. 2001; Suau et al. 1999), notably to kenaf (Visi et al. 2013) and to jute (Munshi and Chattoo 2008) water retting. Therefore, in order to take into account cultivable as well as non-cultivable microorganisms, molecular tools have been chosen to investigate the hemp retting community. Detailed phylogenetic inventories (bacterial and fungal) of six hemp samples cultivated on two types of ground, with two harvested data, and two retting durations on field have been carried out. The results are expected to obtain the best representation of the process. The long-term objective is to optimise retting efficiency and control the process through new strategies. Recently, the water retting process and fibre quality of hemp were improved by simultaneously inoculating water tanks with two selected pectinolytic strains (Di Candilo et al. 2010). Four pectinolytic strains were also selected to improve water retting of jute (Das et al. 2012). These strains belonged to Clostridium and Bacillus genera. Microbial water retting of kenaf was also optimised by adding Bacillus and Paenibacillus strains in experimental tanks (Visi et al. 2013). Development of specific starter strains selected from the molecular inventories could be envisaged for improving dew retting efficiency of hemp and hence the fibre quality.
Materials and methods
Plant material and growth conditions
Hemp (C. sativa L. cv Fedora 17) was grown in 2011 by professional hemp producers in field under standards conditions: sowing in April at 50 kg/ha in a soil fertilised with 120 kg nitrogen/ha (Institut Technique du Chanvre; CETIOM, 2007, Le chanvre industriel, 26 p, http://www.cetiom.fr/). Field trials were performed in the South of Champagne region in France on two types of ground characteristic of this hemp production area: a white “chalky soil” corresponding to the field located at Echemines (48° 38′ N, 3° 83′ E) and a “red soil” corresponding to the field located at Marcilly le Hayer (48° 35′ N, 3° 63′ E). Soil in these sectors is sandy clay of homogeneous texture with less than 1-m depth before the limestone. The clay level is the major difference between these two types of soils; “red soils” are more clayish resulting in their “red colour” compare to “white soil”. These fields being 20 km away from each other, the climatic conditions were similar.
Straw harvest and samples preparation
Two dates of harvest were tested surrounding the seed maturity: the early date (D1) corresponds to the end of flowering stage and the late date (D2) corresponds to the seed maturity stage. From an agronomical point of view, D2 was the normal stage to harvest hemp in this area, farmers harvesting seeds and straw. Considering stem anatomy, tissue organisation was achieved at D1. Whatever the date, harvesting corresponds to stem cut at their base and their disposal on ground. Hemp straw was then swathed to form a long, narrow strip in field, 1 week after cutting. Straw cutting and swathing were mechanically performed. Straw remains on ground during the retting period. According to farmer experience, two levels of retting were tested: a mid-retting corresponding to 3 weeks and a full retting corresponding to 5 or 6 weeks. All these conditions were resumed in Table 1.
After harvest in field, samples of straw were immediately stored in a −20 °C cool box. Five gram of each stem sample were cut into 2-cm pieces in sterile conditions to allow their grinding in the Stomacher® (Lab-blender 80, England) with 45 mL of NaCl 0.9 % for 5 min. Liquid fractions were then recovered in 50-mL sterile tubes and centrifuged during 15 min at 4,000×g, at 4 °C. Pellets were resuspended in 250 μL of NaCl 0.9 %, and total DNA was extracted for each sample. This procedure was repeated twice, the one for culture, the second for total DNA extraction.
Hemp fibre quality control
The quality of hemp fibre was estimated through a measure of linear density of scutched tow. To extract fibre, hemp straw was parallelized and dried then scutched which consists of breaking straw by breaker rollers and “beating” out short fibre and wooden parts into scutched tow. Drying and scutching were proceeded using industrial equipment under the expertise of FRD companies.
Linear density of hemp tow was measured according to Müssig et al. (2006). Hemp tow was cut into 2-cm pieces, then weighed to obtain a measure in grams per kilometre.
Quantification of viable microorganisms
Each sample was analysed using cultivation. Tenfold serial dilutions were carried out on tryptone-salt medium, after which 0.1 mL of each dilution was inoculated onto different media and incubated at the appropriate temperatures. Total aerobic bacteria were cultured at 30 °C in tryptic soy agar (BioMérieux SA, Marcy l’Etoile, France). Anaerobic bacteria were cultured at 30 °C in Brain Heart Infusion agar (BioMérieux SA, Marcy l’Etoile, France) supplemented with yeast extract (5 g/L) and hemin (5 mg/L), under anaerobic conditions (85 % N2, 10 % H2, 5 % CO2). Total fungi were cultured in Sabouraud chloramphenicol agar (BioMérieux SA, Marcy l’Etoile, France) at ambient temperature. Bacteria were numerated after 3 days of incubation, and fungi were quantified after 5 days.
Extraction and purification of total DNA
Total DNA was extracted from 250 μL sample aliquots as previously described (Godon et al. 1997). To extract both bacteria and fungi DNA, a mix of sterile glass beads (diameter 0.1 mm and 0.5 mm) for mechanical extraction was used. The purification was performed using one wash with saturated phenol (pH 8.0) followed by two washes with chloroform: isoamyl alcohol (24:1). Sodium acetate (3 M) and isopropanol 100 % were used to precipitate DNA. Cleaning was achieved with a 70 % ethanol wash. Dried pellets were then resuspended in sterile water.
Clone library construction, screening and sequencing
Twelve libraries of about 50 clones were constructed from the DNA extracts from the bacterial and fungal communities isolated from each hemp sample. Bacterial 16S rRNA genes were amplified with the forward primer S-D-Bact-0339-a-S-20 (5′ CTC CTA CGG GAG GCA GCA GT 3′ (Amann et al. 1990)) and the universal reverse primer S-*-Univ-1385-a-A-18 (5′ GCG GTG TGT ACA AGR CCC 3′ (Magne et al. 2006a)). Fungal 18S rRNA genes were amplified with the forward primer S-D-Fung-0817-a-S-24 (5′ TTA GCA TGG AAT AAT RRA ATA GGA 3′) and the reverse primer S-D-Fung-1536-a-A-20 (5′ ATT GCA ATG CYC TAT CCC CA 3′) (Borneman and Hartin 2000). PCR amplifications were carried out with a standard PCR mix (0.5 U of Taq DNA polymerase (AmpliTaq Gold; Perkin-Elmer Corporation, Foster City, Calif.), 1 x reaction buffer II, 2.5 mM MgCl2, 200 μM of each dNTP, 0.4 μM of each primer and sterile water added to a final volume of 50 μL). Three PCR reactions were made for each of the six samples for both inventories. Initial enzyme activation step was performed at 94 °C for 10 min in a Mastercycler® thermocycler (Eppendorf AG, Hamburg, Germany), followed by 30 cycles of denaturation at 96 °C for 15 s, annealing at 59 °C for bacteria or at 55 °C for fungi for 1 min and elongation at 72 °C for 4 min, which were followed by a final elongation at 72 °C for 15 min. The three PCR products were pooled according to sample, purified and concentrated with a QIAquick spin PCR purification kit (Qiagen, S.A., Courtaboeuf, France) and eluted with 50 μL of sterilised, nuclease-free water. The purified products were ligated into pGEM®-T vector (Promega Corporation, Madison, USA) and used to transform competent Escherichia coli DH10b cells. White recombinant cells were selected on Luria-Bertani medium (Difco, Detroit, USA) supplemented with ampicillin (100 μg/mL), isopropyl-β-d-thiogalactopyranoside (IPTG, 500 μmol/L) and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside, 80 μg/mL). Plasmidic inserts were amplified with appropriate primers then sequenced using primers Bact-0339 or Fung-0817 (Beckman Coulter Genomics, Takeley, UK). The sequences were corrected using Chromas Pro version 1.5. A search of the GenBank nucleotide database was conducted using the BLAST algorithm (www.ncbi.nlm.nih.gov/blast) to determine the closest relative of partial 16S or 18S rRNA gene sequences (Maidak et al. 1999). The sequences were aligned using Clustal Omega 1.1.0. with the profile alignment option in SeaView 4.5.2 (Gouy et al. 2010). A representative of each operational taxonomic unit (OTU or molecular species) was submitted to the GenBank database with the accession numbers KM507203 to KM507290. An OTU or a phylotype consisted of all sequences (clones and reference strains) with less than 2 % divergence (Suau et al. 1999) from 695 to 800 aligned homologous nucleotides and was also generally consistent with the results of the comparison of 16S homology and DNA-DNA re-association values (Stackebrandt and Goebel 1994). The same rate was used for divergence of 18S rRNA gene. Maximum-likelihood trees were built using PhyML 3.1 (Guindon et al. 2010). The stability of phylogenetic relationships was assessed using the bootstrap method based on 100 replications. Chimeric sequences were detected and removed. Generated during PCR, these sequences formed from two or more sequences joined together, branched differently in trees based on 200 aligned bases at the 5′ and at the 3′ ends. Find chimeras from software DECIPHER confirmed the first assessment (Wright et al. 2012). Coverage values were calculated by Good’s method (Good 1953), according to which the percentage of coverage was calculated with the formula [1 − (n/N)] × 100, where n is the number of molecular species represented by one clone (single-clone OTUs) and N is the total number of sequences. This calculation estimates how well the clones accounted for the biodiversity.
Quantification of bacterial and fungal populations by real-time PCR
Reactions were performed in duplicate in a volume of 25 μL within 96-well twin-tech PCR plates, using the Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen, Cergy Pontoise, France). The forward and reverse primers used were Bact-0339 and Bact-0788 for total bacteria (Magne et al. 2006b) and Fung-0817 and Fung-1196 for total fungi (Borneman and Hartin 2000). Amplifications were performed in a Mastercycler ep Realplex4 (exCitation source 470 nm, emission 520/550 nm) (Eppendorf AG, Hamburg, Germany) with the following temperature profile: one cycle at 50 °C (2 min) for uracil-DNA glycosylase action, one cycle at 96 °C (2 min), 40 cycles of denaturation at 96 °C (15 s), primer annealing at 55 °C for bacteria and fungi (1 min) and elongation step at 68 °C (2 min) with fluorescence measure. Finally, the melting curve was made by slowly heating the PCR mixtures from 60 to 96 °C (20 min) with simultaneous measurements of the SYBR Green I signal intensities. A standard curve made from known amounts of plasmid DNA containing a 16S rRNA gene insert from E. coli or a 18S rRNA gene insert from Saccharomyces cerevisiae allowed quantifications.
Data analysis
Quantitative data were presented as log10 16S (or 18S) rRNA gene copies per gram of straw. Each replicate was determined by the average value of two samples. Viable counts were transformed in logarithmic values and then expressed as the mean of the two replicates.
Results
Quality of fibre extract from retted straw
Linear density (weight of a theoretical 1-km strip of tow) gives an estimation of the fineness of tow. Among the parameters used to estimate fibre quality, the fineness of scutched tow seems to be the most global. This parameter has a great impact for textile, composite and building industries. For example, the quality of yarn appears closely linked to the thinness of scutched tow (Sharma and Van Sumere 1992); furthermore, the fineness of fibre added to thermoplastic resin could explain the mechanical properties of composite (Bourmaud 2013).
Retting have an impact on the fineness of hemp scutched tow: a reduction of near 50 % of the linear density could be observed between hemp straw after 6-week retting and raw hemp straw (Fig. 1). No significant difference could be observed between chalky and red soils. The improvement of hemp tow fineness during retting appeared progressive; nevertheless, it was more regular in chalky soil. The substantial standard deviation at 3 weeks for both chalky and red soils came from the heterogeneity of tow and could be an evidence of an unfinished retting process. Finally, these results confirmed that the retting process lead to more thin hemp tow, so the final quality of hemp fibre for technical application was enhanced.

Linear density (g/km) of hemp tow in function of retting. Bars represent standard deviation
Quantification of total bacteria and total fungi using culture
CFU counts ranged for anaerobic bacteria from 5.1 to 8.1 log10 per gram and for aerobic bacteria from 5.5 to 8.7 log10 per gram (Table 2a). Total fungi counts ranged from 5.9 to 7.0 log10 per gram. Red ground had higher counts of total bacteria and fungi after 6 weeks of retting process than after 3 weeks. The same was not observed with chalky ground. Indeed, higher counts of bacteria were found after 3 weeks of retting process than after 5 weeks. Early or normal harvesting data did not change the number of total bacteria or fungi after 3 weeks of retting process on red ground (R2W3 versus R6W3). The number of total bacteria was lower on chalky ground compared to red ground in the same conditions (C6W3 versus R6W3), with similar counts of fungi. Moreover, bacteria and fungi increased on chalky ground between normal harvesting data after 3 weeks of retting process and early harvesting data after 5 weeks of retting process, as attended (C6W3 versus C2W5). These data tend to show that the high bacterial level observed for C2W3 sample is unexpected and may be artefactual.
Quantification of total bacteria and total fungi using qPCR
Quantitatively, we showed that there were 8.1–9.5 log10 16S rRNA gene copies per gram of straw hemp for bacteria and 8.6–9.6 log10 18S rRNA gene copies per gram for fungi (Table 2b). From red ground, the bacteria and fungi quantified after 6 weeks of retting were more numerous than after 3 weeks. However, the microorganisms quantified from chalky ground at 3 and 5 weeks were not different and equivalent to the counts observed on R2W6 sample for bacteria. Fungi counts were higher in R2W6 sample. The number of total bacteria was lower from chalky ground compared to red ground in the same conditions (C6W3 versus R6W3), with similar counts of fungi.
Clone library screening
Analysis of the 16S rRNA clone libraries
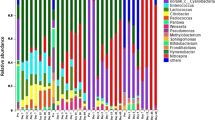
To assess bacterial diversity, 300 16S rRNA genes from the six samples (cultivated on two types of ground, with two harvested data, and two retting durations on field, Table 1), were sequenced and analysed (about 50 per sample). Four sequences were considered as chimera and were removed from analyses. The other sequences were grouped into 66 different phylotypes according to the phylogenetic uniqueness of the closest relative (Supplemental Table S1). The percentages of similarities based on sequence comparison varied from 97 to 100 %. Among the 66 phylotypes, 65 (98.5 %) presented more than 98 % of similarity with previously identified sequences such as Rhizobium huautlense (99–100 %), Pseudomonas graminis (99 %) and Rhodococcus fascians (99 %) (see Supplemental Table S1). The most represented phylum was Proteobacteria (77 % of sequences). Among Proteobacteria, there were mainly γ-Proteobacteria (48 %), α-Proteobacteria (21.5 %) and β-Proteobacteria (7.5 %). Other dominant phyla were Actinobacteria (11 %) and Bacteroidetes (11 %). Firmicutes phylum was represented by only two sequences (1 %). At the phylotype level, the most abundant phylotypes were related to E. coli (JQ907529) (rank 1, 15 % of sequences), Pantoea agglomerans (FJ357815) (rank 2, 12 % of sequences), Pseudomonas rhizosphaerae (GU396285) (rank 3, 10 % of sequences), Rhodobacter sp. CCBAU (5 %), Pseudomonas fulva (4 %), R. huautlense (4 %), Rhodobacter sp. JA745 (3 %) and Massilia timonae (2 %) (Supplemental Table S1). These species were retrieved in almost all libraries.
Analysis of the R2W3 16S rRNA clone library (red ground, early harvesting data, 3 weeks of retting process)
The 49 sequences from the R2W3 library were distributed into 27 phylotypes and three phyla (Supplemental Table S1, Fig. 2). Proteobacteria was the most abundant phylum (80 % of the sequences); they were exclusively γ-Proteobacteria (43 %), α-Proteobacteria (31 %) and β-Proteobacteria (6 %). Other phyla including Actinobacteria (12 %) and Bacteroidetes (8 %) were also identified. Using a threshold of 98 % sequence similarity in 16S rRNA gene to define a molecular species, all sequences were identified to the species level (Supplemental Table S1). Most of them were Pseudomonas species (Pseudomonas psychrotolerans, P. rhizosphaerae, P. fulva, Pseudomonas fluorescens; 12 sequences), Rhodobacter species (eight sequences), E. coli species (five sequences), Pantoea species (P. agglomerans and Pantoea sp.; three sequences) and Rhizobium species (three sequences). The other species were classified within the following genera: Methylobacterium, Rhodococcus, Sphingobacterium and Leifsonia (two sequences each) and Xanthomonas, Massilia, Variovorax, Frigoribacterium, Pedobacter, Chryseobacterium, Brevundimonas, Nocardioides, Achromobacter and Roseomonas (one sequence each). The percentage of coverage ([1 – (n/N)] x 100) was low (65 %). That means that, for this sample, the probability for the next sequence to represent a new phylotype was 35 %.

Phylogenetic distribution in the six clone libraries constructed from bacterial communities of hemp samples
Analysis of the R2W6 16S rRNA clone library (red ground, early harvesting data, 6 weeks of retting process)
The 46 sequences from the R2W6 library were related to 18 unique phylotypes and 4 phyla (Supplemental Table S1, Figs. 2 and 3). The most abundant were Proteobacteria (74 % of the sequences), mostly γ-Proteobacteria (48 %), α-Proteobacteria (24 %) and β-Proteobacteria (2 %). Two other phyla found in abundance were Bacteroidetes (13 %) and Actinobacteria (9 %). Firmicutes phylum was represented only by two sequences (4 %). All sequences had a sequence similarity equal or greater than 98 % with a sequence present in database and were identified as P. agglomerans species (15 sequences), Rhodobacter species (seven sequences), E. coli species (five sequences) and Rhizobium species (Rhizobium huautlense and Rhizobium soli; four sequences). Pseudomonas, Chryseobacterium, Epilithonimonas, Paenibacillus (two sequences), Massilia, Pedobacter, Sphingobacterium, Rhodococcus, Microbacterium, Pseudoclavibacter and Aeromicrobium (one sequence) genera were also found. The percentage of coverage (78 %) was higher than for the sample R2W3.

Phylogenetic tree constructed using bacterial partial SSU rDNA sequences retrieved from R2W6 retting hemp
Analysis of the C2W3 16S rRNA clone library (chalky ground, early harvesting data, 3 weeks of retting process)
The 52 sequences from the C2W3 library corresponded to 21 phylotypes distributed among three phyla (Supplemental Table S1, Fig. 2). The sequences were mainly affiliated to Proteobacteria (77 % of the sequences), mostly γ-Proteobacteria (60 %), α-Proteobacteria (13 %) and β-Proteobacteria (4 %). As previously, Bacteroidetes and Actinobacteria (11.5 % each) were found. Forty-six sequences were identified to cultured species affiliated to species E. coli (13 sequences), P. agglomerans (six sequences), Pseudomonas (P. fulva, P. fluorescens; six sequences), Stenotrophomonas (Stenotrophomonas chelatiphaga, Stenotrophomonas maltophilia; four sequences), Rhizobium (Rhizobium cellulosilyticum, Rhizobium sp.; three sequences), Nocardiopsis (Nocardiopsis synnemataformans, Nocardiopsis prasina; three sequences), Rhodobacter, Achromobacter (two sequences), Aurantimonas, Brevundimonas bullata, Erwinia billingiae, Xanthomonas, Microbacterium oxydans, Agrococcus jejuensis and Saccharopolyspora (one sequence). The six remaining sequences had a sequence similarity equal to 97–98 % with a sequence identified as uncultured Sphingobacterium sp. The percentage of coverage was 77 %.
Analysis of the C2W5 16S rRNA clone library (chalky ground, early harvesting data, 5 weeks of retting process)
The 45 sequences from the C2W5 library represented 25 phylotypes distributed among the already three described phyla (Supplemental Table S1, Fig. 2). Proteobacteria was always the most abundant phylum with 62 % of sequences (γ-Proteobacteria and α-Proteobacteria (26.5 % each), β-Proteobacteria (9 %)). Actinobacteria (20 %) and Bacteroidetes (18 %) phyla were also found in significant abundance. All sequences were identified to the species level and corresponded to E. coli species (nine sequences), Rhizobium species (R. huautlense, R. soli, Rhizobium sp.; six sequences), Rhodobacter sp. (four sequences), Flavobacterium species (Flavobacterium johnsoniae, Flavobacterium sp.; four sequences), Massilia species (M. timonae, Massilia aurea; three sequences), Leifsonia sp. (three sequences), Pseudomonas species (P. rhizosphaerae, P. fulva), Frigoribacterium faeni, R. fascians (two sequences each), Sphingomonas sp., Methylobacterium sp., Achromobacter sp., P. agglomerans, Pedobacter hartonius, Chryseobacterium piscicola, Epilithonimonas lactis, Dyadobacter alkalitolerans, Agrococcus jejuensis and Aeromicrobium erythreum (one sequence each). The percentage of coverage was 67 %.
Analysis of the R6W3 16S rRNA clone library (red ground, normal harvesting data, 3 weeks of retting process)
The 52 sequences from the R6W3 library (13 phylotypes) fell into the same three phyla (Supplemental Table S1, Fig. 2). The sequences were mainly affiliated to Proteobacteria (96 % of the sequences), mostly γ-Proteobacteria (82 %), then α-Proteobacteria (8 %) and β-Proteobacteria (6 %). Actinobacteria and Bacteroidetes were only represented by one sequence (2 % each). All sequences could be identified and were affiliated to Pseudomonas species (P. rhizosphaerae, P. graminis, Pseudomonas syringae; 23 sequences), E. coli (11 sequences), P. agglomerans (nine sequences), R. huautlense, M. timonae (two sequences each), Aurantimonas sp., Methylobacterium sp., Variovorax paradoxus, Hymenobacter sp. and Aeromicrobium ginsengisoli (one sequence each). The percentage of coverage was high (89 %).
Analysis of the C6W3 16S rRNA clone library (chalky ground, normal harvesting data, 3 weeks of retting process)
Fifty-two sequences were also analysed, corresponding to 26 phylotypes and three phyla (Supplemental Table S1, Fig. 2). Proteobacteria (69 % of sequences) were represented by γ-Proteobacteria (25 %), α-Proteobacteria (27 %) and β-Proteobacteria (17 %). Actinobacteria and Bacteroidetes (15.5 % each) were also found in this library. At the species level, species belonging to the Pseudomonas genus were the most abundant (P. rhizosphaerae, P. graminis, P. fulva; eight sequences); then, the following were found: Methylobacterium sp. and Massilia (M. timonae, M. aurea; seven sequences each), Pedobacter (P. hartonius, Pedobacter agri) and F. faeni (six sequences each), E. coli (three sequences), P. agglomerans, Rhodobacter sp., Sphingomonas sp. (two sequences each) and Paracoccus marcusii, Rhizobium sp., Aurantimonas sp., V. paradoxus, Acidovorax sp., Hymenobacter sp., Flavobacterium rivuli, Kineococcus sp., Geodermatophilus sp. (one sequence each). The percentage of coverage was 65 %.
Analysis of the 18S rRNA clone libraries
Three hundred and six 18S rRNA genes were also sequenced and analysed (about 50 per sample). Three sequences were considered as chimera and were removed from analyses. The sequences were grouped into 22 different phylotypes (Supplemental Table S2). The percentage of sequence similarity ranged from 97 to 100 %. Among the 22 phylotypes, 21 (95 %) presented more than 98 % of similarity with previously identified sequences. The best-represented phylum was Ascomycota with 70 % of sequences. The other sequences (30 %) were all affiliated to phylum Basidiomycota. The most abundant phylotypes were related to Cladosporium species (rank 1, 47 % of sequences). However, it was not possible to differentiate species Cladosporium bruhnei (JN397376) from C. herbarum (EU343080) and Cladosporium cladosporioides (JX470336). The second most abundant phylotypes were affiliated to Cryptococcus species (rank 2, 28 % of sequences), notably to Cryptococcus carnescens (24 % of sequences). These phylotypes were shared by all libraries except the C2S3 one, extremely different from the others and dominated by species Stilbella fimetaria (FJ939395).
Analysis of the R2W3 18S rRNA clone library
The 49 sequences from the R2W3 library were distributed into eight phylotypes within two phyla (Supplemental Table S2 and Fig. 4). Ascomycota was the most abundant phylum (61 % of sequences) represented principally by the genus Cladosporium (29 sequences). The second phylum found was Basidiomycota (39 % of sequences) represented principally by the genus Cryptococcus (C. carnescens, Cryptococcus psychrotolerans, Cryptococcus sp., 18 sequences). The other species found belong to Plectosphaerella cucumerina and Rhodotorula marina (one sequence each). The percentage of coverage (94 %) was high. This means that the probability for the next sequence to be already detected was 94 %.

Phylogenetic distribution in the six clone libraries constructed from fungal communities of hemp samples
Analysis of the R2W6 18S rRNA clone library
The 48 sequences from the R2W6 library were related to five unique phylotypes within two phyla (Supplemental Table S2, Figs. 4 and 5). The sequences were mainly affiliated to Ascomycota (90 %), notably to Cladosporium species (40 sequences), then to Basidiomycota (10 %), mostly to C. carnescens (five sequences). Another species belonging to Phoma genera was also retrieved (one sequence). The last species was phylogenetically close to species Endosporium aviarium and Elsinoe sp. (one sequence). The percentage of coverage was 96 %.

Phylogenetic tree constructed using fungal partial SSU rDNA sequences retrieved from R2W6 retting hemp
Analysis of the C2W3 18S rRNA clone library
The 50 sequences from the C2W3 library corresponded to six new phylotypes distributed among the Ascomycota phylum (Supplemental Table S2, Fig. 4). Species S. fimetaria was found in dominance (35 sequences) followed by Aspergillus species (Aspergillus versicolor, Aspergillus glaucus (renamed Eurotium herbariorum), (three sequences each)). The other sequences were affiliated to Cordyceps chlamydosporia, Acremonium sp. and to Penicillium brevicompactum (three sequences each). The percentage of coverage was 100 %.
Analysis of the C2W5 18S rRNA clone library
The 51 sequences were recovered into six phylotypes, affiliated to two phyla already identified (Supplemental Table S2 and Fig. 4). Ascomycota was always the most-represented phylum (86 % of sequences); then, Basidiomycota was found (14 % of sequences). The most important phylotype was attributed to C. herbarum species (33 sequences). Two other important phylotypes corresponded to species P. cucumerina and C. carnescens (seven sequences each). The remaining sequences were attributed to species Elaphocordyceps ophioglossoides (two sequences) and to Geosmithia sp. and E. aviarium/Elsinoe (one sequence each). The percentage of coverage was 96 %.
Analysis of the R6W3 18S rRNA clone library
The 51 sequences from the R6W3 library corresponded to eight phylotypes distributed among two phyla (Supplemental Table S2, Fig. 4). The sequences were mainly affiliated to Basidiomycota (59 % of sequences), particularly to Cryptococcus species (28 sequences). Twenty-one of these sequences were identified as C. carnescens. Ascomycota (41 % of sequences) was abundantly represented by C. herbarum species (19 sequences). Two sequences were phylogenetically close to the species E. aviarium and Elsinoe sp. The remaining sequences were attributed to the species R. marina and Dioszegia hungarica (one sequence each). The percentage of coverage was 94 %.
Analysis of the C6W3 18S rRNA clone library
Fifty-four sequences were also analysed, corresponding to eight phylotypes within two phyla (Supplemental Table S2, Fig. 4). Basidiomycota (56 % of sequences) was the best-represented phylum, and then, Ascomycota was found (44 % of sequences). Species Cryptococcus were found in dominance (C. carnescens, 26 sequences, Cryptococcus sp., one sequence), followed by species Cladosporium (23 sequences). Species represented by only one sequence were also found, E. ophioglossoides, R. marina, Leucosporidium scottii and Dioszegia crocea. The percentage of coverage was 93 %.
Discussion
Industrial use of plant fibres is closely linked to the quality of these fibres (Martin et al. 2013). Several parameters during industrial process from seed to final use could impact on fibre quality. For fibre extracts from plant stems like flax, hemp, jute, kenaf and ramie, the retting process appears to be critical (Tahir et al. 2011). The improvement of hemp fibre tow due to the retting was in line with previous works on flax (Sharma and Van Sumere 1992). According to professional hemp users (farmers, scutching and hackling industrials, composite makers), the quality of the 6-week retted straw and tow obtained in this study was appropriate to thermoplastic composites industries. This quality contrasted with the important heterogeneity of 3-week retted tow which had been described as rough.
Traditionally, hemp retting has been carried out by autochthonous microorganisms present in soil and on plants. However, an industrial application needs standardisation and efficiency of the process. Our goal was to characterise the microbiota implicated in this process. This study represents the first molecular characterisation of bacterial and fungal microbiota in the dew retting of the hemp. Previous studies using culture-dependent methods identified bacterial and fungal species of flax retting (Akin et al. 1998; Henriksson et al. 1997; Sharma 1986b; Sharma et al. 1992). These methods only permitted a partial characterisation of the microbiota, due to specific media utilisation. Employment of culture-independent methods could improve knowledge on microbial composition of an ecosystem, as it is estimated that a large proportion of microorganisms could not be cultivated (Hugenholtz et al. 1998). Changes in bacterial communities in different stages of jute retting were visualised by PCR-denaturing gradient gel electrophoresis from water samples (Das et al. 2013). This method permits a rapid high throughput comparison of bacterial communities over time, but not their identification. Knowledge of the microbial composition is necessary to identify the species that may play a key role in degrading the stems of hemp.
In our study, the 16S or 18S rRNA gene copy number of total bacteria or fungi per gram of hemp straw was very close for the same sample. They were ranging from 8.1 log10 to 9.6 log10 according to sample. It has been shown that bacterial genomes can contain between 1 and 15 ribosomal operons according to the species (Acinas et al. 2004; Klappenbach et al. 2000) whereas it was estimated to be between 50 and more than 200 for fungi (Howlett et al. 1997). A few examples of the number of rRNA operons for microorganisms present in retting: E. coli (seven operons), Pseudomonas putida (six or seven), P. syringae (five), P. fluorescens (five), R. fascians (four or five), Xanthomonas campestris (two), Rhodobacter sphaeroides (three), Variovorax sp. (two), Sphingomonas sp. (one), Aspergillus fumigatus (38 to 91). In consequence, in our samples, since the numbers of rRNA gene were equivalent for bacteria and fungi, there were more bacteria than fungi on straws. This was consistent with the results obtained using culture techniques. From red ground, the bacteria and fungi quantified after 6 weeks of retting process were more numerous than after three. However, the microorganisms quantified from chalky ground after 3 and 5 weeks of retting process were sensibly equivalent together and close to the counts obtained for R2W6. Except for the sample C2W3, higher counts of bacteria and fungi were recovered after 5 or 6 weeks of retting process. And whatever was the method used, fungi were found in higher proportion from red ground after 6 weeks of retting process. It could explain why retting is more efficient on red ground; nevertheless, we have to be cautious since we lack data for chalky ground.
The 296 bacterial clones fell into the phyla Proteobacteria, Bacteroidetes, Actinobacteria and Firmicutes. Proteobacteria was the most abundant phylum with 62–96 % of sequences (mean 77 %). A high redundancy within molecular species was observed between the six samples. This work showed that in each case, the same core of species were present but in different proportions according to ground type, harvest dates and retting duration on field. The most represented bacteria were related to species E. coli, P. agglomerans, P. rhizosphaerae, Rhodobacter sp., P. fulva, R. huautlense and M. timonae. Other Rhizobium species were also found depending on samples (R. soli, R. cellulosilyticum), as well as many other Pseudomonas species (P. psychrotolerans, P. graminis, P. syringae, P. fluorescens, Pseudomonas viridiflava). Genera Methylobacterium, Frigoribacterium and Sphingobacterium were also often retrieved from libraries. Pseudomonas species are particularly important in the decomposition of pectin in plant fibres, both in aerobic and in anaerobic conditions (Betrabet and Bhat 1958; Rosemberg 1965). Two 16S rRNA gene libraries from jute retting in water tanks were recently constructed and analysed. The bulk of clones belong to Proteobacteria phylum (41 %) and a comparatively smaller proportion of clones belonged to the divisions Firmicutes (12 %), Cytophaga-Flexibacter-Bacteroidetes group (7 %), Verrucomicrobia (5 %), Acidobacteria (5 %), Chlorobiales (5 %) and Actinobacteria (2 %) (Munshi and Chattoo 2008). In our study, Proteobacteria represented 77 %, Firmicutes 1 %, Cytophaga-Flexibacter-Bacteroidetes 11 % and Actinobacteria 11 %. In jute retting, Achromobacter, Zoogloea, Erwinia, Pseudomonas, Bacilli, Clostridia, Cytophaga and Verrucomicrobia were found to be common between the two libraries. In hemp retting, P. agglomerans, E. coli, Rhizobium and Pseudomonas species were present in the six libraries. Rhodobacter and Massilia species were present in five libraries and Methylobacterium species in four libraries. In jute retting, 32 bacterial species were isolated on culture media from the two retting environments and affiliated to the attended Bacillus and Clostridium species and some species and genera described in our own libraries like Rhodococcus, Flavobacterium, Chryseobacterium, Achromobacter, P. agglomerans, E. coli, Pseudomonas and Erwinia (Munshi and Chattoo 2008). Similarly, A. parvulus, Aerobacter cloacae, Bacilli, Clostridii, E. coli, P. aeruginosa and Serratia were isolated from flax retting (Rosemberg 1965). Achromobacter, Erwinia and various species of Clostridii and Pseudomonas are known to act as retting agent (Munshi and Chattoo 2008). The species promoting the liberation of flax fibres in the shortest time was P. aeruginosa (Rosemberg 1965). Therefore, the abundance of such species in our libraries is in accordance with these previous studies. Concerning kenaf, the best water retting communities are dominated by members of the order Clostridiales (Visi et al. 2013). In our work, OTU ranged from 13 to 27 depending on samples. The OTU numbers found on chalky ground were not significantly different from those found on red ground (t test, 24 ± 1.5 versus 19 ± 4.1).
The 303 fungal clones fell into the phyla Ascomycota and Basidiomycota. The most represented fungi were mainly identified to Cladosporium and Cryptococcus genera, except for one library (C2W3F), dominated by species S. fimetaria, A. versicolor, A. glaucus, C. chlamydosporia, Acremonium sp. and P. brevicompactum. However, the total fungi counts were not different from the other libraries. The length of cloned sequence did not allow to differentiate species C. bruhnei from C. herbarum and C. cladosporioides. Species of Cladosporium, Penicillium, Rhodotorula and Aspergillus had been previously isolated from flax in the aerobic dew retting process (Fogarty and Ward 1972). These species are known to be retting agents. C. herbarum was notably identified in flax dew retting (Sharma 1986a). It was equally shown that Arthrobacter, Aspergillus, Penicillium genera and S. fimetaria possess a disaccharide specific hydrolase (Mazzaferro et al. 2010). Fungal OTU ranged from five to eight depending on samples. The OTU numbers found on chalky ground were not significantly different from those found on red ground (t test, 6.7 ± 0.7 versus 7 ± 1.7).
In conclusion, the 16S and 18S rRNA gene clone libraries provided a comprehensive sampling of the diversity and clearly demonstrated that a large diversity is present within the examined hemp retting environment. These results indicated that many microorganisms could have a proper role in retting process. The next step would be to study individually the identified species in order to understand their individual impact. For example, successful penetration of plant tissues by Pseudomonas sp., found in all libraries, may require the synergistic action of both pectin and pectate lyases (Hayashi et al. 1997). Aspergillus niger (Aspergillus genus found in one library) is the most commonly used fungal species for industrial production of pectinolytic enzymes (Naidu and Panda 1998). In parallel to molecular techniques, numerous strains have been isolated after culture and stocked. After 16S or 18S rRNA gene identification, and retting tests, combinations of these species which should secrete cellulases, hemicellulases, pectinases and other polysaccharidases should be performed to improve retting efficiency and fibre quality. Enzymatic retting had been initiated with flax but further developments are needed to make it competitive with traditional methods (Hoondal et al. 2002). The enzyme systems used by microorganisms for metabolising and for complete breakdown of pectin are important tools for elaborating the economical, ecofriendly and green chemical technology to use pectin polysaccharides in nature (Hoondal et al. 2002). It could also be carried out for hemp. Identification of pectinolytic and xylanolytic enzymes which are able to degrade specifically the pectin of hemp have to be isolated from the strains selected earlier for their efficiencies. Moreover, since hemp does differ in composition from the most studied plants, such as flax, the enzyme search should be expanded to proteinases, phenolic acid esterases, or even lignin-degrading enzymes. Indeed, some bacteria belonging to Xanthomonadaceae and Rhizobium have been found to be permanently associated with decaying wood, regardless of fungus presence (Tian et al. 2014). These authors described numerous bacteria possessing lignin-degrading enzymes and present in our libraries: the genus Sphingomonas is universally distributed in the biosphere and plays an important role as decomposer. Pseudomonas strains were among the first lignolytic bacteria to be characterized. For example, P. fluorescens was able to produce extracellular lignin peroxidase, a major enzyme involved in lignin degradation. R. fascians was capable of secreting protocatechuate-3, 4-oxygenase, an important downstream enzyme in the lignin degradation pathway. The genus Paenibacillus, which was first known for xylanase production, also includes strains capable of degrading lignocellulosic biomass. Thus, further studies will be needed to select consortia of bacteria and fungi among our libraries to improve dew retting of hemp. In vitro tests could be carried out on the selected strains to determine the best developmental conditions and the effect on retting process. The fibre quality could be estimated with other strategies such as spectrometric approaches (Himmelsbach et al. 2002) or mechanical analysis (Marrot et al. 2013). These methods could be helpful to assess the impact of strain on the retting process.
These studies will help to improve microbial growth in field (depending on pile density of plant stems swathed in the field, on oxygen, on luminosity, on pH, on sugars and water needs), in order to shorten the process. Correlations with harvesting should also be analysed (early/normal) as well as techniques (haying/swathing).
References
Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol 186:2629–2635
Akin DE (2013) Linen most useful: perspective on structure, chemistry and enzyme for the retting flax. ISRN Biotechnology 2013:1–23
Akin DE, Rigsby LL, Henriksson G, Eriksson KL (1998) Structural effects on flax stems of three potential retting fungi. Text Res J 68:515–519
Alkorta I, Garbisu G, Llama MJ, Serra JL (1998) Industrial applications of pectic enzymes: a review. Process Biochem 33:21–28
Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA (1990) Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56:1919–1925
Betrabet SM, Bhat JV (1958) Pectin decomposition by species of Pseudomonas and their role in the retting of malvaceous plants. Appl Microbiol 6:89–93
Borneman J, Hartin RJ (2000) PCR primers that amplify fungal rRNA genes from environmental samples. Appl Environ Microbiol 66:4356–4360
Bourmaud A (2013) Observation of the structure of a composite polypropylene/flax and damage mechanisms under stress. Ind Crop Prod 43:225–236
Cronier D, Monties B, Chabbert B (2005) Structure and chemical composition of bast fibers isolated from developing hemp stem. J Agric Food Chem 53:8279–8289
Das B, Chakrabarti K, Ghosh S, Chakraborty A, Saha MN (2013) Assessment of changes in community level physiological profile and molecular diversity of bacterial communities in different stages of jute retting. Pak J Biol Sci 16:1722–1729
Das B, Chakrabarti K, Ghosh S, Majumdar B, Tripathi S, Chakraborty A (2012) Effect of efficient pectinolytic bacterial isolates on retting and fibre quality of jute. Ind Crop Prod 36:415–417
Di Candilo M, Bonatti PM, Guidetti C, Focher B, Grippo C, Tamburini E, Mastromei G (2010) Effects of selected pectinolytic bacterial strains on water-retting of hemp and fibre properties. J Appl Microbiol 108:194–203
Fogarty M, Ward OP (1972) Pectic substances and pectolytic enzymes. Process Biochem 7:17–31
Godon JJ, Zumstein E, Dabert P, Habouzit F, Moletta R (1997) Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl Environ Microbiol 63:2802–2813
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biom J 40:237–264
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Hayashi K, Inoue Y, Shiga M, Sato S, Takano R, Hirayae K, Hibi T, Hara S (1997) Pectinolytic enzymes from Pseudomonas marginalis MAFF 03-01173. Phytochemistry 45:1359–1363
Henriksson G, Akin DE, Hanlin RT, Rodriguez C, Archibald DD, Rigsby LL, Eriksson KL (1997) Identification and retting efficiencies of fungi isolated from dew-retted flax in the United States and Europe. Appl Environ Microbiol 63:3950–3956
Himmelsbach DS, Khalili S, Akin DE (2002) The use of FT-IR microspectroscopic mapping to study the effects of enzymatic retting of flax (Linum usitatissimum L) stems. J Sci Food Agric 82:685–696
Hoondal GS, Tiwari RP, Tewari R, Dahiya N, Beg QK (2002) Microbial alkaline pectinases and their industrial applications: a review. Appl Microbiol Biotechnol 59:409–418
Howlett BJ, Rolls BD, Cozijnsen AJ (1997) Organisation of ribosomal DNA in the ascomycete Leptosphaeria maculans. Microbiol Res 152:261–267
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Klappenbach JA, Dunbar JM, Schmidt TM (2000) rRNA operon copy number reflects ecological strategies of bacteria. Appl Environ Microbiol 66:1328–1333
Magne F, Abely M, Boyer F, Morville P, Pochart P, Suau A (2006a) Low species diversity and high interindividual variability in faeces of preterm infants as revealed by sequences of 16S rRNA genes and PCR-temporal temperature gradient gel electrophoresis profiles. FEMS Microbiol Ecol 57:128–138
Magne F, Hachelaf W, Suau A, Boudraa G, Mangin I, Touhami M, Bouziane-Nedjadi K, Pochart P (2006b) A longitudinal study of infant faecal microbiota during weaning. FEMS Microbiol Ecol 58:563–571
Maidak BL, Cole JR, Parker CT Jr, Garrity GM, Larsen N, Li B, Lilburn TG, McCaughey MJ, Olsen GJ, Overbeek R, Pramanik S, Schmidt TM, Tiedje JM, Woese CR (1999) A new version of the RDP (Ribosomal Database Project). Nucleic Acids Res 27:171–173
Marrot L, Lefeuvre A, Pontoire B, Bourmaud A, Baley C (2013) Analysis of the hemp fiber mechanical properties and their scattering (Fedora 17). Ind Crop Prod 51:317–327
Martin N, Mouret N, Davies P, Baley C (2013) Influence of the degree of retting of flax fibers on the tensile properties of single fibers and short fiber/polypropylene composites. Ind Crop Prod 49:755–767
Mazzaferro L, Pinuel L, Minig M, Breccia JD (2010) Extracellular monoenzyme deglycosylation system of 7-O-linked flavonoid beta-rutinosides and its disaccharide transglycosylation activity from Stilbella fimetaria. Arch Microbiol 192:383–393
McGovern JN (1983) Fibres (vegetable). In: Grayson M (ed) Encyclopedia of composite materials and components. Wiley-Interscience, New York, pp 497–512
Munshi TK, Chattoo BB (2008) Bacterial population structure of the jute-retting environment. Microb Ecol 56:270–282
Müssig J, Cescutti G, Fischer H (2006) Le management de la qualité appliqué à l’emploi des fibres naturelles dans l’industrie. In: Bouloc P (ed) Le chanvre industriel: production et utilisations. Editions France Agricole, Paris, pp 235–269 [in French]
Naidu GSN, Panda T (1998) Production of pectolytic enzymes: a review. Bioprocess Eng 19:355–361
Pickering KL, Li Y, Farrel RL (2007) Fungal and alkali interfacial modification of hemp fibre reinforced composites. Key Eng Mater 334–335:493–496
Renault D, Vallance J, Deniel F, Wery N, Godon JJ, Barbier G, Rey P (2012) Diversity of bacterial communities that colonize the filter units used for controlling plant pathogens in soilless cultures. Microb Ecol 63:170–187
Rosemberg JA (1965) Bacteria responsible for the retting of Brazilian flax. Appl Microbiol 13:991–992
Sharma HSS, Robinson E (1983) Fungal colonization during glyphosate induced desiccation and dew-retting of flax cultivars. Technical report n 228111
Sharma HSS (1986a) An alternative method of flax retting during dry weather. Ann Appl Biol 109:605–611
Sharma HSS (1986b) Effect of glyphosate treatment on lignifications of fibres of some flax cultivars. Ann Appl Biol 108:114–115
Sharma HSS, Lefevre J, Boucaud J (1992) Role of microbial enzymes during retting and their effect on fibre characteristics. In: Sharma HSS (ed) The biology and processing of flax. M publications, Belfast, pp 199–212
Sharma HSS, Van Sumere CF (1992) In: Sharma HSS (ed) The biology and processing of flax, M publications, Belfast, 576 pp
Song J, Weon HY, Yoon SH, Park DS, Go SJ, Suh JW (2001) Phylogenetic diversity of thermophilic actinomycetes and Thermoactinomyces spp. isolated from mushroom composts in Korea based on 16S rRNA gene sequence analysis. FEMS Microbiol Lett 202:97–102
Stackebrandt E, Goebel BM (1994) Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44:846–849
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Doré J (1999) Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol 65:4799–4807
Tahir P, Ahmed AB, SaifulAzry SOA, Ahmed Z (2011) Retting process of some bast plant fibres and its effect on fibre quality: a review. Bioresources 6:5260–5281
Tamburini E, Gordillo Leon A, Perito B, Di Candilo M, Mastromei G (2004) Exploitation of bacterial pectinolytic strains for improvement of hemp water retting. Euphytica 140:47–54
Tian JH, Pourcher AM, Bouchez T, Gelhaye E, Peu P (2014) Occurrence of lignin degradation genotypes and phenotypes among prokaryotes. Appl Microbiol Biotechnol 98:9527–9544
Vignon MR, Garcia-Jaldon C (1996) Structural features of the pectic polysaccharides isolated from retted hemp bast fibres. Carbohydr Res 296:249–260
Visi DK, D’Souza N, Ayre BG, Webber Iii CL, Allen MS (2013) Investigation of the bacterial retting community of kenaf (Hibiscus cannabinus) under different conditions using next-generation semiconductor sequencing. J Ind Microbiol Biotechnol 40:465–475
Wright ES, Yilmaz LS, Noguera DR (2012) Decipher, a search-based approach to chimera identification for 16S rRNA sequences. Appl Environ Microbiol 78:717–725
Acknowledgments
We thank Ester Pereira for technical assistance in isolating bacterial and fungal strains for the future studies. We thank Antonia Suau for revising the manuscript. This work was financially supported by Fibres Recherche Développement®, France.
Conflict of interest
There is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 135 kb)
Rights and permissions
About this article

Cite this article
Ribeiro, A., Pochart, P., Day, A. et al. Microbial diversity observed during hemp retting. Appl Microbiol Biotechnol 99, 4471–4484 (2015). https://doi.org/10.1007/s00253-014-6356-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6356-5