
Abstract
In recent years, increasing the level of suppressiveness by the addition of antagonistic bacteria in slow filters has become a promising strategy to control plant pathogens in the recycled solutions used in soilless cultures. However, knowledge about the microflora that colonize the filtering columns is still limited. In order to get information on this issue, the present study was carried out over a 4-year period and includes filters inoculated or not with suppressive bacteria at the start of the filtering process (two or three filters were used each year). After 9 months of filtration, polymerase chain reaction (PCR)–single strand conformation polymorphism analyses point out that, for the same year of experiment, the bacterial communities from control filters were relatively similar but that they were significantly different between the bacteria-amended and control filters. To characterize the changes in bacterial communities within the filters, this microflora was studied by quantitative PCR, community-level physiological profiles, and sequencing 16SrRNA clone libraries (filters used in year 1). Quantitative PCR evidenced a denser bacterial colonization of the P-filter (amended with Pseudomonas putida strains) than control and B-filter (amended with Bacillus cereus strains). Functional analysis focused on the cultivable bacterial communities pointed out that bacteria from the control filter metabolized more carbohydrates than those from the amended filters whose trophic behaviors were more targeted towards carboxylic acids and amino acids. The bacterial communities in P- and B-filters both exhibited significantly more phylotype diversity and markedly distinct phylogenetic compositions than those in the C-filter. Although there were far fewer Proteobacteria in B- and P-filters than in the C-filter (22% and 22% rather than 69% of sequences, respectively), the percentages of Firmicutes was much higher (44% and 55% against 9%, respectively). Many Pseudomonas species were also found in the bacterial communities of the control filter. The persistence of the amended suppressive-bacteria in the filters is discussed with regards to the management of suppressive microflora in soilless culture.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The plant diseases caused by zoospore-producing fungi belonging to the genera Pythium and Phythophthora are a major problem in soilless cultures. They result in root alterations and yield losses [15, 26, 30, 31]. Irrigation water taken from diverse sources—lakes, rivers, and wells—is generally colonized by numerous bacteria and fungi including zoosporic plant pathogens. These microorganisms readily spread through the nutrient solutions to the whole cultivation system via the water supply [35]. In soilless cultures, recirculation of nutrient systems allows good control of the release of nutrients into the environment; however, such systems considerably increase the risk of disseminating pathogens and thereby spreading diseases [23]. Therefore, a key element for pathogen control is the removal of pathogenic microorganisms from recirculating nutrient solutions. In the 1990s, the potential of slow filtration to eliminate phytopathogens from re-used solutions has been demonstrated [39, 42]. This method has since been adopted by the horticulture industry for plants grown in greenhouses [13, 28].
Ehret et al. [13] reported that pathogen elimination by slow filters (SF) used in soilless cultures is the consequence of a combination of biological and physical processes. Although studies have shown that microorganisms play a key role in the filtering process, the microbiology of filter-column ecosystems is still considered as a complex “black box”. Brand and Wohanka [6] demonstrated an association between the biological process and the removal of the plant pathogen, Xanthomonas spp. The filtering media of SF provide a large surface area readily colonized by microorganisms that may be responsible for efficient removal of pathogens. Pseudomonas species seem to dominate the cultivable bacterial population [7, 10, 11], Pseudomonas aeruginosa has been detected in sand and rockwool SF [5, 6] and Pseudomonas putida in pozzolana SF [29]. Generally, the most numerous populations have been detected at the top of filters [32]; this observation has been attributed to a higher concentration of organic carbon in the upper layers of SF. The use of molecular tools indicates that conventional plating only reveals a small proportion of the bacterial populations [27]. Less than 2% of the dominant DGGE bands found for populations from SF columns correspond to cultivable species [8], and sequence analysis of DGGE bands indicated that several bacterial species are present in the columns: Bacillus megaterium, Cytophaga, Desulfivibrio, Legionella, and Sphingomonas.
Using bacteria with suppressive activity can prevent colonization of filters by fungal pathogens. Our group found significant better fungal elimination, i.e., Fusarium oxysporum, by a filter unit biologically activated with bacteria with suppressive traits isolated from a mature slow filter than in a none-biologically activated control filter [11]. The 6-month period needed by the control filter to reach its best efficacy against F. oxysporum was shortened to 1 month for the bacteria-amended filter. Fast colonization of the filter unit substrate, i.e., pozzolana grains, by selected bacteria and their subsequent interaction with F. oxysporum are likely responsible for filter efficiency. However, at the end of the cultural season, i.e., 9 months after the start of the filtration process, the control or biologically activated filters displayed similar efficiency in eliminating F. oxysporum during the filtering process.
Since the first studies carried out by Mc Pherson et al. [23] and Tu et al. [38], the management of suppressive microflora in soilless cultures has become a promising way of research to control root pathogens [41]. It was also suggested that the potentially beneficial microflora colonizing the recycled nutrient solutions was responsible for disease suppression. In that context, the SF was considered as a reliable method in the maintenance of the disease suppression because it is harmless to several bacteria [10]. Thus, the strategy that consists in inoculating bacteria with suppressive traits inside the filter units is in line with the global strategy of suppressive microflora management for controlling plant pathogens in soilless culture. However, several information to assess its reliability are still under speculation. For instance, one can wonder if the bacteria inoculated at the start of the filtering process colonize the substrates over a long period. Are they predominant on column substrates? After several months of filtering, are the bacterial communities in control- and bacteria-amended filters still different? Are the communities colonizing the filters the same from one year to another? To answer these questions, the present study was carried out over a 4-year period and includes filters inoculated or not inoculated with suppressive bacteria at the start of the filtering process. Over this 4-year period, the structure of the microbial communities was studied by 16SrRNA-targeted polymerase chain reaction–single strand conformation polymorphism (PCR–SSCP). To fully characterize the bacterial communities inside the filter units, bacterial-amended and control filters from one year were studied by using a polyphasic approach: (1) the bacterial communities on filtering media were quantified by real-time PCR. (2) Community-level physiological profiles (CLPP) generated with sole-carbon-source-utilization tests from Biolog® provided us with physiological data about the microbial population upon filter. (3) Structure of the microbial communities was studied by PCR–SSCP. (4) Bacterial diversity within filter units was studied by libraries construction and sequencing.
Materials and Methods
Filter Units and Bacterial Amendments
Each of the nine filter units used in this study consisted of a plastic pipe (220 cm in length and inner diameter 40 cm) filled with pozzolana particles (1–4-mm diameter) as filtering medium. The nutrient solution flowed through a 100-cm-thick layer of pozzolana deposited above three successive layers of graded gravel stones (2–8, 8–16, 16–32 mm); the overall thickness of gravel was 40 cm. The upper water layer was regulated by a float switch at a distance of 40–50 cm above the pozzolana surface. The filtration rate ranged from 100 to 150 l h−1 m−2.
Each year, the filter units were set at room temperature within the same compartment of an experimental greenhouse (CATE, Saint Pol de Léon, France) used to grow tomato plants (year no. 1—cv. Tradiro, De Ruiter Graines, France; year no. 2—cv. Lemance and Durinta, De Ruiter Graines, France; year no. 3—cv. Durinta, De Ruiter Graines, France; year no. 4—cv. Plaisance, De Ruiter Graines, France and cv. Locatelli, Rijk Zwaan, The Netherlands). For each filter, the drainage solutions flowed in independent recycling circuits. The nutrient solutions that flowed through the various filter units were made of water taken from the same well.
The experiment was conducted over a 4-year period. The first year, three filters were used and two filters were used each other year. Year no. 1: filter P was amended with three strains (L2, L4, and L5) of Pseudomonas putida and filter B was amended with two strains (L1 and L3) of Bacillus cereus group; no bacteria were added to filter 1 C (control). Year no. 2—filter BP was amended with two strains (L1 and L3) of B. cereus group and two strains (L2, L4, and L5) of P. putida, and no bacteria were added in the filter 2 C (control). No bacteria were introduced into the columns in years no. 3 (filters C1 and C2) and no. 4 (filters C1 and C2). Thus, nine filters were studied: three were inoculated with bacteria at the beginning of the cultural season, as described by Déniel et al. [11], and the six others were not. In year no. 1, the efficacy of the three filters in eliminating Pythium spp. and F. oxysporum throughout the cultural season was reported in [11]; after 9 months of filtration (end of the cultural season), their efficacy ranged from 98% to 99%. Relatively similar results were obtained with the filters used in year no. 2 (unpublished data).
The five bacterial strains introduced into the columns were selected for their antagonistic properties [14]. The strains were LMSA3.06.043 (L1), LMSA3.06.044 (L3), LMSA3.06.045 (L2), LMSA3.06.046 (L4), and LMSA3.06.047 (L5), were supplied by the Souchothèque de Bretagne (ESMISAB, Plouzané, France). They were cultured for 24 h in tryptic soy broth (AES Laboratoire) on a rotary shaker (120 rpm) at 30°C. Then, these bacterial suspensions were poured out over the pozzolana grains at the top of the filtering columns. For year no. 1, the total bacterial densities were 9 × 1011 colony forming units (cfu) (8 × 106 cfu g−1 of pozzolana grains) at the first inoculation (late February) and 1 × 1012 cfu (1 × 107 cfu g−1 of pozzolana grains) at the second inoculation (early March). For year no. 2, the total bacterial densities were 5 × 1012 cfu and 5 × 107 cfu g−1 of pozzolana grains at the first (late February) and second (early March) inoculations.
Filters Used in Years No. 1, 2, 3, and 4
Sampling of Microbial Communities
Each year, after 9 months of filtration, i.e., at the end of the cultural season (end of November or early December), the filters were cut into sections and pozzolana samples were collected from the top (0–10 cm), middle (55–65 cm), and bottom (90–100 cm) layers. Then, these grains were fractionated into 7-g fractions; each sample was washed three times with 63 ml of a physiological water (0.85% NaCl)–Tween 80 mixture resuspended in the same solution and sonicated for 90 s at the highest setting in a VibraCell™ bench sonicator (Bioblock Scientific, France); bacteria were collected and samples from each layer pooled. Subsequently, they were used either for community-level physiological profiles analyses or DNA extraction. Aliquots of 200 ml of these pools were filtered through 0.45-μm-pore-size cellulose acetate filters (Sartorious, Germany) and the filters stored at −80°C until DNA extraction.
DNA Extraction
Total DNA was extracted from the filters as reported by Godon et al. [18] with slight changes. Briefly, the frozen filter was ground with a sterile plastic stick and transferred into a 2-ml polypropylene microcentrifuge tube kept frozen in a −20°C freezer rack; then 400 μl of 4 M guanidine thiocyanate–0.1 M Tris (pH 7.5), 120 μl of 10% N-lauroyl sarcosine, and 500 μl of 5% N-lauryl sarcosine–0.1 M phosphate buffer (pH 8.0) were added and the sample heated to 95°C for 10 min. An aliquot of 500 μl of zirconium beads (0.1-mm diameter), sterilized by autoclaving, was added and the mixture was shaken for 10 min at maximal speed in a Vibro shaker (Retsch). Nucleic acids were precipitated with 1 vol of isopropanol added concomitantly with 5 μl of Carrior RNA (Qiagen, Hilden, Germany) to enhance DNA extraction yield. Finally, a QiaAmp DNA microkit (Qiagen) was used for purification of total DNA.
PCR–SSCP Analyses
To analyze the overall structure of the bacterial community as a whole, the V3 region of the 16SrRNA gene was amplified from total DNA with primers W49 (5′-ACGGTCCAGACTCCTACGGG-3′, Escherichia coli position F331) and 5′-fluorescein phosphoramidite-labeled W104 (5′-TTACCGCGGCTGCTGGCAC-3′, E. coli position R533). PCR amplifications were performed with a thermal cycler. The reaction mixtures contained 1× polymerase buffer, 0.2 mM deoxynucleoside triphosphates (dNTPs), 130 ng of each primer, 0.5 U of Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA, USA), 1 μl of genomic DNA, and water added to obtain a final volume of 50 μl. The PCR conditions were an initial denaturation step of 2 min at 94°C; 25 cycles of a three-stage program of 30 s at 94°C, 30 s at 61°C, and 30 s at 72°C; and a final elongation for 10 min at 72°C. The reactions were stopped by cooling the mixture to 4°C.
Amplification product sizes were confirmed by electrophoresis on a 2% (wt/vol) agarose gel. SSCP analysis separates DNA fragments of the same size but with different compositions. One microliter of further diluted PCR product mix was added to 18 μl of formamide and 1 μl of internal size standard Rox 400 HD (Applied Biosystems, Foster City, CA, USA) diluted 1:10. The sample was then denatured for 5 min at 95°C and placed directly on ice for 10 min. SSCP analysis was carried out with an ABI 310 Genetic Analyzer (Applied Biosystems, France) with electrophoresis at 15 kV and 32°C for 30 min per sample. Raw SSCP data were exported into the easily handled csv format with Chromagna shareware (developed by Mark J. Miller at the U.S. National Institutes of Health), and statistical analyses were performed with SAFUM [43] and Matlab 6.5 software (MathWorks).
Filters Used in Year No. 1
Assessment of Community-Level Physiological Profiles
The bacterial mixtures obtained from pozzolana particles sampled from the top, middle, and bottom layers of each filter were distributed in 96-well Biolog® GN2 microplates (AES, France) (150 μl/well) except the one for control, which was filled in with sterile water [17]. The experiment was conducted in hexaplicates, and plates were incubated at 30°C for 48 h. The rate of conversion, in the 96 wells, of tetrazolium violet with the different substrates of the GN2 microtiter plates was determined by measurement of the optical density (OD) at 620 nm with a microplate reader (Titertek Multiscan® MCC1340). To minimize the effect of difference in bacterial mixture densities between plates, data were standardized as follows: the average well color development (AWCD) corresponding to the mean of the blanked absorbance values of the 95 wells was calculated for each plate; then, the blanked absorbance value of each well was divided by the AWCD of the corresponding plate to get a corrected OD value [16, 17]. In accordance with Grove et al. [20], all corrected OD values were set to fall within 0 and 2 (boundary limits).
Quantification of the Bacterial Population by Real-Time PCR
To assess the total bacterial density on the three filters used in year no. 1, a portion of the bacterial 16SrRNA gene was amplified by real-time PCR with the primer-and-probe set BACT2 described by Suzuki et al. [36]. The reactions were performed in 96-well plates (Applied Biosystems), and the concentrations of oligomers and the PCR program were as described by these authors. The final volume of 25 μl contained 12.5 μl of 2× Taqman Universal PCR Master Mix (Applied Biosystems), 5 μl of diluted DNA extract, the forward and the reverse primers, and the TaqMan™ probe labeled with 6-carboxy-fluorescein (FAM) as reporter and 6-carboxy-tetramethyl-rhodamine (TAMRA) as quencher. The data were analyzed with ABI Prism 7000 SDS software, version 1.1. All tests were performed in triplicate. A standard curve was generated for each assay from PCR products of a complete 16SrDNA gene, as described by Rousselon et al. [33].
The PCR product was purified using the QIAquick PCR purification kit (QIAGEN). The concentration of the DNA solutions was determined by spectrophotometry (GeneQuant pro; Amersham Biosciences Europe GmbH, Freiburg, Germany). Standard DNA curves were generated by amplification of serial 10-fold dilutions of these DNA solutions in sterilized water and plotted by ABI Prism 7000 SDS software, version 1.1. For each assay, the cycle threshold (Ct) corresponding to the number of cycles at which the reaction becomes exponential was compared to the standard curve to calculate the number of target samples per PCR. To overcome the inhibitory effect on PCR of compounds present in environmental samples, various dilutions of each sample were amplified and the initial calculated concentrations were compared two by two. The slopes of the standard curves fell within −3.2 and −3.5, which correspond to a PCR reaction efficiency greater than 93%. The coefficient of determination (R 2) was in all cases above 98%. PCR results were converted to mean target genomes per gram of pozzolana grains. Mean values and 95% confidence intervals were calculated from at least six values. Numerical results were compared with the expected values calculated by considering both the inoculum concentration and the dilution factor during the inoculation.
Clone Library Construction, Screening, and Sequencing
Three libraries of about 100 clones per filter, named hereafter Clib, Blib, and Plib, were constructed from the DNA extracts from the bacterial communities isolated from the middle layers of the C-, B-, and P-filters, respectively. Bacterial 16S rRNA genes were amplified by PCR with the universal reverse primer W31 (5′-TTACCGCGGCTGCTGGCAC-3′) and the forward primer W18 (5′-GAGTTTGATCMTGGCTCAG-3′). The final PCR mixture (50 μl) contained 1 μl of DNA template, 2 μl of each primer (100 ng μl−1), 4 μl of dNTP (2.5 mM), 5 μl of 1× Taq reaction buffer, 1 μl of redTaq DNA polymerase (Sigma), and 35 μl of water. An initial DNA denaturation step at 94°C for 2 min was followed by 25 cycles at 94°C for 1 min, 55°C for 1 min, and 72°C at 1 min, and a final 10-min elongation step at 72°C. PCR products were purified with a QIAquick® PCR Purification Kit (Qiagen) in accordance with the manufacturer’s instructions. The purified PCR products were inserted into pCR4-TOPO and used to transform competent Escherichia coli TOP10 cells from the Topo TA cloning kit as described by the manufacturer (Invitrogen, Groningen, The Netherlands). Recombinant cells were selected for kanamycin resistance and ccd gene killer inactivation then grown at 37°C for 24 h in LB2X medium (20 g l−1 tryptone, 10 g l−1yeast extract, 10 g l−1 NaCl). Plasmid DNA was purified with a Montage SEQ96 Sequencing Reaction Cleanup Kit (Millipore) and sequenced in a GeneAmp® PCR System 9700 thermocycler (Applied Biosystem) under the following conditions: a DNA denaturation step at 96°C for 5 min followed with 50 cycles at 96°C for 30 s, 57°C for 30 s, and 60°C for 4 min. Each 5-μl reaction mixture was composed of 1 μl purified DNA template, 1 μl sequencing primer T7 (5′-TAATACGACTCACTATAGGG-3′), 0.5 μl Big Dye Terminator V3.1, 0.75 μl 5× buffer, and 1.75 μl sterile water. DNA sequences were obtained with an Applied Biosystems 3130xl Genetic Analyzer (GIS Ouest Génopole—GENOMER, Roscoff, France). They were trimmed and cleaned with Phred [14] and SeqClean [TGIR, the Institute for Genomic Research, Rockville, MD, USA (http://www.tigr.org/tdb/tgi/software)] software and compared to GenBank databases (www.ncbi.nlm.nih.gov/blast) with the BLAST program [3]. The sequences of 16SrRNA genes were submitted to the GenBank database with the accession numbers JN412138–JN412415.
Various diversity indexes were used to elucidate microbial diversity. Coverage percentage was determined with Good’s coverage estimator (C Good); this non-parametric estimator of the proportion of phylotypes within a library of infinite size that are represented in a smaller library is calculated from the relation: C Good = 1 − (n 1/N), where n 1 is the number of phylotypes appearing only once in the clone library and N is the total number of analyzed clones [19]. Other diversity and similarity estimator were applied with the EstimateS software package (version 8.2; R. K. Colwell. 2009. EstimateS: statistical estimation of species richness and shared species from samples; user’s guide and application published at http://purl.oclc.org/estimates). The total number of phylotypes was estimated by calculating the Chao1 richness estimator (Chao1) and its 95% confidence interval [9].
Global Bacterial Communities Description by Principal Component Analysis (PCA)
For each bacterial community, the results of real-time PCR quantification, community-level physiological profile assessment, and SSCP-structure description were gathered in a single numerical database before being statistically described by a global Principal Component Analysis (PCA) with StatBox® 6.6 software (Grimmer Logiciels, Paris). The final database consisted of nine individuals (the three bacterial communities of the three filters) and a total of 231 variables distributed as follows: the first of them was the bacterial density of the population, 95 other ones corresponded to the 95 carbon sources contained in the Biolog®GN2 microplates, and the last 135 ones were the numerical values measured on the 135 SSCP profile scans. PCA was performed on using the correlation coefficient of Pearson. Variables having a cos2 ≥0.5 on one of the first two principal components (PC1 or PC2) were estimated as sufficiently well represented by the principal plane generated by this PCA.
Results
Filters Used in Years No. 1, 2, 3, and 4: SSCP Analyses of Bacterial Communities
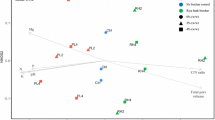
PCA eigenvalues indicated that the first two principal components, PC1 and PC2, explained 70.9% of the total data variance and gave a consistent description of the global database (Fig. 1). With the exception of one point (BP-t, year no. 2), the SSCP profiles of bacterial communities from control filters were clearly separated from those of the bacteria-amended filters. The three filters in year no. 1 were distinguished by PC1, and filter C bacterial communities were separated from those of filters P and B by PC2 (positive versus negative coordinates, respectively). In year no. 2, with the exception of point BP-t, the C-filter bacterial communities were differentiated from those of the BP-filter by PC1 (positive versus negative coordinates, respectively). For year no. 3, for all filter depths, the two control filters were very closed and grouped on the same area on the PCA graph; the same observation was made for the two control filters used in year no. 4 (Fig. 1). Note that the bacterial communities from the control filter used in year no. 1 are markedly separated from those colonizing the filters used in the three other years (Fig. 1).

Distribution of the bacterial communities from the nine filters studied on the principal plane defined by the global Principal Components Analysis. Each bacterial community is identified by two letters. The first indicates the type of filter, control (C): C-filter; Bacillus (B) amended filter: B-filter; Pseudomonas (P) amended filter: P-filter; Bacillus (B) and Pseudomonas (P) amended filter: BP-filter. The second refers to the filter depth—t top, m middle, b bottom. For the control filters in years 3 and 4; C1 and C2 indicates control filter no. 1 and control filter no. 2, respectively. The total variation (%) explained by each Principal Component is given in brackets
Bacteria-amended filters: SSCP profiles for the three filters inoculated with bacteria were substantially different. The three bacteria-amended filters are clearly separated on the PCA graph (Fig. 1).
The SSCP profiles of bacterial communities sampled from the top, middle, and bottom layers exhibited common features for each control filter. The same observation was made for filter inoculated with P. putida strains (filter P, year no. 1) and for filter inoculated with B. cereus group strains (filter B, year no. 1). The only exception was the SSCP profiles for the column inoculated with both P. putida and B. cereus group strains (BP-filter, year no. 2): the SSCP profiles of bacterial communities from the top displayed marked differences from those of the middle and bottom (not shown).
Filters Used in Year No. 1
Global Communities Analysis by PCA
From their bacterial density, their use of Biolog®GN carbon sources, and their SSCP profiles, bacterial communities showed distinctive patterns dependent upon filters (C-, P-, or B-filters) and top, middle, or bottom position in the filter units (Fig. 2). Eigenvalues of PCA indicated that, together, the first two principal components, PC1 and PC2, covered 70.4% of the total data variance and gave a consistent description of the global database. The C-filter bacterial communities were clearly separated from P- and B-filter ones by PC1 (respectively positive versus negative coordinates). Moreover, P-filter communities were distinct from B-filter ones by PC2 (positive versus negative coordinates). Bacterial communities from C-filter were quite close, whatever their position in the filtering column. For P- and B-filters, middle and bottom communities were very similar, but far away from the top layer ones.

Distribution of the nine bacterial communities on the principal plane designed by the global Principal Components Analysis made from bacterial quantification results, Biolog®GN physiological profiles data (95 carbon sources), and numerical values of each SSCP profile (135 scans). The bacterial communities of each filter are identified by two letters: the former indicates the type of filter (C = C-filter; B = B-filter; P = P-filter) whereas the latter refers to the filter stage (t top, m middle, b bottom). The total variation (%) explained by each Principal Component (PC) is given in brackets
PCA permitted us to correlate the position of the bacterial communities on the principal plane with the distribution of variables (Fig. 3). Among the 231 variables under study, 171 were considered as being significantly well represented by one of the first two PCs; the 60 other ones were removed from the principal plane because of their projection that was potentially misrepresented. Comparison of the distribution of individuals (Fig. 2) and variables (Fig. 3) allowed us to evidence four discriminative variable groups more thoroughly described in Table 1. The one denoted c group was highly and positively correlated with PC1, and thus found to determine the position of bacterial communities of C-filter. On the contrary, the i group showed a high negative correlation with PC1, which suggests strong connections with bacterial populations of P- and B-filters. The last two variable groups, denoted p group and b group, were negatively correlated with PC1 and respectively showed positive and negative correlation with PC2; this finding supports the respective position of P-filter and B-filter bacterial communities.

Distribution of the 171 significantly well-represented variables on the principal plane designed by the global PCA made from bacterial quantification results (qPCR) (filled circles), Biolog®GN physiological profile data (filled triangles), and numerical values of SSCP profile scans (multiplication symbols). Four groups of variables are described according to the quality of their representation on the first principal component (PC1) or on the second one (PC2): the c group (c = control) variables have positive coordinates and a cos2 ≥0.5 on PC1; the p group (p = Pseudomonas) variables have negative coordinates on PC1, positive ones on PC2 and a cos2 ≥0.5 on PC2; the i group (i = intermediate) variables have negative coordinates and a cos2 ≥0.5 on PC1; and the b group (b = Bacillus) variables have negative coordinates both on PC1 and PC2 and a cos2 ≥0.5 on PC2. For substrates description, see Table 1
Quantification of Bacterial Population by Real-Time PCR
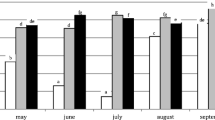
The bacterial densities in the three slow filters were assessed, and the P-filter clearly differed from the others (Fig. 4). In P-filter, the bacteria were more numerous than those in the two other filters at all depths of sampling; there was a quantitative decrease in density from the top to the bottom. In B- and C-filters, the densities of bacteria were similar at all filter depths.

Real-time PCR quantification of total bacteria communities (DNA copy/g pozzolana) in the top (black), middle (gray), and bottom (white) layers of P-, B-, and C-filters. Bars with the same letters are not statistically different according to the Tukey HSD test (α = 0.05)
The description given by PCA showed that the bacterial density determined by real-time PCR was one of the p group (p = Pseudomonas) variables. It was denoted qPCR in Fig. 3 and Table 1.
SSCP Structure of Bacterial Communities
SSCP profiles of bacterial communities indicate marked differences between filters, whatever the filter layer under examination (Fig. 5). A close examination of the contiguous scan parts found in c group (c = control) variables evidenced a common area in SSCP profiles extending from scan s108 to scan s135; the peaks present in this area, denoted c1-domain, were found to be characteristic of the C-filter bacteria communities (Fig. 5 and Table 1). SSCP profiles common to P- and B-filters were correlated with five SSCP domains (i1 to i5) according to the distribution of SSCP scans in the i group variables (i = intermediate) (Fig. 5 and Table 1). P-filter bacteria communities were associated with a large domain extending from scan s4 to s33 (p1-domain, Fig. 5); they belong to the p group variables (p = Pseudomonas) (Table 1). In the same way, three short SSCP domains (b1 to b3, Fig. 5) were associated with B-filter ecosystems; they belong to the b group variables (b = Bacillus) (Table 1).

SSCP profiles obtained from the bacterial communities from the top, middle, and bottom layers of the control filter (Ct, Cm, Cb) and of those amended by Bacillus (Bt, Bm, Bb) or Pseudomonas (Pt, Pm, Pb). t, p, and m refer to the filter depth; t top, m middle, b bottom. According to the global PCA description, SSCP peaks presented in the areas denoted (c1-domain), (p1-domain), (b1-, b2-, and b3-domains), and (i1-, i2-, i3-, i4-, and i5-domains) were specifically correlated to C-filter, P-filter, B-filter, and of the two amended filters, respectively. b Bacillus, c control, i intermediate, p Pseudomonas
Community-Level Physiological Profiles
The description given by PCA of Biolog®GN substrates showed two different trophic behaviors for C-filter bacterial communities and amended filters ones, respectively (Fig. 3 and Table 1). Indeed, 11 of the 17 substrates identified in the c group were carbohydrates (65%) (Table 1). Substrates common to amended filters (P- and B-filters) formed the i group. No carbohydrate were found in the i group which consisted of 27 substrates split into 12 carboxylic acids (44%), 10 amino acids (37%), and five diverse chemicals (Table 1). The microbial populations of C-filter had a metabolic profile oriented toward carbohydrates whereas those from bacteria-amended filters metabolized especially carboxylic acids and amino acids. This metabolic profile was especially marked for B-filter communities: their distribution on the principal plane was highly supported by substrates of both i group and b group (Fig. 3 and Table 1). Among the nine substrates of the b group, five were identified as carboxylic acids and one as amino acid (Table 1).
The comparison of bacterial communities and Biolog®GN substrates distributions on the principal plane (Figs. 2 and 3) indicates that bacterial communities from amended filters metabolized more carboxylic acids and amino acids in the middle and bottom layers than in the top ones. The significance of this location-dependent metabolism was assessed with the LSD test (least significant difference) from StatGraphics Plus 5.1 (Manugistic Inc., Rockville, MD, USA) to determine, for each substrate listed in Table 1, whether differences could be related to the position of bacterial communities within filters (data not shown). For B- and P-filters, there were a statistically significant increase of metabolism (α = 0.05) in the middle and bottom communities for 13 of the i group substrates: eight amino acids (l-aspartic acid, l-glutamic acid, l-histidine, hydroxyl-l-proline, l-proline, l-pyroglutamic acid, d-serine, and γ-aminobutyric acid) and five carboxylic acids (cis-aconitic acid, citric acid, d-galacturonic acid, d-glucuronic acid, and d-saccharic acid).
Analysis of the 16SrRNA Clone Libraries
To assess bacterial diversity, 278 16SrRNA genes from the three filter units were sequenced and analyzed. The sequences were grouped into 147 different phylotypes according to phylogenetic uniqueness of the closest relative (Table 2). The percentage of similarities found by sequence comparison was 81% to 100%. Among the 147 bacterial phylotypes, 26 (18%) presented more than 97% of similarity with previously identified sequences; 118 showed similarity values of 81% to 97% and three were non-identified. Only 44 of the 147 phylotypes had isolated bacteria or species described as closest relatives; this indicates that most of the phylotypes were related to uncultured bacterial taxa (Table 2).
Ten phyla of the 52 described by Rappé and Giovannoni [27] were detected; the best-represented phyla were Proteobacteria (40% of sequences) and Firmicutes (21%). Proteobacteria were mainly α-Proteo (15%), γ-Proteo (13%), and β-Proteo (10%). Other significant phyla were Planctomycetes (10%), Acidobacteria (7%), and Chloroflexi (7%). The remaining minor phyla are reported in Fig. 6. The two most abundant identified phylotypes were related to Clostridium peptidovorans (NR_020519) (no. 1; 9% of sequences) and Clostridium tunisiense (AY187622) (no. 2; 8% of sequences) (Table 2).

Phylogenetic distribution in the three clone libraries constructed from bacterial communities of C-filter (Clib), P-filter (Plib), and B-filter (Blib)
Coverage and Similarity of Libraries Based on Phylotype Diversity
Plib (P-filter) and Blib (B-filter) libraries have more phylotype diversity than Clib (C-filter). In comparison with the control (67%), their percentages of coverage were lower: 54% for P-filter and 55% for B-filter. The Chao1 phylotype richness estimators were higher for P-filter (155) and B-filter (142) than for C-filter (101) (Table 3).
Analysis of the C-Filter 16SrRNA Clone Library
The 104 sequences from the C-filter were distributed into 54 phylotypes and 10 phyla (Table 2 and Fig. 6). Proteobacteria was the most abundant phylum (69% of the sequences); they were exclusively γ-Proteo (32%), β-Proteo (26%), and α-Proteo (11%). Other minor phyla including Firmicutes (9%), Planctomycetes (6%), and Acidobacteria (5%) were also identified.
Using a 97% sequence similarity threshold for 16SrRNA gene sequences of the same species, 44 sequences with described relatives were identified to the species level (Table 2). Most of them were Pseudomonas species (19 sequences) and the others were species of the following genera: Acinetobacter (four sequences), Dyella, Rhizobium (two sequences each), Burkholderia, Nitrobacter, Phaeospirillum, Enterobacter, Alcaligenes, Clostridium, Sphingomonas, and Stenotrophomonas (one sequence each).
Analysis of the P-Filter 16SrRNA Clone Library
The 85 sequences from the P-filter were related to 49 unique phylotypes and 12 phyla (Table 2 and Fig. 6). The most abundant were Firmicutes (55% of the sequences) and Proteobacteria (22%), mostly α-Proteo (17%) and γ- and δ-Proteo (2%); two other phyla found in significant abundance were Planctomycetes (8%) and Chloroflexi (6%).
Of the 31 sequences having a sequence similarity threshold equal or greater than 97% and described relatives with species names, 58% were identified as Clostridium spp. (18 sequences). Sedimentibacter (two sequences), Klebsiella, Streptomyces, Mesorhizobium, and Bdellovibrio (one sequence) species were also found (Table 2 and Fig. 6).
Analysis of the B-Filter 16SrRNA Clone Library
The 89 sequences from the B-filter corresponded to 55 phylotypes distributed among 13 different phyla (Table 2 and Fig. 6). The sequences were mainly affiliated to Firmicutes (44%), Chloroflexi (10%), and α-Proteobacteria (10%). Other significant phyla were Planctomycetes (6%), Acidobacteria (6%), and Actinobacteria (5%).
The 30 sequences identified to the species level were affiliated to species of Clostridium (10 sequences), Sedimentibacter (four sequences), Arcobacter and Variovorax (four sequences each), Curtobacterium, Microbacteriaceae, Stenotrophomonas, and Pseudomonas (one sequence each) (Table 2 and Fig. 6).
Discussion
In the last decade, to protect plants grown in soilless culture, the management of suppressive microflora has been the focus of a growing body of interest. Beside experiments carried out to promote disease suppression on roots, increasing the level of suppressiveness by the addition of antagonistic bacteria in slow filter has become a promising strategy. It was shown that biological activation of filtering columns with bacteria with suppressive traits at the start of the filtering process enhances the elimination of plant pathogenic fungi from water flows [11]. However, knowledge about the effect of bacterial amendments on the whole bacterial ecosystem colonizing the filtering medium is still very limited. It is a key point in the management of this microflora and therefore in determining its future success or failure.
As revealed by PCR–SSCP structural analyses of the bacterial ecosystems, one of the main points of our study is that there are significant differences between the bacterial communities from bacteria-amended and control filters. On the contrary, bacterial communities from control filters set up in the same environmental conditions and in the same year are very similar. In a SSCP study of two biofilters designed for volatile organic compound removal and seeded with two different inocula, Khammar et al. [22] evidenced differences in the composition of microflora both between filters and according to the depth of sampling. Our experiment only points out differences between filters and suggests that bacterial communities are relatively homogeneous inside each filter unit. To determine to which extent the inoculum of suppressive bacteria influenced the microbial communities in the filters, we investigated the densities, functional activity, and composition of bacterial communities from amended and control filters used in year no. 1.
In our study, qPCR showed that the bacterial communities in the P-filter were higher than those in control and B-filters. We observed either a decreasing gradient from top to bottom layers (P-filter) or similar bacterial colonization at all filter depths (B- and C-filters). The higher bacterial densities in the P-filter than in the B-filter may result from differences in the colonization and biofilm-formation capabilities of the bacteria inoculated; the fast growth strategy of Pseudomonas spp. allows quick colonization [21, 34]. Nevertheless, in our experiment, this would not explain the differences in bacterial density between P- and B-filters because sequencing showed that Pseudomonas microflora was not abundant after 9 months in the P-filter. To explain this result, one can assume that bacterial inocula may promote biological activation of biofilters by stimulating the growth of endogenous bacteria on pozzolana grains. In the literature, experiments based on cultured bacterial microflora quantification indicated that large microbial populations develop rapidly and preferentially at the top of filters, and this is generally related to the higher concentration of organic carbon sources in the upper layers of SF [2, 6, 32]. However, according to Calvo-Bado et al. [7], differences between the top, middle, and bottom layers of a sand filter are not always observed. Taken altogether, these reports and our data suggest that the induction of a durable increase in the bacterial population is dependent of at least three factors: the concentration of organic carbon sources in the filtering column, the indigenous microflora colonizing the filtering media, and the early addition of bacterial inoculum to the column. Except for the inoculum, investigations remain to be made to determine how the two others factors may be controlled.
We also evidenced marked differences in (a) the metabolism and (b) in the bacterial populations within the three filters.
-
(a)
Regarding the functional analysis, it was shown that bacteria from the control filter metabolized more carbohydrates than those from the amended filters whose trophic behaviors were more targeted towards carboxylic acids and amino acids; among them, five carboxylic acids (acetic acid, citric acid, malonic acid, succinic acid, and propionic acid) and five amino acids (l-aspartic acid, hydroxy-l-proline, l-serine, γ-aminobutyric acid, and l-asparagine) are known as constituents of root exudates [8]. The report by Alsanius et al. [1] of a strong metabolization of amino acids by Pythium spp. led us to assume the existence of some competition for amino acids between microbial communities of bacteria-amended filters and pathogenic fungi, e.g., Pythium spp. This assumption could partially explain the filtration efficacy of amended filter towards pathogenic fungi.
-
(b)
Sequencing the 16SrRNA gene libraries confirmed the differences between the bacterial populations within the three filters. The bacterial communities in P- and B-filters both exhibited significantly more phylotype diversity and markedly distinct phylogenetic compositions than those in the C-filter. Indeed, although there were far fewer Proteobacteria in B- and P-filters than in the C-filter (22% and 22% rather than 69% of sequences, respectively), the percentages of Firmicutes was much higher (44% and 55% against 9%, respectively). Many Pseudomonas species were also found in the bacterial populations of the control filter. This is consistent with previous reports [5–7, 29] of cultivable Pseudomonas spp. in the microflora colonizing substrates of slow filters. Clostridium species were abundant in the bacterial populations of the two amended filters. The two major phylotypes recovered in P- and B-filters were affiliated to strains of the Clostridium genus: C. tunisiense sp. nov. [37] and C. peptidivorans sp. nov. [24]; their growth requires an environment with a sufficiently low redox potential. Presumably, within bacteria-amended columns, the consumption of nutrient sources by amended and/or endogenous bacteria generated more reducing compounds and lowered the redox potential (relative to the control column) and thereby favored the further growth of Clostridium strains. Thus, although the filter efficacies of the control and bacteria-amended filters were similar at the end of the cultural season [11], they resulted from the pozzolana colonization by microbial communities that differed both in structural and in functional properties in each filter.
After 9 months of filtration, the Pseudomonas and Bacillus bacteria used to inoculate P- and B-filters were not recovered in significant numbers on substrates, i.e., pozzolana grains, from these filtering columns. Indeed, only one out of 85 clones randomly sampled from the P-filter was affiliated to the Pseudomonas genus. This proportion is far lower than that for the control filter (32%). Similarly, none of the Bacillus was recovered from the B-filter. It was noticeable that despite the scarcity and non-persistence of the inoculated bacteria on pozzolana grains, inoculation of bacteria caused a significant shift in the overall microbial populations in the filtering columns. Data available in the literature about inoculum strategies report that addition of exogenous microbial strains, e.g., bacteria, fungus, or oomycete, to an existing community structure does not necessarily affect the global ecosystem significantly. For instance, addition of a strain of Arthrobacter protophormiae to a pesticide-contaminated soil had only a small effect on the structure of the existing bacterial community [25]. Similarly, it was reported that the biocontrol fungus Fusarium oxysporum strain 47 is able to establish in different soil environments without perturbing the bacterial and fungal communities [12]. In soilless culture, our group has recently demonstrated that the biocontrol oomycete Pythium oligandrum was able to colonize and persist in the rhizosphere of tomato plants without significantly modifying the indigenous fungal populations, other than a reduction of the population of pathogenic Pythium dissotocum [40]. In the present experiment, the microbial communities of the filter units are less complex than in soils or rhizospheres and are therefore more easily shifted. One can assume that the inoculated microorganisms create ecological parameters favoring the installation of indigenous microbial populations, mainly representatives of the Firmicutes in our system. To support our hypothesis, Bomo et al. [4] reported that the introduction of the fish-pathogenic bacterium Yersinia ruckeri into sand-filter columns stimulated the growth of other microorganisms, which in turn caused a shift in the composition of the microbial community.
To conclude, our data suggest that the introduction of suppressive bacteria in filter units dramatically changed the microbial status of the columns. Because of the non-persistence of the inoculated bacteria on the filtering media, i.e., pozzolana grains, it is really difficult to predict which communities will colonize the filter units. In our opinion, this point is of particular importance because it raises questions on the management of the microflora, particularly the suppressive microflora, in soilless culture. Further investigations aimed at determining the key factors responsible for the microbial shifts would help in elucidating this issue.
References
Alsanius BW, Khalil S, Hultberg M (1998) Biochemical and chemical characterization of Pythium ultimum. Med Fac Landbouww Univ Gent 63/3a:891–897
Alsanius BW, Nilsson L, Jensén P, Wohanka W (2001) Microbial communities in slow filters. Acta Hortic 548:591–601
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bomo AM, Ekeberg D, Stevik TK, Hanssen JF, Frostegård A (2004) Retention and removal of the fish pathogenic bacterium Yersinia ruckeri in biological sand filters. J Appl Microbiol 97:598–608
Brand T (2000) Beurteilung der bakterienflora von langsamfiltern als biologische filtrationskomponente in geschlossenen bewässerungssystemen des gartenbaus. Technischen Universität München, Ph.D. thesis
Brand T, Wohanka W (2001) Importance and characterization of the biological component in slow filters. Acta Hortic 554:313–321
Calvo-Bado LA, Morgan JAW, Sergeant M, Pettitt TR, Whipps JM (2003) Molecular characterization of Legionella populations present within slow sand filters used for fungal plant pathogen suppression in horticultural crops. Appl Environ Microb 69:533–541
Campbell CD, Grayston SJ, Hirst DJ (1997) Use of rhizosphere carbon sources in sole carbon source tests to discriminate soil microbial communities. J Microbiol Meth 30:33–41
Chao A, Chazdon RL, Colwell RK, Shen TJ (2005) A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecol Lett 8:148–159
Déniel F, Renault D, Tirilly Y, Barbier G, Rey P (2006) A dynamic biofilter to remove pathogens during tomato soilless culture. Agron Sustain Dev 26:185–193
Déniel F, Rey P, Chérif M, Guillou A, Tirilly Y (2004) Inoculations of filter unit with antagonistic- and PGPR-bacteria improve slow filtration efficiency in soilless culture. Can J Microbiol 50:499–508
Edel-Hermann V, Brenot S, Gautheron N, Aimé S, Alabouvette C, Steinberg C (2009) Ecological fitness of the biocontrol agent Fusarium oxysporum. FEMS Microb Ecol 68:37–45
Ehret DL, Alsanius B, Wohanka W, Menzies JG, Utkhede R (2001) Disinfestation of recirculating nutrient solutions in greenhouse horticulture. Agronomie 21:323–339
Ewing B, Hillier L, Wendl M, Green P (1998) Basecalling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res 8:175–185
Favrin RJ, Rahe JE, Mauza B (1988) Pythium spp. associated with crown rot of cucumbers in British Columbia greenhouses. Plant Dis 72:683–687
Garland JL (1996) Analytical approaches to the characterization of samples of microbial communities using patterns of potential C source utilization. Soil Biol Biochem 28:213–221
Garland JL, Mills AL (1991) Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Appl Environ Microb 57:2351–2359
Godon JJ, Zumstein E, Dabert P, Habouzit F, Moletta R (1997) Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl Environ Microb 63:2802–2813
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264
Grove JA, Kautola H, Javadpour S, Moo-Young M, Anderson WA (2004) Assessment of changes in the microorganism community in a biofilter. Biochem Eng J 18:111–114
Juteau P, Larocque R, Rho D, LeDuy A (1999) Analysis of the relative abundance of different types of bacteria capable of toluene degradation in a compost biofilter. Appl Microbiol Biot 52:863–868
Khammar N, Malhautier L, Degrange V, Lensi R, Godon JJ, Fanlo JL (2005) Link between spatial structure of microbial communities and degradation of a complex mixture of volatile organic compounds in peat biofilters. J Appl Microbiol 98:476–490
McPherson GM, Harriman MR, Pattison D (1995) The potential for spread of root diseases in recirculating hydroponic systems and their control with disinfection. Med Fac Landbouww Univ Gent 60/2b:371–379
Mechichi T, Fardeau ML, Labat M, Garcia JL, Verhé F, Patel BKC (2000) Clostridium peptidivorans sp. nov., a peptide-fermenting bacterium from an olive mill wastewater treatment digester. Int J Syst Evol Microbiol 50:1259–1264
Paul D, Pandey G, Meier C, van der Meer JR, Jain RK (2006) Bacterial community structure of a pesticide-contaminated site and assessment of changes induced in community structure during bioremediation. FEMS Microbiol Ecol 57:116–127
Rafin C, Tirilly Y (1995) Characteristics and pathogenicity of Pythium spp. associated with root rot of tomatoes in soilless culture in Brittany, France. Plant Pathol 44:779–785
Rappé MS, Giovannoni SJ (2003) The uncultured microbial majority. Annu Rev Microbiol 57:369–394
Renault D (2007) Characterization of bacterial communities colonising the biofilters, nutrients solutions and Lycopersicon esculentum roots in soilless cultures. Université de Bretagne Occidentale, Ph.D. thesis, 194 pp
Renault D, Tirilly Y, Benizri E, Sohier D, Barbier G, Rey P (2007) Characterization of Bacillus and Pseudomonas strains with suppressive traits isolated from tomato hydroponic-slow filtration unit. Can J Microbiol 53:784–797
Rey P, Benhamou N, Tirilly Y (1998) Ultrastructural and cytochemical investigation of asymptomatic infection by Pythium spp. Phytopathology 88:234–244
Rey P, Nodet P, Tirilly Y (1997) Pythium F induce a minor but ubiquitous disease in tomato soilless cultures. J Plant Pathol 79:173–180
Rey P, Déniel F, Guillou A, Le Quillec S (2005) Management of bacteria to improve slow filtration efficiency in tomato soilless culture. Acta Hortic 691:349–356
Rousselon N, Delgenès JP, Godon JJ (2004) A new real time PCR (Taqman® PCR) system for detection of the 16S rDNA gene associated with fecal bacteria. J Microbiol Meth 59:15–22
Roy S, Gendron J, Delhoménie MC, Bibeau L, Heitz M, Brzezinski R (2003) Pseudomonas putida as the dominant toluene-degrading bacterial species during air decontamination by biofiltration. Appl Microbiol Biot 61:366–373
Stanghellini ME, Rasmussen SL (1994) Hydroponics, a solution for zoosporic pathogens. Plant Dis 78:1129–1138
Suzuki MT, Taylor LT, deLong EF (2000) Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5’-nuclease essays. Appl Environ Microb 66:4605–4614
Thabet OB, Fardeau ML, Joulian C, Thomas P, Hamdi M, Garcia JL, Ollivier B (2004) Clostridum tunisiense sp. nov., a new proteolytic, sulfur-reducing bacterium isolated from an olive mill wastewater contaminated by phosphogypse. Anaerobe 10:185–190
Tu JC, Papadopoulos AP, Hao X, Zheng J (1999) The relationship of Pythium root rot and rhizosphere microorganisms in a closed circulating and an open system in rockwool culture of tomato. Acta Hort 481:577–583
van Os EA, Amsing JJ, van Kuik AJ, Willers H (1999) Slow filtration: a potential method for the elimination of pathogens and nematodes in recirculating nutrient solutions from glasshouse-grown crops. Acta Hortic 481:519–526
Vallance J, Le Floch G, Deniel F, Barbier G, Levesque CA, Rey P (2009) Pythium oligandrum biocontrol in the rhizosphere: influence on fungal and oomycete populations dynamics. Appl Environ Microbiol 75:4790–4800
Vallance J, Le Floch G, Déniel F, Guerin-Dubrana L, Blancard D, Rey P (2011) Pathogenic and beneficial microorganisms in soilless cultures. Agron Sustain Dev 31:191–203
Wohanka W (1995) Disinfection of recirculating nutrient solutions by slow sand filtration. Acta Hortic 382:246–255
Zemb O, Haegeman B, Delgenes JP, Lebaron P, Godon JJ (2007) SAFUM: statistical analysis of SSCP fingerprints using PCA projections, dendrograms and diversity estimators. Mol Ecol Notes 7:767–770
Acknowledgments
This project was supported by the Regional Councils of Brittany and Pays de la Loire and by the French Ministry of Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Renault, D., Vallance, J., Déniel, F. et al. Diversity of Bacterial Communities that Colonize the Filter Units Used for Controlling Plant Pathogens in Soilless Cultures. Microb Ecol 63, 170–187 (2012). https://doi.org/10.1007/s00248-011-9961-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-011-9961-1