
Abstract
Interest in sustainable development has led to efforts to replace petrochemical-based monomers with biomass-based ones. Itaconic acid, a C5-dicarboxylic acid, is a potential monomer for the chemical industry with many prospective applications. cis-aconitate decarboxylase (CadA) is the key enzyme of itaconate production, converting the citric acid cycle intermediate cis-aconitate into itaconate. Heterologous expression of cadA from Aspergillus terreus in Escherichia coli resulted in low CadA activities and production of trace amounts of itaconate on Luria-Bertani (LB) medium (<10 mg/L). CadA was primarily present as inclusion bodies, explaining the low activity. The activity was significantly improved by using lower cultivation temperatures and mineral medium, and this resulted in enhanced itaconate titres (240 mg/L). The itaconate titre was further increased by introducing citrate synthase and aconitase from Corynebacterium glutamicum and by deleting the genes encoding phosphate acetyltransferase and lactate dehydrogenase. These deletions in E. coli’s central metabolism resulted in the accumulation of pyruvate, which is a precursor for itaconate biosynthesis. As a result, itaconate production in aerobic bioreactor cultures was increased up to 690 mg/L. The maximum yield obtained was 0.09 mol itaconate/mol glucose. Strategies for a further improvement of itaconate production are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Itaconic acid is a C5-dicarboxylic acid that can be used as a building block for the production of a diverse set of isomeric lactones, diols, cyclic ethers (Geilen et al. 2010) and polymers (Hughes and Swift 1993). The polymers are potential substituents for many acrylic-based materials such as resins or synthetic fibres (Okabe et al. 2009; Willke and Vorlop 2001).
Since the 1960s, itaconic acid has been commercially produced using natural mutants of the filamentous fungus Aspergillus terreus (Klement and Büchs 2013; Willke et al. 2001). Kuenz et al. (2012) recently reported one of the best performing itaconic acid producing A. terreus cultivations, which had a productivity of 0.51 g/L/h, a maximum titre of 86.2 g/L and a yield of 86 mol%. Better achievements seem possible because the productivity is relatively low and is caused by the low oxygen transfer rates that can be achieved with the filamentous growth form (Klement et al. 2012); the yield is only 65 % of the maximum theoretical yield of 1.33 mol itaconic acid, and titres over 200 g/L citric acid—a precursor of itaconic acid—have been achieved by closely related A. niger. Other disadvantages of A. terreus are sensitivity of the filament pellets to hydro-mechanical stress, laborious handling of spores (Klement et al. 2012), low reproducibility of fermentations (Kuenz et al. 2012) and the fact that an interruption of oxygen supply strongly decreases itaconic acid production (Klement et al. 2012).
Several groups have searched for other microorganisms able to produce itaconic acid. Ustilago sp. and Candida sp. (Tabuchi et al. 1981) have also been found to produce itaconic acid, but with titres below 55 g/L. Recombinant production hosts such as Escherichia coli have been proposed for cheaper production of itaconic acid (Yu et al. 2011). As a facultative anaerobic bacterium, E. coli has many advantages as a production host, like rapid growth under both aerobic and anaerobic conditions, simple medium requirements and well-established protocols for genetic modification.
Wild-type E. coli does not produce itaconate, because it misses cis-aconitate decarboxylase (CadA), which catalyses the conversion of cis-aconitate to itaconate (Yahiro et al. 1995) and is the key enzyme for itaconate biosynthesis in A. terreus. It has been successfully expressed in E. coli (Li et al. 2011), but product titres remained low.
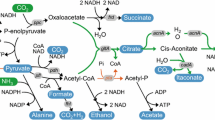
In this paper, we investigate the potential of E. coli to produce itaconate by introducing cis-aconitate decarboxylase (CadA) from Aspergillus terreus and the effects of the introduction of the heterologous enzymes citrate synthase and aconitase from C. glutamicum and the elimination of the native phosphate acetyltransferase and lactate dehydrogenase activities, which could stimulate itaconate production by enhancing the availability of precursors. The proposed itaconate pathway in E. coli is shown in Fig. 1.

Itaconate production pathway in E. coli. The bold arrows indicate the introduced pathway consisting of genes encoding citrate synthase (gltA) and aconitase (acnA) from C. glutamicum and cis-aconitate decarboxylase (cadA) from A. terreus. The dotted lines indicate that phosphate acetyltransferase (pta) and lactate dehydrogenase (ldhA) were deleted
Materials and methods
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Table 1.
Deletion of phosphate acetyltransferase (pta) and lactate dehydrogenase (ldhA) genes
The gene encoding lactate dehydrogenase (ldhA) was inactivated in E. coli BW25113 Δpta by using the Lambda red-mediated gene replacement method described by (Datsenko and Wanner 2000). Shortly, E. coli BW25113 Δpta was transformed with pKD46 and cultured in the presence of l-arabinose to induce λ-red recombinase expression, which is an inducer for recombination. The target gene ldhA was replaced by a kanamycin resistance gene flanked by flippase recognition target (FRT) sites. For this, a deletion cassette containing a kanamycin resistance gene with FRT sites was amplified from pKD4 by using Phusion high-fidelity DNA polymerase (Thermo Scientific) and primers that contain 50 bp targeting flanks to the ldhA region in the genome (Table 2) and transformed into E. coli BW25113 Δpta (pKD46). Transformants were screened for their proper genotype by selecting for kanamycin resistance and colony PCR (GoTaq Green polymerase, Promega) using primers that flank the target gene. The phenotype was verified in liquid cultures. The kanamycin resistance gene was subsequently eliminated by using the temperature-sensitive helper plasmid pCP20 encoding the flippase (FLP), followed by curing of the temperature-sensitive plasmids by culturing strains at 42 °C for 16 h.
Site-specific integration of the λDE3 prophage
Site-specific integration of the λDE3 prophage into E. coli BW25113 and into its derivative E. coli BW25113 Δpta ΔldhA was done using the λDE3 Lysogenization Kit (Novagen). The integration of the λDE3 prophage and expression of T7 polymerase in strains were verified according to the protocol in the kit. Besides, the functional expression of T7 polymerase was confirmed by transforming the strains with pET101/D/lacZ. The transformants were able to cleave 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) after induction with isopropyl-beta-D-thiogalactopyranoside (IPTG).
Construction of pACYC expression vectors
The expression vector pACYCDuet-1 (Novagen) was used to express the genes cadA (NCBI reference sequence ID: BAG49047.1), acnA (ID: NP_600755.1) and gltA (ID: NP_600058.1) under the transcriptional control of the T7 promoter. All genes were codon optimized according to the algorithm of OptimumGeneTM (GenScript) and synthesized by GenScript, USA. The sequences of the codon-optimized cadA, acnA and gltA can be retrieved from GenBank (ID: KM464677, ID: KM464678, ID: KM464679, respectively. cadA was ligated between the NcoI-HindIII restriction sites in MCS1 of pACYCDuet-1, yielding pKV-C. acnA and gltA were respectively ligated between the NdeI-XhoI and XhoI-PacI sites in MCS2 of pKV-C, yielding pKV-CGA, Table 1. An additional ribosomal binding site (rbs), identical to those in pACYCDuet-1, was introduced upstream of gltA gene.
Cultivation conditions
Culture media
For plasmid construction and gene expression analysis, E. coli strains were cultured on Luria-Bertani (LB) agar plates or in LB liquid medium at either 30 or 37 °C. Recombinants harbouring temperature-sensitive plasmids were cultured at either 30 °C for cultivation or 42 °C to cure the selection markers. Expression of lacZ was detected by blue/white screening in agar plates on top of which 40 μL of 20 mg/mL X-gal in dimethyl sulfoxide and 40 μL of 1 M IPTG were spread on top of the plates. When needed, medium and plates were supplemented with ampicillin (50 μg/mL) or chloramphenicol (35 μg/mL). Induction of gene expression in liquid cultures was started by the addition of 1 mM of IPTG when the optical density at 600 nm (OD600) of the culture reached approximately 0.4.
The other cultivations were done in M9 minimal medium (MM), which contained per 1 L: 200 mL 5 × M9 minimal salts (BD Difco) supplemented with 50 mmoles of glucose, 2 mmoles of MgSO4, 0.1 mmoles of CaCl2, 15 mg of thiamine and 0.30 mg of selenite. Medium was buffered with 0.1 M 3-(N-morpholino) propanesulfonic acid (MOPS), and the pH was adjusted to 6.9 with NaOH.
Cultivation in bioreactors
E. coli BW25113 (DE3) and E. coli BW25113 (DE3) Δpta-ΔldhA containing either pEV, pKV-C or pKV-CGA were cultivated at 30 °C in 0.5 L mini bioreactors, connected to myControl controller units (Applikon, The Netherlands) with a working volume of 400 mL. The pH was maintained at 6.9 by the automated addition of 2 M NaOH. Cultures were continuously stirred at 1200 rpm and sparged with medical air at 400 mL/min. Bioreactors were inoculated with 5 % (v/v) of a pre-culture that was grown in a 250-mL Erlenmeyer flasks with 50 mL of MM at 250 rpm and 30 °C for 24 h. Samples of 2 mL were regularly taken to determine the OD600 of the cultures and the concentrations of substrate and products.
Enzymatic assays
For enzymatic assays, 50 mL of bioreactor culture was harvested by centrifugation (5 min, 7745 × g) after 17 h of cultivation in the presence of IPTG and washed with MM. Cell free extracts (CFE) were made according to the Y-PER Yeast Protein Extraction Reagent kit instructions (Thermo Scientific). Protein concentrations were determined by using the Total Protein Kit, Micro Lowry, Peterson’s Modification (Sigma Aldrich).
The activity of cis-aconitate decarboxylase (CadA) was measured by using a method adapted from Dwiarti et al. (2002) and Li et al. (2011): CFE’s were incubated with 17 mM of cis-aconitate in 200-mM sodium phosphate buffer (pH 6.2) for 10 min at 30 °C. Reactions were terminated by adding 1 M HCl. Supernatants were analysed for itaconate formation by HPLC.
The activity of aconitase was measured by monitoring the formation of cis-aconitate at 240 nm in a UV-vis spectrophotometer (UV-1650PC SHIMADZU) using an extinction coefficient for cis-aconitate of 3.5 mM−1 cm−1 (Baumgart and Bott 2011). The assays were performed at 30 °C in 100-mM Tris–HCl buffer (pH 8.0) and 20-mM trisodium citrate as a substrate.
Citrate synthase activity was determined by monitoring the hydrolysis of the thioester of acetyl coenzyme A (acetyl-CoA), which results in the formation of CoA. The thiol group of CoA reacts with 5,5′-dithiobis-(2-nitrobenzoic acid (DTNB) in the reaction mixture to form 5-thio-2-nitrobenzoicacid (TNB). The formation of TNB was measured at 412 nm by using 13.6 mM−1 cm−1 as extinction coefficient at 30 °C according to Morgunov and Srere (1998) with minor adjustments. The reaction mixture contained 0.31-mM acetyl-CoA, 0.5-mM oxaloacetate, 0.1-mM DTNB and ca. 0.25 % Triton X-100 in 100-mM Tris–HCl (pH 8.0).
Analytical methods
The cell density was determined by measuring the OD600 by using a spectrophotometer (Dr. Lange XION 500).
The concentrations of glucose and organic acids were determined by using HPLC by using a Dionex Ultimate 3000 (Thermo Fisher) equipped with an RI detector (Shodex, RI-101) and a UV detector (Dionex, 3400 RS at 210 nm). The samples were separated on a Micro-Guard Cation H pre-column (30×4.6 mm, Biorad) and an Aminex HPX-87H column (300×7.8 mm, Biorad) at 35 °C using 0.6 mL/min of 5 mM H2SO4 as an eluent.
Results
Heterologous expression of cadA, acnA and gltA
Gene cadA from A. terreus was codon optimized and expressed in E. coli to enable itaconate production. Small amounts of itaconate were produced (below 10 mg/L) when E. coli BW25113 (DE3) (pKV-C) was cultivated in LB in shake flask cultures at 37 °C, but no detectable CadA activity was found in CFE of these cultures. SDS-PAGE analysis showed that almost all CadA was present in the form of inclusion bodies (data not shown). As inclusion bodies are often associated with fast and high-level expression of heterologous proteins (Jurgen et al. 2010), two measures were taken to reduce these rates: cultivation in MM instead of LB and cultivation at lower temperatures. When E. coli BW25113 (DE3) (pKV-C) was grown in MM in pH-controlled bioreactors at 37°, CadA could be detected in CFE with a specific activity of 0.03 U/mg. The activity was further increased to 0.38 U/mg when the cultivation temperature was lowered to 30 °C (Fig. 2). SDS-PAGE analysis showed that the amount of soluble protein had increased by these measures (data not shown).

Conversion of cis-aconitate to itaconate in CFE of E. coli BW25113 (DE3) (pKV-C) that was cultured in bioreactors at either 30 °C (circles) or 37 °C (squares). Protein concentrations in the assay mixtures were 5.8 and 4.3 mg/mL, respectively. The activities of CadA were derived from the slopes of the lines
To channel more acetyl-CoA to itaconate, the codon-optimized genes encoding citrate synthase (gltA) and aconitase (acnA) from C. glutamicum were overexpressed in E. coli together with cadA, yielding E. coli BW25113 (DE3) (pKV-CGA). The expression levels of the heterologous genes were determined by measuring the activities of the corresponding enzymes in CFE of E. coli BW25113 (DE3) strains containing either pEV, pKV-C or pKV-CGA. The activities of citrate synthase and aconitase in CFEs of E. coli BW25113 (DE3) (pKV-CGA) increased 4 and 40 times, respectively, compared to the native activities measured in E. coli BW25113 (DE3) (pKV-C) (Fig. 3). It appeared that expression of cadA increased the native citrate synthase and aconitase activities in E. coli BW25113 (DE3) (pKV-C), as the activities of these enzymes were lower in E. coli BW25113 (DE3) (pKV-EV), which might be due to an activating effect of itaconate. Simultaneous expression of gltA and acnA together with cadA resulted in a lower specific CadA activity compared to the E. coli BW25113 (DE3) (pKV-C), which is probably due to a dilution effect caused by the overexpression of the two additional genes.

Specific enzymatic activities (U/mg) of cis-aconitate decarboxylase (hatched lines), citrate synthase (horizontal lines) and aconitase (diagonal lines) in CFE of E. coli BW25113 (DE3) containing either pEV, pKV-C or pKV-CGA. Strains were cultured in bioreactors at 30 °C on MM medium. The average values and standard deviations (SD) of duplicate parallel studies are given
Itaconate production in E. coli BW25113 (DE3)
Itaconate production by E. coli BW25113 (DE3) containing either pEV, pKV-C or pKV-CGA was monitored in pH-controlled bioreactors in MM at 30 °C for 72 h. Itaconate was produced up to 1.9 mM with both E. coli BW25113 (DE3) (pKV-C) and BW25113 (DE3) (pKV-CGA), but not in the control strain (Fig. 4). Overexpression of gltA and acnA together with cadA had no significant impact on itaconate production under these conditions as the production profiles were similar with both pKV-C and pKV-CGA plasmids. This suggests that the availability of precursors is limiting itaconate production.

Batch cultivation of E. coli BW25113 (DE3) containing pEV (left panel), pKV-C (middle panel) and pKV-CGA (right panel) in pH-controlled bioreactors on MM at 30 °C. The OD600 (diamond) and the concentrations of glucose (squares), acetate (triangles) and itaconate (circles) are indicated
During growth on glucose, acetate was observed in all cultures after 1 day of cultivation, which accumulated up to 55 mM. When glucose was depleted from the medium, the cells started consuming acetate. In addition, low concentrations (below 5 mM) of ethanol, citrate, pyruvate, lactate, succinate and formate were detected in the medium during cultivation of all strains, and some cis-aconitate (<5 mM) was formed by E. coli BW25113 (DE3) (pKV-C) and BW25113 (DE3) (pKV-CGA) (data not shown). Most of these compounds were only intermediary products and disappeared over time.
Itaconate production in E. coli BW25113 (DE3) Δpta-ΔldhA
To increase the availability of precursors, E. coli BW25113 (DE3) was made deficient in acetate and lactate production. Deletion of pta, encoding phosphate acetyltransferase, is known to result in accumulation of pyruvate in the cells, which may be redirected to itaconate. As Δpta strains have been reported to convert pyruvate to lactate (Castano-Cerezo et al. 2009), this conversion was eliminated as well by deleting ldhA. To test the effect of these eliminations, the resulting strain E. coli BW25113 (DE3) Δpta-ΔldhA, containing either pEV, pKV-C or pKV-CGA, was cultivated in pH-controlled bioreactors in MM at 30 °C.
E. coli BW25113 (DE3) Δpta-ΔldhA (pKV-CGA) produced three times more itaconate than its wild-type equivalent. Overexpression of gltA and acnA was essential to improve itaconate production, as production was not enhanced in E. coli BW25113 (DE3) Δpta-ΔldhA (pKV-C) (Fig. 5). In all E. coli BW25113 (DE3) Δpta-ΔldhA cultivations, pyruvate accumulated up to 30 mM, after which it was consumed. Acetate was still produced in the double knockout strain, but with a significant delay. Citrate and/or cis-aconitate were also observed, but at trace levels without clear correlations with strain and growth conditions (results not shown).

Batch cultivation of E. coli BW25113 (DE3) Δpta-ΔldhA containing pEV (left panel), pKV-C (middle panel) and pKV-CGA (right panel) in pH-controlled bioreactors on MM at 30 °C. The OD600 (diamond) and the concentrations of glucose (squares), acetate (triangles), pyruvate (crosses) and itaconate (circles) are indicated
The results show that the simultaneous elimination of pta and ldhA and the heterologous expression of gltA and acnA increased the flux through CadA, resulting in higher itaconate titers of up to 690 mg/L, which corresponds to an itaconate yield from glucose of 0.09 mol/mol,
Discussion
In recent years, several microorganisms have been investigated for their ability to produce itaconic acid, besides the well-known itaconic acid producer A. terreus. Some studies have focused on non-conventional natural producers of itaconic acid, such as Pseudozyma antarctica (Levinson et al. 2006) and Ustilago maydis (Klement et al. 2012). Besides, several heterologous production hosts, like A. niger (van der Straat et al. 2014), Yarrowia lipolytica (Wang et al. 2011) and potato (Koops et al. 2011) were studied since cadA was identified as the gene responsible for itaconic acid biosynthesis in A. terreus (Kanamasa et al. 2008).
E. coli has been widely studied for the production of chemicals, such as lactic acid (Zhou et al. 2003), succinic acid (Lee et al. 2005), 1,3-propanediol (Tong et al. 1991) and 1,4-butanediol (Yim et al. 2011); (Yu et al. 2011). So far, E. coli has not been studied extensively for itaconic acid production. Only one study is published (Li et al., 2011), in which E. coli was used as a control strain for the identification of enzymes from A. terreus that are relevant for itaconic acid production. Overexpression of cadA in E. coli resulted in itaconate production, but at low titres (56 mg/L).
We focused on improving E. coli’s potential for itaconate production. Also in our work, the introduction of cadA resulted in low levels of itaconate (<10 mg/L). Expression of heterologous genes in E. coli often causes problems that lead to the synthesis of inactive enzymes, such as protein misfolding and inclusion body formation (Baneyx 1999). This was also observed in our study, but we could significantly enhance the heterologous production of active CadA by optimizing expression conditions (temperature and culture medium), which resulted in higher itaconate titres up to 240 mg/L.
The metabolic flux to itaconate does not only depend on CadA activity, but also on the availability of precursors. This availability is the resultant of the flux to itaconate and the fluxes to by-products. E. coli excretes acetate under aerobic conditions as an overflow metabolite when glucose is in excess (Castano-Cerezo et al. 2009). This indicates that the capacity of the glycolytic pathway is higher than the capacity of the TCA cycle. Citrate synthase is known to exert a strong control on the TCA flux, due to inhibition by high NADH concentrations (Holms 1996), which is at least one of the reasons for acetate overflow, even at aerobic conditions (Vemuri et al. 2006). So, a feasible approach to enhance itaconate production is to reduce acetate production and to diminish the control on the TCA flux.
As the citrate synthase of C. glutamicum (GltA) is not inhibited by high NADH concentrations (Eikmanns et al. 1994), we overexpressed gltA and simultaneously introduced the aconitase gene (acnA) from the same organism in E. coli. The corresponding enzymatic activities increased significantly in E. coli, but did not result as such in higher itaconate titres.
Acetate is produced from acetyl-CoA by phosphate acetyltransferase (pta), which is constitutively expressed under both aerobic and anaerobic conditions (Chang et al. 1999). Deletion of pta reduces the formation of acetate and results in the accumulation of pyruvate (Chang et al. 1999; Diazricci et al. 1991); (Tarmy and Kaplan 1968). Acetate formation is still possible in E. coli Δpta strains, due to the direct oxidation of pyruvate to acetate that is catalysed by pyruvate oxidase (poxB) (Abdel-Hamid et al. 2001).
Castano-Cerezo et al. (2009) reported a significant transient accumulation of lactate during cultivation of a Δpta strain on glucose. We therefore decided to knock out both pta and ldhA, which resulted in a strain that accumulated pyruvate but not lactate, and showed a delayed acetate production. Overexpression of cadA in this Δpta-ΔldhA strain did not result in enhanced itaconate production, but itaconate production was significantly improved up to 690 mg/L when a combination of cadA, gltA and acnA was overexpressed in the Δpta-ΔldhA strain.
The production of acetate was delayed in our Δpta-ΔldhA strain, which is in line with studies that showed that expression of poxB is repressed in the early exponential phase (Castano-Cerezo et al. 2009) and is mainly activated at low growth rates (Abdel-Hamid et al. 2001). Still, acetate remained a dominant by-product in our process. Elimination of poxB is an obvious strategy to decrease acetate formation, although Phue et al. (2010) showed that acetate still accumulated when both pta and poxB were deleted in E. coli, indicating that more metabolic pathways are involved in acetate formation. An alternative approach is to overexpress the acetyl-CoA synthetase gene (acs), which is known to reduce acetate production and increase the intracellular acetyl-CoA concentration during aerobic growth on glucose (Lin et al. 2006).
A. terreus produces itaconic acid at pH values around 3, below the low pKa value of 3.85. Therefore, itaconic acid is largely in its fully protonated form, which is also the required final product. E. coli growth is generally applied at pH values higher than 5.5, above the high pKa value of 5.45. Under these conditions, itaconic acid will be largely in the fully deprotonated form. This has consequences for both fermentation and downstream process. Itaconic acid production by E. coli will require titration with a base to maintain a constant pH. During downstream processing, the pH will have to be decreased by adding acid to form the final fully protonated product, however, with concomitant production of salts. Disposal or recycling of the salts contributes significantly to the overall process costs. On the other hand, organic acids are generally more toxic to microorganisms in their fully protonated form. Product inhibition will therefore be stronger at low-pH itaconic acid production by A. terreus. A similar situation occurs with the production of lactic acid, which has been more extensively studied. Downstream processes at both low and high pH are being developed and improved (Abdel-Rahman et al. 2013), also in combination with in situ product removal to prevent product inhibition (Dafoe and Daugulis 2014). At the moment, it seems unclear if low or neutral pH processes will be the best option for production of organic acids.
To our best knowledge, 690 mg/L is the highest itaconate titre produced by metabolically engineered E. coli strains published in peer-review journals. This was realized without maximizing sugar concentrations and without employing growth limiting conditions. We obtained this titre by increasing the synthesis of soluble CadA and the availability of precursors for itaconate by eliminating pta and ldhA and by overexpressing the genes that are responsible for the conversion of acetyl-CoA to the direct precursor cis-aconitate. The maximum itaconate yield from glucose was 0.09 mol/mol, which is only 7 % of the theoretical maximum yield of 1.33 mol/mol. A significant improvement is therefore required before the production of itaconate with E. coli can become economically feasible. Further optimization of cadA expression and reduction of acetate formation are obvious strategies to achieve this.
References
Abdel-Hamid AM, Attwood MM, Guest JR (2001) Pyruvate oxidase contributes to the aerobic growth efficiency of Escherichia coli. Microbiology 147:1483–1498
Abdel-Rahman MA, Tashiro Y, Sonomoto K (2013) Recent advances in lactic acid production by microbial fermentation processes. Biotechnol Adv 31(6):877–902. doi:10.1016/j.biotechadv.2013.04.002
Baneyx F (1999) Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol 10(5):411–421. doi:10.1016/S0958-1669(99)00003-8
Baumgart M, Bott M (2011) Biochemical characterisation of aconitase from Corynebacterium glutamicum. J Biotechnol 154(2–3):163–170
Castano-Cerezo S, Pastor JM, Renilla S, Bernal V, Iborra JL, Canovas M (2009) An insight into the role of phosphotransacetylase (pta) and the acetate/acetyl-CoA node in Escherichia coli . Microbial Cell Factories 8 doi:54 10.1186/1475-2859-8-54
Chang DE, Shin S, Rhee JS, Pan JG (1999) Acetate metabolism in a pta mutant of Escherichia coli W3110: importance of maintaining acetyl coenzyme a flux for growth and survival. J Bacteriol 181(21):6656–6663
Dafoe JT, Daugulis AJ (2014) In situ product removal in fermentation systems: improved process performance and rational extractant selection. Biotechnol Lett 36(3):443–460. doi:10.1007/s10529-013-1380-6
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97(12):6640–6645. doi:10.1073/pnas.120163297
Diazricci JC, Regan L, Bailey JE (1991) Effect of alteration of the acetic-acid synthesis pathway on the fermentation pattern of Escherichia coli. Biotechnol Bioeng 38(11):1318–1324. doi:10.1002/bit.260381109
Dwiarti L, Yamane K, Yamatani H, Kahar P, Okabe M (2002) Purification and characterization of cis-aconitic acid decarboxylase from Aspergillus terreus TN484-M1. J Biosci Bioeng 94(1):29–33. doi:10.1263/jbb.94.29
Eikmanns, B.J., Thumschmitz, N., Eggeling, L., Ludtke, K.U. and Sahm H. (1994) Nucleotide sequence, expression and transcriptional analysis of the Corynebacterium glutamicum gltA gene encoding citrate synthase. Microbiology-Sgm 140, 1817–1828
Geilen FMA, Engendahl B, Harwardt A, Marquardt W, Klankermayer J, Leitner W (2010) Selective and flexible transformation of biomass-derived platform chemicals by a multifunctional catalytic system. Angew Chem Int Ed 49(32):5510–5514. doi:10.1002/anie.201002060
Holms H (1996) Flux analysis and control of the central metabolic pathways in Escherichia coli. FEMS Microbiol Rev 19(2):85–116. doi:10.1111/j.1574-6976.1996.tb00255.x
Hughes KA, Swift G (1993) Process for polymerization of itaconic acid. Patent US5223592 A
Jurgen B, Breitenstein A, Urlacher V, Buttner K, Lin H, Hecker M, Schweder T, Neubauer P (2010) Quality control of inclusion bodies in Escherichia coli. Microb Cell Fact 9(1):41. doi:10.1186/1475-2859-9-41
Kanamasa S, Dwiarti L, Okabe M, Park E (2008) Cloning and functional characterization of the cis-aconitic acid decarboxylase (CAD) gene from Aspergillus terreus. Appl Microbiol Biotechnol 80:223–229. doi:10.1007/s00253-008-1523-1
Klement T, Büchs J (2013) Itaconic acid—a biotechnological process in change. Bioresour Technol 135(0):422–431. doi:10.1016/j.biortech.2012.11.141
Klement T, Milker S, Jager G, Grande P, Dominguez de Maria P, Buchs J (2012) Biomass pretreatment affects Ustilago maydis in producing itaconic acid. Microb Cell Fact 11(1):43
Koops AJ, De Graaff LH, Van Der Meer IM, Van Den Berg WAM (2011) Nucleotide sequences coding for cis-aconitic decarboxylase and use thereof US 20110099670 A1
Kuenz A, Gallenmuller Y, Willke T, Vorlop KD (2012) Microbial production of itaconic acid: developing a stable platform for high product concentrations. Appl Microbiol Biotechnol 96(5):1209–1216. doi:10.1007/s00253-012-4221-y
Lee SJ, Lee DY, Kim TY, Kim BH, Lee JW, Lee SY (2005) Metabolic engineering of Escherichia coli for enhanced production of succinic acid, based on genome comparison and in silico gene knockout simulation. Appl Environ Microbiol 71(12):7880–7887. doi:10.1128/aem.71.12.7880-7887.2005
Levinson WE, Kurtzman CP, Kuo TM (2006) Production of itaconic acid by Pseudozyma antarctica NRRL Y-7808 under nitrogen-limited growth conditions. Enzyme Microb Technol 39(4):824–827. doi:10.1016/j.enzmictec.2006.01.005
Li A, van Luijk N, ter Beek M, Caspers M, Punt P, van der Werf M (2011) A clone-based transcriptomics approach for the identification of genes relevant for itaconic acid production in Aspergillus. Fungal Genet Biol 48(6):602–611. doi:10.1016/j.fgb.2011.01.013
Lin H, Castro NM, Bennett GN, San K-Y (2006) Acetyl-CoA synthetase overexpression in Escherichia coli demonstrates more efficient acetate assimilation and lower acetate accumulation: a potential tool in metabolic engineering. Appl Microbiol Biotechnol 71(6):870–874. doi:10.1007/s00253-005-0230-4
Morgunov I, Srere PA (1998) Interaction between citrate synthase and malate dehydrogenase—substrate channeling of oxaloacetate. J Biol Chem 273(45):29540–29544. doi:10.1074/jbc.273.45.29540
Okabe M, Lies D, Kanamasa S, Park E (2009) Biotechnological production of itaconic acid and its biosynthesis in Aspergillus terreus. Appl Microbiol Biotechnol 84:597–606. doi:10.1007/s00253-009-2132-3
Phue J-N, Lee SJ, Kaufman JB, Negrete A, Shiloach J (2010) Acetate accumulation through alternative metabolic pathways in ackA(-) pta(-) poxB(-) triple mutant in E. coli B (BL21). Biotechnol Lett 32(12):1897–1903. doi:10.1007/s10529-010-0369-7
Tabuchi T, Sugisawa T, Ishidori T, Nakahara T, Sugiyama J (1981) Itaconic acid fermentation by a yeast belonging to the genus Candida. Agric Biol Chem 45(2):475–479
Tarmy EM, Kaplan NO (1968) Kinetics of Escherichia coli B D-lactate dehydrogenase and evidence for pyruvate-controlled change in confirmation. J Biol Chem 243(10):2587
Tong IT, Liao HH, Cameron DC (1991) 1,3-Propanediol production by Escherichia coli expressing genes from the Klebsiella pneumoniae dha regulon. Appl Environ Microbiol 57(12):3541–3546
van der Straat L, Vernooij M, Lammers M, van den Berg W, Schonewille T, Cordewener J, van der Meer I, Koops A, de Graaff L (2014) Expression of the Aspergillus terreus itaconic acid biosynthesis cluster in Aspergillus niger. Microb Cell Fact 13(1):11. doi:10.1186/1475-2859-13-11
Vemuri G, Altman E, Sangurdekar D, Khodursky A, Eiteman M (2006) Overflow metabolism in Escherichia coli during steady-state growth: transcriptional regulation and effect of the redox ratio. Appl Environ Microbiol 72(5):3653–3661. doi:10.1128/AEM.72.5.3653-3661.2006
Wang JH, Tsai SH, Teng K (2011) Producing Itaconic Acid in Yeast Using Glycerol as the Substrate. US Patent US20110053232 A1
Willke T, Vorlop K-D (2001) Biotechnological production of itaconic acid. Appl Microbiol Biotechnol 56:289–295. doi:10.1007/s002530100685
Willke T, Welter K, Vorlop KD (2001) Biotechnological production of itaconic acid from sugar. Zuckerindustrie 126(6):444–447
Yahiro K, Takahama T, Park YS, Okabe M (1995) Breeding of Aspergillus terreus mutant TN-484 for itaconic acid production with high yield. J Ferment Bioeng 79(5):506–508. doi:10.1016/0922-338x(95)91272-7
Yim H, Haselbeck R, Niu W, Pujol-Baxley C, Burgard A, Boldt J, Khandurina J, Trawick JD, Osterhout RE, Stephen R, Estadilla J, Teisan S, Schreyer HB, Andrae S, Yang TH, Lee SY, Burk MJ, Van Dien S (2011) Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol 7(7):445–452. doi:10.1038/nchembio.580
Yu C, Cao YJ, Zou HB, Xian M (2011) Metabolic engineering of Escherichia coli for biotechnological production of high-value organic acids and alcohols. Appl Microbiol Biotechnol 89(3):573–583. doi:10.1007/s00253-010-2970-z
Zhou SD, Causey TB, Hasona A, Shanmugam KT, Ingram LO (2003) Production of optically pure D-lactic acid in mineral salts medium by metabolically engineered Escherichia coli W3110. Appl Environ Microbiol 69(1):399–407. doi:10.1128/aem.69.1.399-407.2003
Acknowledgments
This work has been carried out with a grant from the BE-BASIC program FS 01.002 itaconic/fumaric acids: Novel Economic and eco-efficient processes for the production of itaconic and fumaric acid.
We thank Annemarie Hage for her assistance with the bioreactor experiments, Susan Witte for setting up an HPLC method and Sven Keuris for constructing the DE3 lysogenized strains.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vuoristo, K.S., Mars, A.E., Sangra, J.V. et al. Metabolic engineering of itaconate production in Escherichia coli . Appl Microbiol Biotechnol 99, 221–228 (2015). https://doi.org/10.1007/s00253-014-6092-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6092-x