
Abstract
SNF1/AMPK protein kinase plays important roles in fungal development and activation of catabolite-repressed genes. In this study, we characterized the role of the Beauveria bassiana SNF1 ortholog. The vegetative growth of the ΔBbSNF1 mutant was reduced by 16 to 50 % on diverse carbon/nitrogen sources. Transcriptional analysis revealed a collection of proteases and chitinases that were not induced when the mutant was grown on complex carbon/nitrogen sources. BbSNF1 also contributes to extracellular acidification. The ΔBbSNF1 mutant had enhanced production of lactic, pyruvic, and citric acid, but oxalic acid production was partially repressed. Transcriptional analysis showed that a set of genes involved in acid biosynthesis and secretion was changed in the disruption mutant, indicating that BbSNF1 controls the production of different organic acids with different mechanisms. Deletion of BbSNF1 resulted in a significant reduction in conidiation (57–75 %) and blastospore yield (80–95 %) in the mutant. Additionally, BbSNF1 regulates the morphology of blastospore-forming structures and the in vitro blastospore size. Insect bioassays revealed that the ΔBbSNF1 strain exhibited an approximately doubled median lethal time in topical bioassays, but the decreased virulence in intrahemocoel assays (~20 % change) was not as great as in the topical bioassays. These data suggest that BbSNF1 is important in penetration through the host cuticle. Moreover, a series of genes regulated by BbSNF1 and associated with blastospore formation were primarily involved in metabolism, cell cycle, and transportation. In conclusion, the SNF1/AMPK kinase contributes to the biocontrol potential of B. bassiana by mediating cellular differentiation and utilization of carbon/nitrogen sources.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Beauveria bassiana, a well-known entomopathogenic fungus with extensive host range, has long been regarded as an alternative strategy with lower environmental risk to control insect pests with economical and medical importance (Fan et al. 2012; Feng et al. 1994). Under natural conditions, B. bassiana infection begins with the adhesion of infective propagules, including conidia or blastospores, to the host cuticle (Holder and Keyhani 2005; Holder et al. 2007). During fungal penetration of the host cuticle, the entomopathogenic fungus secretes various hydrolases to accelerate the penetration process (Cho et al. 2006; Fang et al. 2005; Pedrini et al. 2013; Zhang et al. 2008). Upon entering the host hemocoel, the fungus produces yeast-like cells, termed blastospores, or hyphal bodies via a dimorphic conversion from mycelia (Lewis et al. 2009; Wanchoo et al. 2009), a process that is similar to that of Candida albicans during its infection of the host, which improves the invasion efficiency of the pathogen. The single-cell form facilitates the distribution of the pathogen in the bloodstream and adherence to surfaces of endothelial cells (Han et al. 2011). Likewise, hyphal bodies of the entomopathogen could overcome the insect immune system and successfully propagate by easily assimilating the nutrient constituents of the hemolymph, e.g., sugars and proteins (Pendland et al. 1993). Prior to reaching the hemocoel, B. bassiana must also overcome the nutrient-poor microenvironment on insect cuticles and perform the dimorphic change in insect hemocoels (Zhang et al. 2013a).
Fungi can adopt mechanisms of carbon catabolite repression to preferentially use the simple carbon sources (e.g., glucose) by depressing genes involved in metabolizing other unfavorable and complex carbon sources (Strauss et al. 1999). The SNF1/AMPK protein kinases, which are highly conserved, are found in all eukaryotic creatures including filamentous fungi (SNF1), yeasts (SNF1), insects (AMPK), mammals (AMPK), and plants (SnRK1). This family of kinases was also regarded as an important regulator that is involved in fungal development and activation of catabolite-repressed genes (Ghillebert et al. 2011; Lee et al. 2009). The activated SNF1/AMPK could phosphorylate downstream targets, such as Mig1 and Msn2, to strengthen catabolic pathways and increase ATP production (Hedbacker and Carlson 2008). In yeast, SNF1 functions as a heterotrimeric kinase complex containing an α-subunit (catalytic subunit, Snf1), two β-subunits (regulatory subunit, Sip1, Sip2, or Gal83), and a γ-subunit (Snf4). The activity of the SNF1 complex can be initiated by three upstream kinases (Sak1, Elm1, and Tos3) via phosphorylation (Hedbacker and Carlson 2008; Seip et al. 2013). In Saccharomyces cerevisiae, SNF1 is an important regulator in glucose starvation during growth, permitting yeast cells to assimilate alternative or unfavorable carbon sources, such as sucrose and ethanol. Additionally, it also plays an important function in various processes related to development, including fungal meiosis, sporulation, filamentous growth, and biofilm formation (Ghillebert et al. 2011). For a number of filamentous pathogenic fungi, the SNF1 gene has been shown to be critical for fungal development and virulence. In the plant pathogen Gibberella zeae, the GzSNF1 deletion mutant is unable to form the perithecia and ascospores (Lee et al. 2009). In Penicillium digitatum, ablation of PdSNF1 also resulted in impaired conidiation and the defective conidiophore structures (Zhang et al. 2013b). Furthermore, disruption of SNF1 generally resulted in repressed expression of cell-wall-degrading enzymes and reduced virulence in phytopathogenic fungi, such as Cochliobolus carbonum, Fusarium oxysporum, G. zeae, P. digitatum, and Verticillium dahliae (Lee et al. 2009; Ospina-Giraldo et al. 2003; Tonukari et al. 2000; Tzima et al. 2011; Zhang et al. 2013b). However, little is known about the function of SNF1 in the development and pathogenicity of entomopathogenic fungi, particularly in the dimorphic transition from filamentous growth to the yeast-like form (blastospore).
B. bassiana transcriptomic analysis of blastospore development has been adopted to identify phase-specific genes and genes involved in cell development that are mediated by a G protein signaling pathway (Cho et al. 2006; Ying et al. 2013). In this study, we identified the B. bassiana SNF1 homolog, BbSNF1. Using a homologous recombination strategy, the disruption mutant of BbSNF1 was constructed, and the role of BbSNF1 in fungal development and virulence was characterized. The disruption mutant appeared to have the defective cell differentiation into conidia and blastospores, and the blastospore size was reduced. Insect bioassays revealed a significant decrease in the virulence of the disruption mutant. These results indicated that BbSNF1 plays an important role in fungal development and virulence. Additionally, a number of BbSNF1 target genes were identified by transcriptional and transcriptomic analysis. These results revealed that genes that are influenced by BbSNF1 during blastospore formation are highly enriched in the functional catalogs of metabolism, cell cycle, and transport.
Materials and methods
Microbial strains and basic media
Bacterial and fungal strains were cultured and maintained according to previous methods (Ying et al. 2013). The wild-type strain, B. bassiana ARSEF2860 (Bb2860 hereafter), was routinely maintained on Sabouraud dextrose agar (SDAY: 4 % glucose, 1 % peptone, and 1.5 % agar plus 1 % yeast extract), and its conidia were kept at −76 °C as a mixture with sterile sands (Şahil and Otag 2013). Escherichia coli DH5α (Invitrogen, Carlsbad, CA, USA) was used for plasmid propagation and cultured in Luria-Bertani (LB) medium containing ampicillin (100 μg/ml) or kanamycin (50 μg/ml) based on plasmid resistance. Agrobacterium tumefaciens AGL-1 (kindly provided by Professor Chengshu Wang, Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, Shanghai, China) was used in fungal transformation and grown at 28 °C on yeast extract broth medium (w/v: 0.5 % sucrose, 1 % peptone, 1.5 % agar plus 0.1 % yeast extract and 0.05 % MgSO4) (Ying et al. 2013).
Phylogenetic analysis of BbSNF1
The sequence of SNF1p (GenBank No. EAA29429) in Neurospora crassa, a model filamentous fungus, was used to query the database of predicated proteins of the Bb2860 genome (Xiao et al. 2012), leading to the identification of the B. bassiana homolog, BbSNF1p (locus tag BBA_04261). The complementary DNA (cDNA) sequence of BbSNF1p was used to query the B. bassiana genome, generating its upstream and downstream flanking sequences as well as the whole open reading frame of BbSNF1.
Additional fungal SNF1 orthologs present in the NCBI protein database were identified via online BLAST analysis (http://www.ncbi.nlm.nih.gov/BLAST/) using the keyword “SNF1.” After manual correction, the deduced SNF1 amino acid sequence was compared with other SNF1 orthologs using ClustalW, and a phylogenetic relationship was established with MEGA version 5 (Tamura et al. 2011).
Generation of BbSNF1 gene knockout and complemented strains
Using the genomic DNA of the wild-type strain as template, upstream (~1.53 kb) and downstream (~1.58 kb) flanking sequences of the BbSNF1 open reading frame (ORF, 2,230 bp) were obtained by PCR using primers P1/P2 and P3/P4 (Table S1 in Supplementary Material), respectively. The P1/P2- and P3/P4-amplified PCR fragments were ligated into the EcoRI/BamHI and BglII/SpeI sites of p0380-bar (Xie et al. 2012), respectively. The resultant plasmid was designated as pKO-BbSNF1 for gene disruption. For complementation of the disruption mutant, a plasmid was constructed by amplifying the full-length BbSNF1 ORF sequence plus 1.63 kb of upstream and 0.3 kb of downstream sequence using primers P7/P8 (Table S1 in Supplementary Material). The resultant PCR fragment was recombined into plasmid p0380-sur-gateway using Gateway® BP Clonase™ II Enzyme Mix (Invitrogen) (Xie et al. 2012), yielding vector p0380-sur-BbSNF1 with the sulfonylurea resistance gene (sur).
Plasmids were proliferated in E. coli DH5α and transformed into B. bassiana using Agrobacterium-mediated transformation (Fang et al. 2004). Putative BbSNF1 gene disruption mutants were screened on Czapek-Dox agar (CZA) supplemented with phosphinothricin (200 μg/ml), and complemented mutants were screened on CZA containing chlorimuron ethyl (15 μg/ml). To prove correct recombination in transformants, the genomic DNA was extracted from transformants cultured on SDAY plates, and PCR screening was performed with primers P5/P6 (Table S1 in Supplementary Material). Putative transformants of the disruption mutant and complemented transformants were confirmed by additional Southern blotting. The EcoRI-digested genomic DNA (10 μg) of the indicated strain was resolved in a 1 % agarose gel and transferred to a Biodyne B nylon membrane (Gelman Laboratory, Shelton, WA, USA). The target fragment was probed with a 527-bp probe amplified with primers P9/P10. Probe preparation, membrane hybridization, and visualization were performed according to operation manual of DIG High Prime DNA Labeling and Detection Starter Kit II, using the chemiluminescent detection method (Roche, Penzberg, Germany).
Phenotypic assays
Phenotypic analysis, such as vegetative growth, spore development, and virulence, were performed among the wild-type, the disruption mutant, and the complemented strains.
Vegetative growth
The vegetative growth of three strains on different culture media was examined as previously described (Ying et al. 2013). Conidia of indicated strains were harvested from SDAY plates and suspended in solution of 0.02 % Tween 80. Aliquots of 1-μl conidial suspensions (1 × 106 cells/ml) were dotted onto plates (9-cm diameter) containing SDAY or minimal salts medium (CZA minus sucrose or NaNO3) supplemented with different carbon/nitrogen sources. Carbon-modified media were prepared by replacing the 3 % sucrose in CZA with 3 % glucose, sucrose, trehalose, or glycerol, 0.5 % sodium acetate, 0.5 % olive oil, or 0.2 % oleic acid. Nitrogen-modified media were prepared using CZA minimal salts (lacking 0.3 % NaNO3) supplemented with either 0.5 % N-acetyl-glucosamine (GlcNAc), gelatin, or colloidal chitin. Plates were incubated for 7 days at 25 °C using a 12:12 h (light/dark) light cycle and the diameters of the fungal colonies were measured.
Conidial yield
The conidial yield on different culture media was quantified as previously described (Xie et al. 2012). In brief, conidial suspensions (100 μl of 106 conidia ml−1) were uniformly smeared on CZA and SDAY agar plates and incubated for 7 days at 25 °C. The conidia on mycelial disks (5 mm) were suspended in 0.02 % Tween 80 by vortexing, and the conidial concentration was converted to the number of conidia per square centimeter.
Analysis of pH and organic acids
Czapek-Dox minimal broth (i.e., free of sucrose) was supplemented with either glucose, sucrose, trehalose, maltose, or fructose (final concentration of 3 %), generating CZGLU, CZSUC, CZTRE, CZMAL, and CZFRU media. Conidia harvested from SDAY plates were inoculated into different culture broths as listed above (50 ml) at the final concentration of 106 conidia/ml and were incubated with shaking (180 rpm) at 25 °C. During the 4-day incubation, the pH values of the culture were examined at 2, 3, and 4 days. Mycelia and medium of 3-day-old cultures were separated by filtering the culture through filter paper. The resulting supernatant was submitted to analysis for extracellular organic acids. Mycelia were lyophilized to extract intracellular organic acids. Mycelia (0.2 g) were ground in liquid nitrogen and suspended in 10 ml ultrapure water (Milli-Q Academic, Millipore Corporation, MA, USA). The acid solution was collected by centrifuging to remove cellular debris, and the proteins in solution were removed by an equal volume of deproteinization agent (chloroform/isopropanol, 25:1). The resultant solution was used as the sample for acid analysis.
Organic acids were analyzed via ion-exchange chromatography on a DIONEX ICS-2000 (Dionex Corporation, Sunnyvale, CA, USA). Detected organic acids included citrate, pyruvate, oxalate, and lactate. Briefly, the sample was filtered through Dionex OnGuard™ II RP and H cartridge (Thermo, Sunnyvale, CA, USA) to remove interfering substances and was loaded into the instrument according to the operator’s manual. Organic acids in culture media were calculated with the standard solution (10 ppm) of each organic acid.
Blastospore development in liquid culture
The yield and development of blastospores was quantified in different culture broths as previously described (Ying et al. 2013). The nitrogen source (0.3 % NaNO3) of Czapek-Dox medium was replaced by 0.5 % peptone, and the carbon source was substituted with glucose, sucrose, trehalose, maltose, or fructose (3 % final concentration) to yield PEPGLU, PEPSUC, PEPTRE, PEPMAL, and PEPFRU media, respectively. Conidia grown on SDAY plates were inoculated in the above culture broth media (50 ml) at the final concentration of 106 conidia/ml and were cultured with continuous shaking (180 rpm) at 25 °C. After a 3-day incubation, the blastospore concentration in the culture was examined by counting the number of blastospores with a hemocytometer under a microscope; meanwhile, mycelia were collected and quantified. The blastospore yield was calculated as the number of spores per milliliter of medium. To observe blastospore development, the mycelia or/and blastospores were sampled from the PEPTRE medium at 12, 24, 36, and 48 h, respectively, and their morphology were examined under a microscope.
Flow cytometry was performed to examine the morphologic change of blastospores. The cells were harvested from 3-day-old liquid cultures by filtering the culture broth through lens paper to remove the mycelia. Blastospore suspensions were adjusted to a concentration of 105 cells/ml and loaded into a flow cytometer (model FC500, Beckman Coulter, Brea, CA, USA) to analyze cell size using a forward scatter detector (FSc), and cell complexity using a side scatter detector (SSc) (Zhang et al. 2013a).
Fungal pathogenicity and in vivo development
Bioassays on Galleria mellonella larvae (~300 mg per capita) were used to evaluate fungal virulence by two different bioassay methods, topical application and intrahemocoel injection (Ying et al. 2013; Zhang et al. 2012). In the topical application assay, the larvae were dipped in conidial solution (107 conidia/ml in 0.02 % Tween 80) for 10–15 s, and the treated larvae were dried on a paper towel. Larvae treated with 0.02 % Tween 80 were used as a blank control. For intrahemocoel injection, 5-μl conidial suspension (105 conidia/ml in 0.02 % Tween 80) was injected into the larvae, and 0.02 % Tween 80 without conidial suspension was used as a control. For each strain, bioassays were performed in triplicate, using 30 to 35 larvae per replicate. Treated larvae were maintained in large Petri dishes (15-cm diameter) for 7 days at 25 °C. The number of dead larvae was recorded each day, and the median lethal time (LT50) was calculated by Probit analysis as previously described (Ying et al. 2013). All phenotypic experiments were performed in triplicate. Cadavers from the topical bioassay were kept in humid Petri plates for 7 days, and fungal growth on cadavers was recorded by photography.
To observe the development of in vivo blastospores (hyphal bodies), the hemolymph was collected and examined according to previous methods (Lewis et al. 2009; Ying et al. 2013). At the time point of the estimated LT50 for each strain, the hemolymph was bled from five larvae (still alive) infected with the wild-type, ΔBbSNF1 mutant, and the complement strains. The fresh hemolymph was dropped into 0.2 ml of anticoagulant solution (0.14 M NaCl, 0.1 M glucose, 26 mM citric acid, 30 mM trisodium citrate, 10 mM EDTA, pH 4.6). The morphology of in vivo cells was examined via microscopy. All the steps in the experiment, including bleeding, centrifugation, and washing, were performed at 4 °C.
Transcriptional analysis of cuticle-degrading enzymes and acid biosynthesis and secretion genes
The transcriptional analysis of the genes involved in cuticle degradation and carboxylate biosynthesis and transportation was performed as previously described (Ying et al. 2013). Total RNA was isolated from the wild-type and ΔBbSNF1 mutant strains grown under four conditions. With the assimilation of protein (1) or chitin (2), wild type and ΔBbSNF1 (100 μl of 107 conidia/ml) were evenly spread onto cellophane-overlaid CZA-derived plates, using gelatin or chitin as the single nitrogen source (prepared as described in section of “Vegetative growth”). After 2 days of incubation at 25 °C and 12:12 h, the mycelia were harvested and ground into powder in liquid nitrogen. In carboxylate biosynthesis and secretion, conidia of each strain were inoculated into 50 ml of CZSUC (3) and CZTRE (4) broth at the final concentration of 106 conidia/ml. After 3 days of incubation with aeration (180 rpm) at 25 °C, mycelia were collected by centrifugation. Total RNA was extracted from the different cell samples with RNAisoTM Plus Reagent (TaKaRa, Dalian, China), and cDNA was synthesized using the PrimeScript® RT Reagent Kit (TaKaRa). Resultant cDNA samples (tenfold dilution) were used as templates to measure transcriptional levels of indicated genes via quantitative real-time PCR (qRT-PCR) using SYBR® Premix Ex TaqTM (TaKaRa) on a Mastercycler® Ep-Realplex (Eppendorf, Hamburg, Germany) cycler. The list of genes examined and their primers are listed in Table S2 in Supplementary Material. The relative transcript level of each gene was calculated as the transcript ratio of ΔBbSNF1 to WT using the fungal 18S rRNA as an internal standard and the 2−ΔΔCt method (Livak and Schmittgen 2001).
Construction and analysis of BbSNF1-mediated transcriptome
In order to probe the BbSNF1-mediated transcription during blastospore development, a genome-wide expression analysis was performed on the wild-type and the ΔBbSNF1 mutant strains using the previously described methods (Ying et al. 2013). Total RNA was extracted from 2-day-old mycelia of the wild-type and ΔBbSNF1 strains cultured in PEPGLU and PEPTRE media as described above, resulting in four RNA libraries, i.e., WTGLU, WTTRE, ΔBbSNF1 GLU, and ΔBbSNF1 TRE. RNA samples were further purified using the Qiagen RNeasy Mini kit plus on-column treatment with DNase I; then, samples were sequenced at Beijing Genomics Institute (Shenzhen, China, Illumina HiSeqTM 2000 platform). Sequence data have been deposited in NCBI’s Gene Expression Ominibus and are accessible through GEO Series Accession No. GSE53582. All samples were replicated two times in the independent experiments.
All clean reads of each library were mapped onto the genome sequence of Bb2860 (Xiao et al. 2012). All mapped genes were normalized in terms of the number of reads per kilobase of exon region per million mapped reads (RPKM) and functionally annotated with known or putative gene annotation information in the NCBI protein databases (or expressed as “hypothetical proteins” if no information was available) at the cutoff error level of <10−5.
For the two biological replicates of each experiment, we used on the pretreated data the NOISeq approach (Tarazona et al. 2011) to search differentially expressed genes (DEGs) between the paired libraries, i.e., WTGLU/ΔBbSNF1 GLU and WTTRE/ΔBbSNF1 TRE. The mean value of RPKMs from two replicates was calculated as the representative gene expression in each library. The differential expression statistics from the NOISeq approach are the log ratio (M) and the absolute value of the difference (D) between two conditions, and these two statistics were used to calculate the probability (P) of a gene being differentially expressed. The DEGs between the indicated comparison set were screened at the threshold of P > 0.8, using the standard of log2 ratio (fold change) >1 or < −1.
For each comparison set, the potential DEGs were submitted for Gene Ontology (GO) mapping and enrichment analysis (Benjamini and Yekutieli 2001). Preliminary analysis showed low GO records for enrichment analysis; therefore, the Functional Catalogue (FunCat) method was used to analyze the potential function of DEGs (Ruepp et al. 2004). To identify the orthologs of B. bassiana genes in the model fungus N. crassa genome, all genes were searched against the N. crassa protein database downloaded from the Broad Institute (http://www.broad.mit.edu/annotation/genome/neurospora/Downloads.html), using E-value cutoffs of <10−5. All of the obtained N. crassa orthologous locus tags were submitted in the online MIPS Functional Catalogue portal (http://mips.helmholtz-muenchen.de/proj/funcatDB/) and used to perform an enrichment analysis using a threshold of corrected P < 0.05.
Data and sequence analyses
All data, including radial growth (colony diameter), concentration of organic acid, conidial production, blastospore yield, and insect bioassay, were subjected to one-way ANOVA. The protein domains of all the SNF1ps were identified via searches using the pfam website (http://pfam.sanger.ac.uk/). The potential secreted hydrolases (protease and chitinase) were predicted using the CBS Prediction Servers (http://www.cbs.dtu.dk/services/). The subset of downregulated genes in the ΔBbSNF1 mutant (overlapped between WTGLU/ΔBbSNF1 GLU and WTTRE/ΔBbSNF1 TRE) was queried in the Pathogen Host Interaction (PHI) database (http://www.phi-base.org/), using a BLAST score cutoff E-value of 10−20 to identify the potential genes involved in pathogen-host interactions.
Results
Sequence analysis and construction of targeted gene knockout and complementation strains
A translated ORF annotated as an ortholog of SNF1p was identified in a B. bassiana genome sequence (locus tag BBA_04261). The genomic sequence of the BbSNF1 ORF was 2,230 bp long with two introns, and it encoded for a 702-amino acid protein that contained a Pkinase domain (Table S3 in Supplementary Material), which is found in all SNF1p orthologs. An additional domain, UBA_2, could be found in all examined SNF1p orthologs of filamentous fungi. Phlyogenetic analyses showed that all of the SNF1ps could be separated into four groups (Fig. S1 in Supplementary Material). Group I was derived from filamentous fungi, group II consisted of SNF1p members found in yeasts, groups III consisted of SNF1p from plants and slime, and group IV consisted of SNF1p from animals and protists. Phylogenetic analysis indicated that BbSNF1 is much more closely related to the SNF1 orthologs of the filamentous fungi than to those of yeasts, plants, or animals. No clustering of sequences by fungal lifestyle, i.e., saprobes, plant, animal, or insect pathogens, was observed. BbSNF1 displayed the closest homology to that of entomopathogenic fungi, i.e., Cordyceps militaris and two Metarhizium species.
In order to further investigate the function of BbSNF1, a gene disruption strain was constructed by homologous recombination strategy. Homologous recombination led to the insertion of the phosphinothricin resistance marker (bar) into the ORF of the target gene as mentioned in the “Materials and methods” section (Fig. S2a in Supplementary Material). A complemented strain was constructed by ectopic insertion of the full-length BbSNF1, including 1,109 bp of upstream sequence into the ΔBbSNF1 strain using the sulfonylurea resistance gene (sur) as a selection marker. The correct gene disruption and complementation strains were confirmed by PCR and Southern blotting (Fig. S2b, c in Supplementary Material).
BbSNF1 contributes to hyphal growth and conidial production
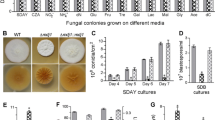
Radial growth rates were examined on different medium types (Fig. S3 in Supplementary Material). Compared to the wild-type strain, no significant difference was observed in the growth rate of the ΔBbSNF1 mutant on rich media (SDAY) (Fig. 1a). However, a slight to moderate reduction (16–50 %) in growth was seen in minimal medium supplemented with the indicated carbon and nitrogen sources, with gelatin causing the greatest reduction (50 %) (Fig. 1a). Despite the lack of effect on the vegetative growth rate (as shown in colony diameters) on SDAY medium, the conidial yield of the ΔBbSNF1 mutant was remarkably reduced (74.5 %), producing only 1.82 ± 0.13 × 108 conidia/cm2, whereas the wild-type strain yielded 7.14 ± 0.25 × 108 conidia/cm2. On Czapek-Dox medium, the ΔBbSNF1 strain also showed a significant reduction (56.6 %) in conidial yield, with the wild-type and disruption strain yielding 0.83 ± 0.04 and 0.36 ± 0.04 × 108 conidia/cm2, respectively.

Phenotypic analysis of Beauveria bassiana wild-type, ΔBbSNF1, and complemented strains. a Fungal growth on various media. The indicated strains were grown on Sabouraud dextrose agar or minimal medium supplemented with various carbon/nitrogen sources. Colony diameters were examined after 7 days of growth at 25 °C. b pH values in culture media. The indicated strains were grown in minimal medium with either glucose (Glu), sucrose (Suc), trehalose (Tre), maltose (Mal), or fructose (Fru) as the sole carbon source. The pH values of media were measured after 2, 3, and 4 days of incubation, respectively. Analysis of organic acids in c liquid media and in d mycelia. The acid types and concentrations were analyzed in the culture medium and mycelia grown in broth containing sucrose or trehalose as the sole carbon source. Asterisks above bars indicate a significant difference between the ΔBbSNF1 mutant and either the wild-type or complemented strain (Tukey’s honestly significant difference (HSD): P < 0.05). Error bars: standard deviation
BbSNF1 is involved in extracellular acidification
To probe the effect of BbSNF1 on extracellular acidification, the pH values of defined media (modified CZ media) were examined (Fig. 1b). During the whole culture process, the disruption of BbSNF1 did not cause a significant change in the pH value of CZMAL and CZFRU media compared with the wild-type strain. However, after a 3-day incubation, the disruption of BbSNF1 resulted in a significant decrease in the pH value of CZGLU, CZSUC, and CZTRE media, with decreases of 1.17, 1.22, and 0.56 pH units, respectively. These differences were not observed 2 days post-incubation, but they persisted at 4 days.
To determine whether the secretion of any organic acids was influenced after the disruption of BbSNF1, aliquots from 3-day-old liquid culture in CZSUC and CZTRE media were analyzed by ion chromatography (Fig. 1c). A significant difference was noted between the wild-type strain and the ΔBbSNF1 mutant in the secretion of organic acids, and the trends of the change are similar between the two media. Compared with the wild-type strain, the ΔBbSNF1 mutant produced more lactic acid, pyruvic acid, and citric acid. Particularly, the wild-type strain secreted hardly any pyruvic acid, but its concentration in CZSUC and CZTRE media when culturing the mutant was 9.05 ± 0.10 and 5.55 ± 0.10 ppm, respectively. For secretion of oxalic acid, the ΔBbSNF1 strain displayed a significant reduction in CZSUC (68 %) and CZTRE (34 %) media compared with the wild-type strain. Meanwhile, the acid content in mycelia also showed a similar trend for the change between two culture media (Fig. 1d). No significantly detectable citrate was found in mycelia of the ΔBbSNF1 mutant. However, the lactate and oxalate contents in the ΔBbSNF1 mutant mycelia were significantly higher than those of the wild-type and the complemented strains. In addition, no significance in the intracellular content of pyruvic acid was noted between the wild-type and ΔBbSNF1 strains.
BbSNF1 contributes to blastospore development
In liquid culture, B. bassiana produces the single yeast-like cells termed blastospores. The blastospore yield was measured as spore number per milliliter of medium (mean ± SD) (Fig. 2a). The blastospore yield of the wild-type strain differed with medium, ranging from 6.23 ± 0.33 (PEPFRU medium) to 10.22 ± 0.46 × 106 (PEPSUC medium). The disruption of BbSNF1 caused a significant reduction (80–94 %) in blastospore production, with the greatest reduction in PEPMal medium. Additionally, the ΔBbSNF1 mutant had a significant reduction in biomass in each medium compared with the wild-type and complemented strains (Fig. 2b). Microscopic examination of the blastospores of the ΔBbSNF1 mutant indicated some alterations in the morphology of these cells (Fig. S4 in Supplementary Material). To examine any such changes more quantitatively, the size and complexity of blastospore were examined by flow cytometry using FSc and SSc detectors, respectively. These data indicated that blastospores in each medium could be sorted into two groups, i.e., group A (small) and group B (large). The BbSNF1-disrupted strain showed a significant increase in percentage of group A cells grown in PEPGLU, PEPTRE, and PEPFRU media, compared with the wild-type strain and complemented strains, indicating that the size of blastospores had decreased. However, no morphologic difference between wild-type and mutant blastospores was noted from PEPSUC and PEPMAL media (Fig. 2c).

Blastospore development of Beauveria bassiana wild-type, ΔBbSNF1, and complemented strains. a Blastospore yield. The indicated strains were grown in minimal medium (using 0.5 % peptone as the nitrogen source) containing glucose, sucrose, trehalose, maltose, or fructose as the sole carbon source. Blastospore production was measured after 3 days of growth in liquid medium, and data are presented as the number of blastospores per milliliter. b The biomass of mycelia grown in the each medium as listed in Fig. 2a. c Distribution of the blastospore population. On the basis of their size, the blastospore population from different media could be sorted into two groups (groups A and B) by flow cytometry analysis. The percentage of small blastospores (group A) was used as a representative index to evaluate the change in blastospore size. Asterisks on bars indicate a significant difference between the ΔBbSNF1 mutant and wild-type or complemented strain (Tukey’s HSD: P < 0.05). Error bars: standard deviation
Microscopic observations of conidial germination and hyphal growth within the first 24 h of cultivation in PEPTRE medium indicated that both the wild-type and ΔBbSNF1 strains had similar germination and initial development (Fig. 3a). After 36–48 h of growth, however, the ΔBbSNF1 mutant produced an attenuated blastospore-forming structure with distorted budding sites (the point between the blastospore and blastospore-forming structure). After 60–72 h of growth, the wild-type strain produced abundant numbers of blastospores, whereas few blastospores were observed in mutant cultures, and abnormal structures were observed. Hyphal bodies (also known as in vivo blastospores) are cells that are produced during fungal growth inside the insect hemocoel and can be isolated from infected hosts as described in the “Materials and methods” section. The conidia of both the wild-type and ΔBbSNF1 strains could germinate and form hyphal bodies in host hemocoel (Fig. 3b). No significant differences were seen in morphology of hyphal bodies from the wild-type and ΔBbSNF1 strains. However, fewer budding hyphal bodies (with one or two daughter cells at the end of the hyphal body) were observed in the mutant strain than in the wild-type and complement strains. Additionally, melanized points could be seen in some hemocytes from ΔBbSNF1 mutant samples, but few hemocytes with melanotic points were found in hemolymph challenged by the wild-type or complement strain.

Microscopic view of blastospore development in the wild-type, ΔBbSNF1, and complemented strains. a Development of in vitro blastospores. Conidia from indicated strains were inoculated into Czapek-Dox medium with the nitrogen source replaced by 0.5 % peptone and the carbon source was trehalose (the PEPTRE medium) and grown for 3 days at 25 °C. The growing cultures were sampled every 12 h and examined under light microscopy. The blastospore-producing structures are noted with arrows. b Development of in vivo blastospores. Hyphal bodies were harvested from the hemocoel of greater wax moth challenged by injecting conidia from each indicated strain. The insect hemocytes are designated with arrows, and the letter “M” indicates the melanized points in insect hemocytes. Scale bar: 10 μm
Ablation of BbSNF1 reduces fungal virulence
The effects of BbSNF1 on virulence were determined with two types of insect bioassays. Mortality was measured over a 7-day period, and the results showed that the mortality percentage increased with time post-inoculation (Fig. 4a, b).The LT50 values of the wild-type, ΔBbSNF1, and complement strains were calculated by Probit analysis (Fig. 4c, d).

Virulence bioassay of Beauveria bassiana wild-type, ΔBbSNF1, and complemented strains. Mortality trends of the three strains in a greater wax moth model are shown for the topical (a) and intrahemocoel injection (c) bioassays. The median lethal time (LT50) values in the topical application (b) and intrahemocoel injection (d) assays were estimated by Probit analysis for the indicated strains. Significant differences between the strains are indicated with asterisks on column (Tukey’s HSD: P < 0.05). Error bars: standard deviation. e Representative images for mycosis on cadavers, which were kept in a moist chamber for 1 week at 25 °C. Scale bar: 1 cm
In the topical bioassay, which represents the fungal infection route in natural conditions, the ΔBbSNF1 strain displayed a significant increase in LT50 (7.1 days), as compared with the wild-type (3.7 days) and complemented strains (3.9 days) (Fig. 4c). After maintenance in moist conditions for 1 week, the ΔBbSNF1 mutant produced few mycelia on cadavers compared with the dense growth of the wild-type and complemented strains (Fig. 4e). Intrahemocoel injection bioassays, which represent an infection route without cuticle penetration, were used to examine pathogen-host interactions in the host hemocoel. The LT50 of the ΔBbSNF1 mutant increased to 5.2 days, whereas that of the wild-type strain and complemented strains was 4 and 4.2 days, respectively, indicating a ~25 % decrease in the time to kill hosts after injection (Fig. 4d).
BbSNF1 regulates gene transcription of cuticle-degrading enzymes and genes in acid biosynthesis and secretion
Quantitative RT-PCR was used to examine the transcriptional regulation of cuticle-degrading genes, including 17 chitinases and 48 proteases. In order to see the change in expression levels, the data were shown for each gene as the ratio wild type/ΔBbSNF1 (Fig. 5). In the presence of chitin as the sole nitrogen source, the following kinds of changes in gene expression could be observed (Fig. 5a). The first class included genes whose expression levels were unaffected in the mutant strain. This only included ChiV8. The second class included genes downregulated in the mutant, including (1) genes with a slight to moderate reduction (50–90 % of the wild-type levels): Chi1, Chi3, ChiEn, ChiIII1, ChiIII3, ChiIII4, ChiV5, and ChiV6; and (2) genes expressing <50 % of the wild-type levels: Chi2, ChiD, ChiV1, ChiV2, and ChiV4. Third, three genes, ChiIII2, ChiV3, and ChiV7, were upregulated in the mutant compared with the wild-type strain, with a fold change from 1.17 to 6.68. On medium with gelatin as the sole nitrogen source, all tested proteases were either upregulated or downregulated (Fig. 5b). The upregulated set (fold change ranging from 1.18 to 3.29) included Asp10, Asp11, Asp13, Asp4, Asp5, Asp7, Cln2a, Mep1b, Mep3, Mep6, Pr1A, Pr1C, Sep2, Tsp2, and Tsp4. Slightly to moderately (expression 50–90 % of the wild-type expression levels) depressed genes included Asp2, Asp3, Asp8, Asp9, Cln2b, Mep1c, Mep1e, Mep5, Pr1F1, Sep1, Sep3, and S53p. The strongly repressed proteases (expression <50 % of the wild-type expression level) included Asp1, Asp12, Asp6, AspA, BssI, Chy, Mep1a, Mep1d, Mep1f, Mep2, Mep4, Pr1F2, Pr1F3, Sep5, Sep6, Tsp1, Tsp3, and Tsp5, with minimal expression of BssI (8.82 %).

The transcriptional analysis of Beauveria bassiana genes encoding hydrolases and genes involved in acid biosynthesis and secretion. Relative transcript levels (RTLs) of genes encoding proteases (a) and chitinases (b) were analyzed in the wild-type and ΔBbSNF1 mutant strains after 3 days of growth on the medium using gelatin and chitin as the sole nitrogen source, respectively. RTL values of genes encoding monocarboxylic acid transporters (MCTs) (c) and genes involved in acid biosynthesis (d) were analyzed in the wild-type and ΔBbSNF1 mutant strains after 3 days of growth in the minimal broth containing either sucrose or trehalose as the sole carbon source
To probe the potential target transporters under the control of BbSNF1, the transcriptional level of 14 monocarboxylate transpoter genes (Mct) was evaluated in mycelia grown in CZSUC and CZTRE media (Fig. 5c). Except Mct2, whose expression levels were unaffected in the two media, the expression of transporters was influenced in at least one of the two media. Six transporter genes, i.e., Mct1, Mct3, Mct4, Mct5, Mct13, and Mct14, were upregulated (1.10- to 5.79-fold) in the mutant compared with the wild-type strain in two media. In CZSUC medium, Mct6, Met7, and Mct11 were downregulated, and the expression of the Mct8 and Mct10 was enhanced to 1.59 and 4.26 times the wild-type levels, respectively; however, all five of these genes were downregulated in CZTRE medium. The expression of Mct9 and Mct12 in the mutant stain was upregulated in CZTRE medium but repressed in CZSUC medium. As shown in Fig. 5d, Ldh1 expression in the ΔBbSNF1 mutant was notably induced approximately eight times in the two media, and the gene Ldh2 and Oah expression levels were enhanced in at least one of the two media. However, the expression of gene Cts did not change in the two media.
Global expression analysis
In order to explore global expression consequences of the loss of BbSNF1 and to identify potential gene targets regulated by BbSNF1 during blastospore development, two sets of genome-wide expression analyses using high-throughput sequencing (RNA-seq) were performed, i.e., comparative analysis of the ΔBbSNF1 mutant to wild-type strains grown in PEPGLU (set 1) and PEPTRE (set 2) media, using glucose and trehalose as the carbon sources, respectively. In PEPGLU medium, the loss of BbSNF1 resulted in altered expression of 228 genes, with 165 upregulated (~1.6 % of the genome) and 63 downregulated (~0.6 % of the genome) genes in the mutant compared with the wild-type strain (Fig. 6a and Tables S4 and S5 in Supplementary Material). In PEPTRE medium, the disruption of BbSNF1 caused changed expression of 286 genes, including 220 upregulated (2.1 % of the genome) and 66 downregulated (~0.6 % of the genome) genes compared with the wild-type strain (Fig. 6a and Tables S6 and S7 in Supplementary Material). Between the two comparison sets, 106 overlapping genes were upregulated and 24 genes were downregulated. One gene (BBA_08131) was downregulated in PEPGLU but upregulated in PEPTRE (Fig. 6b).

Summary of RNA-seq data and Functional Catalog (FunCat) analysis. a Differentially expressed genes (DEGs) were determined by comparing paired libraries, i.e., WTGLU/ΔBbSNF1 GLU and WTTRE/ΔBbSNF1 TRE. b Venn diagram illustrating the number of overlapping genes between library pairs, i.e., WTGLU/ΔBbSNF1 GLU and WTTRE/ΔBbSNF1 TRE. Green indicates the downregulated genes and red indicates upregulated genes. c FunCat sorting of DEGs. FunCat analysis was performed for all DEGs from each comparison set, i.e., WTGLU/ΔBbSNF1 GLU and WTTRE/ΔBbSNF1 TRE (Color figure online)
During growth in PEPGLU medium, the loss of BbSNF1 resulted in 63 downregulated genes (log2 ratio changes from −4.41 to −1.60); among these genes, orthologs for 46 genes could be identified in N. crassa (Table S4a, b in Supplementary Material), and 43 could be sorted into FunCat categories. Of the 165 upregulated genes, orthologs for 67 genes were identified in N. crassa, and 57 could be sorted into FunCat categories. The FunCat analysis showed that downregulated genes included those associated with metabolism, energy, cell transport, defense and virulence, interactions with the environment, and cell type differentiation (Fig. 6c and Table S4c in Supplementary Material). Eight downregulated transcripts displayed log2 ratio (ΔBbSNF1/wild type) changes of >3, i.e., greater than an eightfold decrease in gene expression levels. These genes included two hydrolases, one transporter, and an alcohol dehydrogenase. Overall, downregulated transcripts were involved in the generation of cell energy, including a number of genes crucial for glycolysis and gluconeogenesis (e.g., malate synthase, alcohol dehydrogenase, and oxalate decarboxylase); cell defense and virulence, including a number of acid protease, multicopper oxidase and alcohol dehydrogenase; cellular transport, including a number of ZIP Zinc transporter and Na/K ATPase alpha 1 subunit; and cell differentiation, including a number of cell wall proteins (e.g., cell surface protein, Mmc protein, and allergen-like protein) and development regulatory proteins (e.g., cyclin-domain-containing protein and FoabaA-like protein). In addition, several genes involved in lipid metabolism were also found to be downregulated, including lipase, coenzyme A transferase, and lipocalin. Upregulated genes in the ΔBbSNF1 mutant were largely represented within metabolism, as well as protein fate, and cell death in FunCat analysis (Fig. 6c and Table S5a, b, c in Supplementary Material). These included a large number of proteins involved in amino acid metabolism (e.g., tyrosinase, aminotransferase, and asparagine synthetase), and secondary metabolite production (e.g., oxaloacetate acetylhydrolase, steroid monooxygenase, cytochrome P450, and kynurenine 3-monooxygenase), as well as several hydrolases (e.g., lipase, glucanase, and chitinase) and a few proteins involved in signal perception and transduction (e.g., vacuolar protease and serine/threonine protein kinase).
Comparative analysis of the wild-type and ΔBbSNF1 strains in PEPTRE uncovered significantly altered expression of 286 genes, including 220 upregulated and 63 downregulated genes. The log2 ratio changes of all the repressed genes ranged from −4.22 to −1.37, and orthologs for 50 genes could be identified in N. crassa (Table S6a, b in Supplementary Material). On the basis of FunCat analysis, the downregulated transcripts were mainly associated with metabolism, energy regeneration, transcription, cell transport, and cell rescue; numerous genes are involved in amino acid metabolism and secondary metabolite production (Fig. 6c and Table S6c in Supplementary Material). The genes involved in cell transport included genes that are crucial for electron transport and transport ATPase (e.g., NADPH dehydrogenase, flavohemoglobin, and ATP synthase protein), and these genes were also sorted into energy regeneration. Additionally, another large downregulated gene set included a number of cell-wall-associated proteins (e.g., allergen-like protein, Mmc protein, hydrophobin, and covalently linked cell wall protein), two transcription factors, and two cyclin-like proteins. Genes that were upregulated in the ΔBbSNF1 mutant were largely represented in metabolism, protein fate, and interaction with the environment (Fig. 6c and Table S7a, b, c in Supplementary Material). Genes that were sorted into metabolism consisted of a large number of proteins involved in amino acid metabolism (e.g., tyrosinase and asparagine synthetase) and lipid metabolism (e.g., two short-chain dehydrogenases, two cytochrome P450s, lipase, and steroid monooxygenase), as well as secondary metabolite production (genes that were also involved in lipid metabolism). Moreover, several upregulated transcripts were overrepresented in interaction with environment, including two hydrophobins, heterokaryon incompatibility protein, frequency clock protein, and a vacuolar protease.
Overlapping downregulated genes in the two culture conditions were involved into different functional catalogs (Table 1), including three cell-wall-associated proteins (e.g., allergen-like protein, Mmc protein, and covalently linked cell wall protein), three regulators of development (FoabaA-like protein, cyclin, and serine/threonine-protein kinase), an aquaporin, and several metabolic enzymes (e.g., dienelactone hydrolase and glucanase). BLAST analysis of the overlapping genes against the PHI database identified four genes that are potentially involved in fungal pathogenicity (Table 1).
Discussion
Similar to other SNF1 orthologs, the B. bassiana sucrose non-fermenting protein kinase (BbSNF1) contains two major domains, a catalytic (kinase) domain at its N-terminus region and a regulatory domain at its C-terminus. SNF1 has been called a “fuel gauge and energy regulator” with both nuclear and cytoplasmic targets (Ghillebert et al 2011). In the present study, we probed the function of BbSNF1 using the gene disruption strategy. Our results demonstrate that BbSNF1 plays important roles in regulating B. bassiana carbon/nitrogen utilization, extracellular acidification, development of spores (conidia, in vitro blastospore, and, to a lesser extent, in vivo hyphal body development), and virulence.
In the initial steps of fungal infection, B. bassiana is thought to be under nutrient-limiting stress (Ortiz-Urquiza and Keyhani 2013). BbSNF1 plays an important role in regulating the assimilation of carbon/nitrogen sources. One of the best understood roles of SNF1 is to regulate the expression of the metabolic genes that are repressed by glucose in yeast (Ghillebert et al. 2011). Although BbSNF1 has a high sequence identity with other SNF1 genes, its roles are not the same as those in S. cerevisiae and some filamentous fungi. As in yeast, SNF1 controls the ability to produce the invertase for sucrose utilization, and the yeast SNF1 mutant cannot grow on sucrose as the sole carbon source (Carlson et al. 1981). However, our data show that the B. bassiana ΔBbSNF1 mutant could still grow on sucrose. This result was similar with the observations in the Magnaporthe oryzae and G. zeae SNF1 mutants (two plant pathogens) (Lee et al. 2009; Yi et al. 2008). Moreover, BbSNF1 was required for vegetative growth on the defined medium with glucose as the sole carbon source. This similar result was observed in M. oryzae, where the growth rate was reduced by half on glucose-containing medium (Yi et al. 2008). Furthermore, SNF1 appears to differentially control the utilization of complex carbon sources (e.g., pectin and cellulose) in different filamentous phytopathogenic fungi (Ospina-Giraldo et al. 2003; Yi et al. 2008; Zhang et al. 2013b). In P. digitatum, the ablation of PdSNF1 resulted in a significant decrease in vegetative growth on medium with pectin as the sole carbon source (Zhang et al. 2013b). For two other plant pathogens, V. dahliae and C. carbonum, however, SNF1 mutants only displayed partial defects in growth rate on pectin (Tonukari et al. 2000; Tzima et al. 2011). Our results show that the BbSNF1 is also required for growth on the media with the chitin or gelatin as the sole nitrogen source. This agreed with further transcriptional analysis of genes encoding cuticle-degrading enzymes and indicated that the BbSNF1 is crucial for transcriptional activation of most proteases and chitinases. Because the ΔBbSNF1 mutant produced fewer cuticle-degrading enzymes than the wild-type strain, it was suggested that the reduced ability of the mutant to use complex nitrogen sources in insect cuticles is related to its reduced fungal virulence. Similar suggestions have been made for SNF1 mutants of the plant pathogens F. oxysporum, G. zeae, C. carbonum, and V. dahliae when they were grown on the medium containing complex carbon sources (Lee et al. 2009; Ospina-Giraldo et al. 2003; Tonukari et al. 2000; Tzima et al. 2011 ). These data suggest a general metabolic impairment in the absence of SNF1 that is needed for fungal infection of a host.
In addition to its role in regulating the assimilation of carbon/nitrogen sources, BbSNF1 also regulates the extracellular acidification. The ablation of BbSNF1 resulted in a reduced pH value in culture media. Chemical analysis indicated that the secretion of lactic, pyruvic, and citric acid was enhanced in the ΔBbSNF1 mutant, but the secretion of oxalic acid was repressed, indicating that the BbSNF1 controls the secretion of organic acids with different mechanisms. Oxalate had been characterized as an important toxic factor in the virulence of B. bassiana (Kirkland et al. 2005). This result indicated that the reduced ability of the mutant to secrete oxalic acid might be another factor accounting for the reduced fungal virulence. SNF1 might be a negative regulator of lactate production in B. bassiana. Similar results were observed in rat skeletal muscle cells in which activated AMPK (ortholog of SNF1 in animals) could reduce the lactate production (Ceddia and Sweeney 2004). Filamentous fungi could quickly acidify the medium and significantly lower its pH value when cultured in medium without buffering agents. The mechanism underlying the acidification is mainly due to organic acid excretion via carboxylic transporters (Vrabl et al. 2012). As seen with the MCTs, a general enhancement of most MCTs was observed in the ΔBbSNF1 mutant with the exception of the MCT6 gene. These results suggest that the MCT6 is a potential transporter for oxalate secretion and that the other enhanced MCTs are involved in the transport of lactic, pyruvic, and citric acids. The enhancement and repression of transporters might result in the enhanced secretion (citrate) and accumulation (oxalate) of intracellular acids, respectively. The pathway involved in the secretion of organic acids in fungi is still not completely elucidated (Vrabl et al. 2012), but our results at least partly account for SNF1 regulation of extracellular acidification in B. bassiana by transcriptional control of monocarboxylate transporters. Regarding virulence, the ΔBbSNF1 strain displayed a double LT50 in topical bioassays, which represent the natural route of infection. Our data suggested that the main cause for the decreased virulence in the mutant is likely due to the impaired fungal ability to penetrate the cuticle (defective in the secretion of cuticle-degrading enzymes), and repressed oxalate secretion.
Another well-known role of SNF1 in filamentous fungi is the regulation of conidial development, which was established in several phytopathogenic fungi. Deletion mutants of G. zeae SNF1 lost the ability to form the perithecia and ascospores (Lee et al. 2009), and the ablation of P. digitatum SNF1 also resulted in impaired conidiation (Zhang et al. 2013b). As expected, the BbSNF1 is also involved in conidiation on rich and defined media. Although aspects of conidial development might be shared with other filamentous fungi, little is known about the role of BbSNF1 in the regulation and development of the blastospores. In addition, upon entering into the insect hemocoel, the B. bassiana undergoes dimorphic conversion from mycelia, and this morphologic change facilitates fungal propagation in early stages of invasion (Lewis et al. 2009; Wanchoo et al. 2009). In C. albicans (human pathogen), Magnaporthe grisea (plant pathogen), and Ustilago maydis (plant pathogen), however, dimorphic transitions from budding to filamentous growth have been considered as principle factors determining fungal invasion and colonization (Palecek et al. 2002). Our results show that BbSNF1 is critical to blastospore development of B. bassiana and fungal virulence. BbSNF1 activates the pathway for blastospore development, maintaining normal morphology of blastospore-producing structures and blastospore size. Little is known about the pathway(s) leading to blastospore development, although a G-protein-coupled receptor is also required to maintain the normal morphology and function of blastospore-forming structures (Ying et al. 2013). Whether BbSNF1 is a downstream target of this signaling pathway remains to be determined. However, the morphology of the in vivo hyphal bodies was unaffected in the ΔBbSNF1 mutant, indicating that BbSNF1 does not regulate the morphology of these cells, although a decrease in the number of in vitro blastospores was evident which could be simply due to the slower growth rate of the mutant. This compromised ability might be responsible for the small decrease in virulence that was observed in the intrahemocoel assays (~20 %); the effect was not as great as that in the topical bioassay (~75 % reduction).
Global gene expression analysis (RNA-seq), performed in two culture media (PEPGLU and PEPTRE), was used to develop a map of varied gene expression and obtain clues for potential target genes of BbSNF1 during blastospore development. In terms of physiology, BbSNF1 may contribute to blastospore development via mediation of the metabolic process, energy regeneration, transcription, and cellular transport. Polyamine is required for dimorphism in the human pathogenic fungus, P. marneffei. The disruption of the spermidine biosynthetic pathway blocked the production of the yeast form from the filamentous form (Kummasook et al. 2013). Similarly, ornithine decarboxylase, a committed enzyme in polyamine synthesis, was also downregulated in the BbSNF1 mutant grown in PEPGLU, but not in PEPTRE. Other affected genes included those associated with cell cycle and the cell wall and membrane function, some of which are potential virulence-related genes. One transcription factor, AbaA, which is potentially involved in blastospore development, was affected by BbSNF1. The transcription factors AbaA and Tec1 belong to the same family, and they play crucial roles in filamentous growth in S. cerevisiae and conidiophore formation in Aspergillus nidulans, respectively. In yeast, Ste12 controls Tec1 transcription, and their collaboration is required for pseudohyphal growth. As in A. nidulans, AbaA expression is regulated by StuA (Liu 2001). In S. cerevisiae, SNF1 has been linked to transcription of FLO11, which encodes a cell surface glycoprotein that is indispensable in filamentous invasive growth (Kuchin et al. 2002; Vyas et al. 2003). Similarly, BbSNF1 regulates the transcription of several cell wall proteins in blastospore formation. As mentioned, previous studies indicated a G protein signaling pathway and proteins involved in autophagy to play important roles in regulating blastospore formation in B. bassiana (Ying et al. 2013; Zhang et al. 2013a); however, no overlapping genes were noted between G-protein-coupled receptor and SNF1 transcriptomic data. These data suggest that BbSNF1 may contribute to blastospore development via a distinct pathway.
In conclusion, our studies show that SNF1 regulates development, extracellular acidification, and virulence in B. bassiana. BbSNF1 contributes to the transcriptional regulation of extracellular hydrolases and genes involved in extracellular acidification. In addition to conidiation, we also dissected the roles of BbSNF1 in blastospore development. BbSNF1 modulates production and cell size of in vitro blastospores, but the morphology of the in vivo hyphal bodies was unaffected in the mutant, suggesting that the morphology of these cells might be controlled by a distinct pathway. Because SNF1 is a protein kinase, further work recognizing the direct phosphorylated targets of BbSNF1 will reveal the mechanism behind the diverse functions of the SNF1/AMPK kinase.
References
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188
Carlson M, Osmond BC, Botstein D (1981) Mutants of yeast defective in sucrose utilization. Genetics 98:25–40
Ceddia RB, Sweeney G (2004) Creatine supplementation increases glucose oxidation and AMPK phosphorylation and reduces lactate production in L6 rat skeletal muscle cell. J Physiol 555:409–421
Cho EM, Liu L, Farmerie W, Keyhani NO (2006) EST analysis of cDNA libraries from the entomopathogenic fungus Beauveria (Cordyceps) bassiana. I. Evidence for stage-specific gene expression in aerial conidia, in vitro blastospores and submerged conidia. Microbiology 152:2843–2854
Fan YH, Pereira RM, Kilic E, Casella G, Keyhani NO (2012) Pyrokinin β-neuropeptide affects necrophoretic behavior in fire ants (S. invicta), and expression of β-NP in a mycoinsecticide increases its virulence. PLoS One 7:e26924
Fang WG, Zhang YJ, Yang X, Zheng X, Duan H, Li Y, Pei Y (2004) Agrobacterium tumefaciens-mediated transformation of Beauveria bassiana using an herbicide resistance gene as a selection marker. J Invertebr Pathol 85:18–24
Fang WG, Leng B, Xiao YH, Jin K, Ma JC, Fan YH, Feng J, Yang XY, Zhang YJ, Pei Y (2005) Cloning of Beauveria bassiana chitinase gene Bbchit1 and its application to improve fungal strain virulence. Appl Environ Microbiol 71:363–370
Feng MG, Poprawski TJ, Khachatourians GG (1994) Production, formulation and application of the entomopathogenic fungus Beauveria bassiana for insect control: current status. Biocontrol Sci Tech 4:3–34
Ghillebert R, Swinnen E, Wen J, Vandesteene L, Ramon M, Norga K, Rolland F, Winderickx J (2011) The AMPK⁄SNF1⁄SnRK1 fuel gauge and energy regulator: structure, function and regulation. FEBS J 278:3978–3990
Han T-L, Cannon RD, Villas-Bôas SG (2011) The metabolic basis of Candida albicans morphogenesis and quorum sensing. Fungal Genet Biol 48:747–763
Hedbacker K, Carlson M (2008) SNF1/AMPK pathway in yeast. Front Biosci 13:2408–2420
Holder DJ, Keyhani NO (2005) Adhesion of the entomopathogenic fungus Beauveria (Cordyceps) bassiana to substrata. Appl Environ Microbiol 71:260–5266
Holder DJ, Kirkland BH, Lewis MW, Keyhani NO (2007) Surface characteristics of the entomopathogenic fungus Beauveria (Cordyceps) bassiana. Microbiology 153:3448–3457
Kirkland BH, Eisa A, Keyhani NO (2005) Acid as a fungal acaracidal virulence factor. J Med Entomol 42:346–351
Kuchin S, Vyas VK, Carlson M (2002) Snf1 protein kinase and the repressors Nrg1 and Nrg2 regulate FLO11, haploid invasive growth, and diploid pseudohyphal differentiation. Mol Cell Biol 22:3994–4000
Kummasook A, Cooper CR Jr, Sakamoto A, Terui Y, Kashiwagi K, Vanittanakom N (2013) Spermidine is required for morphogenesis in the human pathogenic fungus, Penicillium marneffei. Fungal Genet Biol 58–59:25–32
Lee S-H, Lee J, Lee S, Park E-H, Kim K-W, Kim M-D, Yun S-H, Lee Y-W (2009) GzSNF1 is required for normal sexual and asexual development in the ascomycete Gibberella zeae. Eukaryot Cell 1:116–127
Lewis MW, Robalino IV, Keyhani NO (2009) Uptake of the fluorescent probe FM4-64 by hyphae and haemolymph-derived in vivo hyphal bodies of the entomopathogenic fungus Beauveria bassiana. Microbiology 155:3110–3120
Liu H (2001) Transcriptional control of dimorphism in Candida albicans. Curr Opin Microbiol 4:728–735
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT. Methods 25:402–408
Ortiz-Urquiza A, Keyhani NO (2013) Action on the surface: entomopathogenic fungi versus the insect cuticle. Insects 4:357–374
Ospina-Giraldo MD, Mullins E, Kang S (2003) Loss of function of the Fusarium oxysporum SNF1 gene reduces virulence on cabbage and Arabidopsis. Curr Genet 44:49–57
Palecek SP, Parikh AS, Kron SJ (2002) Sensing, signalling and integrating physical processes during Saccharomyces cerevisiae invasive and filamentous growth. Microbiology 148:893–907
Pedrini N, Ortiz-Urquiza A, Huarte-Bonnet C, Zhang S, Keyhani NO (2013) Targeting of insect epicuticular lipids by the entomopathogenic fungus Beauveria bassiana: hydrocarbon oxidation within the context of a host-pathogen interaction. Front Microbiol 4:24
Pendland JC, Hung SY, Boucias DG (1993) Evasion of host defense by in vivo-produced protoplast-like cells of the insect mycopathogen Beauveria bassiana. J Bacteriol 175:5962–5969
Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Güldener U, Mannhaupt G, Münsterkötter M, Mewes HW (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res 32:5539–5545
Şahil D, Otag F (2013) Filamentous fungi isolated from clinical samples stored for a long time in the sand. Clin Microb 2:e104
Seip J, Jackson R, He H, Zhu Q, Hong S-P (2013) Snf1 is a regulator of lipid accumulation in Yarrowia lipolytica. Appl Environ Microbiol 79:7360–7370
Strauss J, Horvath HK, Abdallah BM, Kindermann J, Mach RL, Kubicek CP (1999) The function of CreA, the carbon catabolite repressor of Aspergillus nidulans, is regulated at the transcriptional and post-transcriptional level. Mol Microbiol 32:169–178
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A (2011) Differential expression in RNA-seq: A matter of depth. Genome Res 21:2213–2223
Tonukari NJ, Scott-Craig JS, Walton JD (2000) The Cochliobolus carbonum SNF1 gene is required for cell wall-degrading enzyme expression and virulence on maize. Plant Cell 12:237–247
Tzima AK, Paplomatas EJ, Rauyaree P, Ospina-Giraldo MD, Kang S (2011) VdSNF1, the sucrose nonfermenting protein kinase gene of Verticillium dahliae, is required for virulence and expression of genes involved in cell-wall degradation. Mol Plant-Microbe Interact 24:129–142
Vrabl P, Fuchs V, Schinagl CW, Burgstaller W (2012) Organic acid excretion in Penicillium ochrochloron increases with ambient pH. Front Microbiol 3:121
Vyas VK, Kuchin S, Berkey CD, Carlson M (2003) Snf1 kinases with different β–subunit isoforms play distinct roles in regulating haploid invasive growth. Mol Cell Biol 23:1341–1348
Wanchoo A, Lewis MW, Keyhani NO (2009) Lectin mapping reveals stage-specific display of surface carbohydrates in in vitro and haemolymph-derived cells of the entomopathogenic fungus Beauveria bassiana. Microbiology 155:3121–3133
Xiao G, Ying SH, Zheng P, Wang ZL, Zhang S, Xie XQ, Shang Y, St. Leger RJ, Zhao GP, Wang C, Feng MG (2012) Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci Rep 2:483
Xie XQ, Li F, Ying SH, Feng MG (2012) Additive contributions of two manganese-cored superoxide dismutases (MnSODs) to antioxidation, UV tolerance and virulence of Beauveria bassiana. PLoS One 7:e30298
Yi M, Park J-H, Ahn J-H, Lee Y-H (2008) MoSNF1 regulates sporulation and pathogenicity in the rice blast fungus Magnaporthe oryzae. Fungal Genet Biol 45:1172–1181
Ying S-H, Feng M-G, Keyhani NO (2013) A carbon responsive G-protein coupled receptor modulates broad developmental and genetic networks in the entomopathogenic fungus, Beauveria bassiana. Environ Microbiol 15:2902–2921
Zhang YJ, Feng MG, Fan YH, Luo ZB, Yang XY, Wu D, Pei Y (2008) A cuticle-degrading protease (CDEP-1) of Beauveria bassiana enhances virulence. Biocontrol Sci Tech 18:551–563
Zhang S, Widemann E, Bernard G, Lesot A, Pinot F, Pedrini N, Keyhani NO (2012) CYP52X1, representing a new cytochrome P450 subfamily, displays fatty acid hydroxylase activity and contributes to virulence and growth on insect cuticular substrates in the entomopathogenic fungus Beauveria bassiana. J Biol Chem 287:13477–13486
Zhang L, Wang J, Xie XQ, Keyhani NO, Feng MG, Ying SH (2013a) The autophagy gene BbATG5, involved in the formation of the autophagosome, contributes to cell differentiation and growth but is dispensable for pathogenesis in the entomopathogenic fungus Beauveria bassiana. Microbiology 59:243–252
Zhang T, Sun X, Xu Q, Zhu C, Li Q, Li H (2013b) PdSNF1, a sucrose non-fermenting protein kinase gene, is required for Penicillium digitatum conidiation and virulence. Appl Microbiol Biotechnol 97:5433–5445
Acknowledgments
We thank Prof. Nemat O. Keyhani (University of Florida, USA) for his kind help with the manuscript preparation. We are grateful to Dr. She-long Zhang (Equipment and Technology Service Platform, College of Life Sciences, Zhejiang University) and Mrs. Li-juan Mao (985-Institute of Agrobiology and Environmental Sciences, Zhejiang University) for their kind assistance in taking micrographs and using ion-exchange chromatography, respectively. This study was supported jointly by two grants (31371990 and 31171898) from the Natural Science Foundation of China.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, XX., He, PH., Feng, MG. et al. BbSNF1 contributes to cell differentiation, extracellular acidification, and virulence in Beauveria bassiana, a filamentous entomopathogenic fungus. Appl Microbiol Biotechnol 98, 8657–8673 (2014). https://doi.org/10.1007/s00253-014-5907-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5907-0