
Abstract
Fusarium oxysporum pathogenicity is believed to require the activity of cell wall-degrading enzymes. Production of these enzymes in fungi is subject to carbon catabolite repression, a process that in yeast is mostly controlled by the SNF1 (sucrose non-fermenting 1) gene. To elucidate the role of cell wall-degrading enzymes in F. oxysporum pathogenicity, we cloned and disrupted its SNF1 homologue (FoSNF1). The fosnf1 mutants had a reduced expression of several genes encoding cell wall-degrading enzymes and grew poorly on certain carbon sources. Infection assays on Arabidopsis thaliana and Brassica oleracea revealed that progression of wilt symptoms in plants infected by fosnf1 mutants was considerably delayed, in comparison with those infected by a wild-type strain. In conclusion, mutations in FoSNF1 prevent F. oxysporum from properly derepressing the production of cell wall-degrading enzymes, compromise the utilization of certain carbon sources, and reduce its virulence on A. thaliana and B. oleracea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diseases caused by fungal pathogens pose a serious challenge to modern agriculture. One such pathogen is Fusarium oxysporum, an economically important soil-borne phytopathogen that causes vascular wilt disease in over 100 cultivated species throughout the world (Armstrong and Armstrong 1981). Following penetration into the host root system, the fungal hyphae advance towards the vascular tissues (Deese and Stahmann 1962). Mycelia remain exclusively within the xylem, migrating upwards toward the stem and crown of the host, leading to a severe restriction in vascular system function (Agrios 1997). Factors such as vessel clogging by mycelium, spores, and tyloses generate a water imbalance that gives rise to a rapid water loss, stomata closure, and leaf death, which is typically followed by plant collapse (Agrios 1997). Alternatively, the fungus can cause rotting of seeds and seedlings and rotting of roots, stems, crowns, and underground organs (Johnston et al. 1979; Moorman and Klemmer 1980). During the progression of disease, it is likely that degradative enzymes such as cellulase and polygalacturonase play an important role, individually or as a group, by assisting the fungus in breaching the plant′s major physical barrier, the cell wall.
Although the importance of cell wall-degrading enzymes for fungal pathogenicity has been hypothesized, direct evidence supporting such hypothesis is limited (Walton 1994). Due to the genetic redundancy of fungal genes encoding cell wall-degrading enzymes (Sweigard et al. 1992; Gao et al. 1996; Wu et al. 1997; Scott-Craig et al. 1998; García-Maceira et al. 2000), mutagenesis of individual genes may not cause obvious defects in pathogenicity. For example, deletion mutants in pgx4, a gene encoding exopolygalacturonase in F. oxysporum, maintained a highly pathogenic phenotype and possessed residual exopolygalacturonase activity (García-Maceira et al. 2000). Furthermore, the disruption of xylanase genes in Cochliobolus carbonum and Magnaporthe grisea did not affect their ability to cause disease (Apel-Birkhold and Walton 1996; Wu et al. 1997).
In systems where glucose is the preferred carbon source, the expression of genes involved in the metabolism of alternative carbon sources (e.g., arabinose, sucrose, galactose, maltose) or non-fermentable carbon sources is inhibited when glucose is available. This process is known as carbon catabolite repression or glucose repression; and it appears to control the expression of most genes encoding cell wall-degrading enzymes (Tonukari et al. 2000). Thus, disruption of the elements that control the derepression mechanism could lead to the loss of (or reduced) expression of cell wall-degrading enzymes, potentially permitting a test of the role of cell wall-degrading enzymes as a whole in fungal pathogenicity (Tonukari et al. 2000).
The SNF1 (sucrose non-fermenting 1) gene has been shown to play a central role in carbon catabolite repression in Saccharomyces cerevisiae (Carlson et al. 1981) and is required for invasive growth during glucose starvation (Palecek et al. 2002). In the absence of glucose, SNF1 causes the derepression of SUC2, a gene encoding a secreted invertase that hydrolyzes sucrose to fructose and glucose, thus allowing yeast to utilize sucrose as a carbon source. SNF1 is a protein kinase that phosphorylates MIG1, a DNA-binding transcriptional repressor (Treitel et al. 1998), leading to the derepression of all genes under MIG1 control. A recent report (Lo et al. 2001) showed that SNF1 also regulates the transcription of INO1, a gene involved in inositol biosynthesis, in concert with the histone acetyltransferase, GCN5. Phosphorylation of histone H3 serine-10 by SNF1 promotes GCN5-mediated acetylation of lysine-14. Consequently, both events lead to INO1 transcription enhancement. Even under derepressive conditions, mutations in the SNF1 homologue in C. carbonum caused significant reduction in cell wall-degrading enzyme activities and their transcripts (Tonukari et al. 2000). These mutants also exhibited reduced virulence on maize, supporting a role for cell wall-degrading enzymes in virulence. In addition, the mutations also caused reduced growth on certain simple and complex carbon sources, suggesting that the SNF1 homologue is also important for the utilization of these carbon sources.
As part of our efforts to develop the F. oxysporum–Arabidopsis thaliana system as a model for studying soil-borne fungal diseases, we characterized the role of the SNF1 homologue in F. oxysporum pathogenicity and growth. Consistent with the data from C. carbonum (Tonukari et al. 2000), mutations in the SNF1 homologue in F. oxysporum diminish the transcription of genes encoding cell wall-degrading enzymes and reduce its virulence on A. thaliana and Brassica oleracea.
Materials and methods
Fungal cultures, media, and growth conditions
F. oxysporum strain O-685 was obtained from the Fusarium Research Center (FRC) at The Pennsylvania State University and maintained as a monoconidial culture at −80 °C. Growth reactivation was done by culturing the fungus on 1% water/agar supplemented with irradiated carnation leaf-pieces (Fisher et al. 1982). The fungus was grown in potato/dextrose broth (PDB) for DNA extraction and on potato/dextrose agar (PDA) plates for single-spore isolation. The radial growth rate (diameter of colonies) on different carbon sources was measured by culturing the fungus (three replicates) on minimal medium containing 1.5% agar (Difco, Detroit, Mich.) and supplemented with 2% (w/v) arabinose, fructose, galactose, glucose, pectin, trehalose, xylan, or xylose (Sigma, St. Louis, Mo.). The biomass of fungal cultures in liquid minimal medium (50 ml) supplemented with 2% (w/v) of the same carbon sources (as listed above) was measured after 5 days of growth. Plates and flasks were inoculated with 5 µl of a conidial suspension (104 conidia ml−1) and incubated at 28 °C (plates) or room temperature (flasks). For gene expression analysis, fungal cultures were grown in PDB for 4 days at room temperature with mild agitation (50 rpm). The fungal mycelium was harvested by filtration, washed several times with deionized water, fragmented in a blender, transferred to liquid minimal medium supplemented with 1% (w/v) polygalacturonic acid (Sigma), and grown for an additional 40 h.
Nucleic acid manipulations
Total RNA from fungal mycelium was extracted with TRI reagent (MRC, Cincinnati, Ohio), following the provider′s protocol, and then separated on a gel and transferred onto Hybond-N+ hybridization membranes (Amersham Pharmacia Biotech, Amersham, UK). Northern hybridizations were done at 65 °C, using a modified Church and Gilbert (1984) buffer. Probes for exopolygalacturonase, endopolygalacturonase, and pectate lyase were obtained by PCR, using primers designed after sequences available in the databases. Membranes were washed at 65 °C three times (2× SSC, 0.1% SDS, then 1× SSC, 0.1% SDS, finally 0.1× SSC, 0.1% SDS) and exposed to Biomax MS X-ray film (Kodak, Rochester, N.Y.). Fungal DNA was obtained following a modified cetyltrimethylammonium bromide miniprep method (O′Donnell et al. 1997). For constructing a F. oxysporum genomic library, DNA was further purified using Genomic Tip 100/G (Qiagen, Valencia, Calif.). Purified DNA was partially digested with Sau3AI to result in an average size of 15 kb, treated with Klenow fragment in the presence of dGTP and dATP to fill-in the first two nucleotides of the 5′ overhanging ends generated by Sau3AI, and ligated to LambdaGEM-12 XhoI half-site arms (Promega, Madison, Wis.). The library was packaged using the Gigapack III Gold packaging extract (Stratagene, La Jolla, Calif.). The titer of the unamplified library was >1×105 plaques.
DNA sequencing reactions were performed using the BigDye terminator cycle sequencing kit (Applied Biosystems, Foster City, Calif.) and analyzed using an ABI 377 sequencer (Applied Biosystems). Sequence data were analyzed with the Lasergene software package (DNASTAR, Madison, Wis.). Sequence alignment was done following the Clustal W algorithm (Thompson et al. 1994) and phylogenetic relationships were determined following the neighbor-joining algorithm (Saitou and Nei 1987). Phylogenetic trees were validated by a bootstrap test of 1,000 replications and all gaps were excluded from the analysis. Sequence similarity was determined using BLASTX (Altschul et al. 1997) and motifs were identified using PSORT, a computer program for the prediction of protein localization within cells (Nakai and Kanehisa 1992).
Isolation of the SNF1 homologue from F. oxysporum
A portion of the SNF1 gene homologue in F. oxysporum (FoSNF1) was isolated by PCR, using the following degenerate primers based on conserved domains of SNF1 and its homologues in other fungi: SNF1F (5′-CAYCCNCAYATHATHAA-3′, forward primer) and SNF1R (5′-TCNGGNGCNGCRTATTT-3′, reverse primer). PCR conditions were: one cycle of 94 °C for 4 min, 50 °C for 10 s, and 72 °C for 15 s and then 33 cycles of 91 °C for 30 s, 50 °C for 10 s, and 72 °C for 30 s , followed by one cycle of 72 °C for 30 s. The PCR product was cloned into the pGEM-T Easy cloning vector (Promega) for sequence analysis. The lambda genomic library was screened using a PCR fragment corresponding to FoSNF1 as a probe (Sambrook and Russell 2001). DNA from positive lambda clones was prepared using the Lambda kit (Qiagen), following the manufacturer′s directions.
Mutagenesis of FoSNF1
A 2.1-kb EcoRI fragment (Fig. 3), containing part of FoSNF1 [1 kb containing the putative promoter region, plus 1.1 kb of the open reading frame (ORF)], was subcloned into pDHt (Mullins et al. 2001) to generate pSK746. This FoSNF1 fragment in pSK746 was disrupted using an in vitro transposon-mediated insertional mutagenesis technique. The 1.4-kb HpaI fragment of pCB1004 (Carroll et al. 1994), carrying the modified hygromycin B resistance gene (HPH) under the control of the Aspergillus nidulans trpC promoter, was ligated to the SmaI site of the pMOD<MCS> vector (Epicentre Technologies, Madison, Wis.) to construct pSK597. The Tn5 transposon carrying the HPH gene was released from pSK597 by digestion with PvuII and subsequently mobilized to mutagenize pSK746 using EZ::TN transposase (Epicentre Technologies), according to the manufacturer′s directions. Following the transposon mutagenesis, pSK746 was transformed into Escherichia coli, and the resulting transformants were screened by hybridization with the HPH gene as a probe. Approximately 2–3% of the transformants contained pSK746 tagged by the transposon. Tagged pSK746 in these transformants was initially screened by restriction analysis to identify those clones that had an insertion in FoSNF1. Two clones, pSK800 (allele fosnf1-1) and pSK801 (allele fosnf1-2), were sequenced to determine the exact location of insertion by the transposon.
Fungal transformation with pSK800 and pSK801 using Agrobacterium tumefaciens was performed as described by Mullins et al. (2001). Cells of A. tumefaciens AGL-1 carrying each mutant allele were mixed with F. oxysporum O-685 spores and cocultivated on a 0.45-µm sterile membrane filter (Whatman, London, UK) on cocultivation medium for 40 h at 28 °C. After this period, the membrane was transferred to minimal medium containing hygromycin B (50 µg ml−1) as a selection agent for fungal transformants and cefotaxime (200 µM) and moxalactum (100 µg ml−1) to eliminate A. tumefaciens cells. Individual transformants were recovered and single-spore cultures were obtained as described by Mullins et al. (2001). Genomic DNA from the purified transformants was analyzed by Southern hybridization, using the 2.1-kb EcoRI fragment as a probe, to determine whether the FoSNF1 gene had been disrupted.
Plants and growth conditions
Arabidopsis thaliana seeds (Lehle Seed Co., Round Rock, Tex.) were sown onto soil (Redi-Earth Peat Lite Mix; Scotts–Sierra Horticultural Products Co., Marysville, Ohio), pre-wetted with sterile water, in compartmentalized plug trays (Landmark Plastic, Akron, Ohio) and incubated at 3 °C for 3–5 days. Trays were subsequently transferred to a growth chamber with conditions set at 24 °C and ~80% humidity, with a 14-h photoperiod under 150 mol s−1 m−2 light intensity. After germination and before pathogen inoculation, the seedlings were thinned to one seedling per compartment. Seeds of B. oleracea (cabbage) var. Grenadier (Rogers NK Seed Co., Boise, Idaho) were sown under the surface of the same type of soil used for Arabidopsis and grown under identical growth conditions.
Pathogenicity assays
Two inoculation methods were adopted to assess the virulence of O-685 and its fosnf1 mutants on Arabidopsis and cabbage. For the root-dip inoculation (RDI) protocol, 10- to 11-day-old cabbage seedlings were removed from the soil and immersed in a conidial suspension (106 conidia ml−1). While the seedlings were submerged in the suspension, their roots were slightly trimmed using scissors to allow the spores to directly enter into the vascular system. After 1 min immersion in the conidial suspension, the plants were placed back into soil. The soil-injection (SI) method (Halila and Strange 1997; Kroes et al. 1998; Sink and Grey 1999) involved pipetting the inoculum (0.5 ml of a suspension of 106 conidia ml−1) into the soil at the base of the stem of 10- and 23-day-old Arabidopsis and cabbage seedlings, respectively. In both methods, control plants were inoculated with sterile water. Seedlings were not watered for 24 h post-inoculation, to prevent the inoculum being washed from the root zone, but following this period plants were watered every 48 h with sterile water. Disease progress was evaluated by monitoring the plants daily with the aid of the following arbitrary disease index (ADI): class 0 has no visual symptoms, class 1 has <25% of leaves exhibiting typical wilt symptoms, class 2 has 25–50% of leaves exhibiting symptoms, class 3 has 51–75% of leaves exhibiting symptoms, and class 4 has 76–100% of leaves exhibiting symptoms.
All pathogenicity assays were repeated at least twice with a minimum of five seedlings tested per interaction. To determine the degree of fungal colonization within cabbage, plants treated with water, an ectopic transformant, and a fosnf1 mutant were removed, washed free of soil, and surface-sterilized by treating with bleach for 3 min, double-distilled (dd)H2O for 30 s, and 70% ethanol for 2 min, before finally rinsing in ddH2O for a further 30 s. Sections of root, stem, and leaf were placed on PDA plates supplemented with 50 µg hygromycin B ml−1. The combination of hygromycin B and sterilization successfully prevented the growth of other microbes. After 3 days of incubation at 23 °C, plates were scored for fungal growth.
Results
Isolation of a SNF1 homologue (FoSNF1) from F. oxysporum
The conserved regions in S. cerevisiae SNF1 (Celenza and Carlson 1984) and its homologues from C. carbonum (Tonukari et al. 2000), Candida glabrata (Petter and Kwon-Chung 1996), and C. tropicalis (Kanai et al. 1999) were used to design two degenerate primers. Amplification of F. oxysporum genomic DNA by PCR with these primers produced a 357-bp product that exhibited strong similarity to SNF1 and its homologues. This PCR product was subsequently used to isolate the F. oxysporum SNF1 homologue, termed FoSNF1, from a genomic library.
Southern analysis of EcoRI-digested genomic DNA using the 357-bp PCR product as a probe produced a single hybridizing band, suggesting that FoSNF1 is a single-copy gene. FoSNF1 contains a 2,307-bp ORF interrupted by three introns (77, 55, 52 bp), located at positions 393, 1,290 and 1,984, respectively. This ORF encodes a protein (FoSNF1) that consists of 706 amino acids (Fig. 1) and has a predicted molecular mass of 79 kDa. FoSNF1 exhibits its strongest similarity to homologues from the ascomycetes Hypocrea jecorina, Neurospora crassa, and Sclerotinia sclerotiorum. FoSNF1 shares with these proteins 84.2%, 78.3%, and 76.2% identical amino acids, respectively, with the most conserved region located near the N-terminus. This part of FoSNF1, encompassing amino acid residues 67–314, includes the kinase and activation domains (Hardie et al. 1998). The activation domain, where phosphorylation putatively activates the kinase, is 100% identical to the corresponding domains in its homologues. The degree of similarity is lower at the C-terminus, with only a few blocks showing identical residues (Fig. 1). A predictive analysis of protein-sorting signals and localization sites in FoSNF1 using PSORT (Nakai and Kanehisa 1992) showed that FoSNF1 contains three putative nuclear localization signals (Fig. 1) and has a probability of 69.6% of being localized in the nucleus. While SNF1 homologues from N. crassa (NcSNF1), S. sclerotiorum (SsSNF1), and Cochliobolus carbonum (CcSNF1) also have a high probability of being localized in the nucleus (76.7%, 76.7%, 78%, respectively), other SNF1 proteins, including the homologue from H. jecorina (HjSNF1) appeared to lack obvious nuclear localization signals. The high probability that FoSNF1 could be localized in the nucleus is consistent with its likely site of action. One distinct feature of FoSNF1 is the presence of a stretch of glutamine residues (24–36) in its N-terminus (Fig. 1). The glutamine-rich domain usually mediates transcriptional activation by several eukaryotic transcription factors, such as Sp1 or Oct1, by interacting with other components of the transcription machinery (Escher et al. 2000). At the similar location, SNF1 proteins from Saccharomyces cerevisiae (ScSNF1) and Candida spp contain a histidine-rich domain. The remaining SNF1 homologues lack such repeats.

Sequence comparison of FoSNF1 with four of its fungal homologues reported in GenBank. Residues identical in all proteins are indicated with an asterisk. Residues for the kinase domain in FoSNF1 are in bold and those for the activation segment are underlined. One of the three putative nuclear localization signals (amino acids 521–527) is italicized. Dashes indicate gaps introduced in the amino acid sequences during the Clustal alignment. The nucleotide sequence of FoSNF1 has been deposited in GenBank under accession number AF420488. C. Cochliobolus, F. Fusarium, H. Hypocrea, S. (upper) Saccharomyces, S. (lower) Sclerotinia
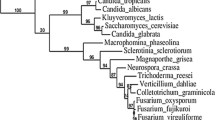
A phylogenetic analysis using the neighbor-joining method revealed that SNF1 from F. oxysporum and H. jecorina share the most recent ancestor among all the SNF1 homologues of fungal origin included in the protein alignment (Fig. 2). CcSNF1 appears to be more distantly related, although it is still part of the same cluster. A sister group is constituted by ScSNF1 and the homologues from Candida spp. This tree topology is supported by bootstrap values greater than 99%.

Phylogenetic analysis of fungal SNF1 proteins. The phylogenetic tree was obtained by the neighbor-joining method. The total number of amino acid residues used for the phylogenetic inference was 525, after gaps were deleted from the analysis. The numbers at nodes represent the values obtained after a bootstrap test with 1,000 replications
Targeted mutagenesis of FoSNF1
To construct mutant alleles of FoSNF1 for gene replacement, we employed a mutagenesis protocol in which FoSNF1 cloned in the Agrobacterium binary vector pDHt (Mullins et al. 2001) was randomly tagged by a modified Tn5 transposon carrying the hygromycin B resistance gene. Two different insertional mutant alleles of FoSNF1 (Fig. 3A), designated fosn1-1 and fosnf1-2, were generated using this technique, with transposon insertion occurring 136 bp and 222 bp downstream, respectively, from the start codon of FoSNF1. Both mutant alleles were introduced via A. tumefaciens-mediated transformation (Mullins et al. 2001), into O-685, a F. oxysporum strain pathogenic on the cruciferous plants A. thaliana and B. oleracea. A total of 52 hygromycin B-resistant transformants (35 with fosnf1-1, 17 with fosnf1-2) were isolated and subsequently purified through single-spore isolation. To determine whether the FoSNF1 gene had been disrupted in these transformants, genomic DNA from individual transformants was digested with EcoRI and probed with the 2.1-kb EcoRI fragment (Fig. 3A) that was used to construct these alleles. Southern analysis revealed that 71% and 65% of the transformants with fosnf1-1 and fosnf1-2, respectively, had undergone gene replacement by homologous recombination at the FoSNF1 locus. As expected, the untransformed O-685 showed only one band of approximately 2.1 kb (Fig. 3B). Mutants carrying a disrupted fosnf1 gene contained two hybridizing bands, of approximately 1.2 kb and 2.4 kb, due to the presence of an EcoRI site in the inserted transposon. Ectopic transformants still contained the 2.1-kb fragment, in addition to the 1.2-kb and 2.4-kb fragments (Fig. 3B).

Construction and analysis of fosnf1 mutant alleles. A Map of the FoSNF1 gene. The triangle indicates the transposon insertion site in fosnf1-1. The transposon insertion site in fosnf1–2 is located 85 bp downstream from that of fosnf1-1 and is denoted by an arrow. The 2.1-kb EcoRI fragment used to generate insertional mutant alleles is denoted by an open rectangle underneath the map. B Southern blot hybridization of genomic DNA from wild-type F. oxysporum (WT), fosnf1-1 (lane 1), fosnf1-2 (lane 2), and an ectopic transformant (lane 3). DNA was digested with EcoRI and the blot was probed with the 357-bp PCR product containing part of FoSNF1. Bar 1 kb
Transcripts of FoSNF1 in O-685 and its ectopic transformants were detected by Northern hybridization and no disparity was observed in their accumulation patterns (data not shown). In contrast, FoSNF1 transcripts could not be detected in the fosnf1 mutants using the same technique.
Growth characteristics of the fosnf1 mutants in the presence of simple and complex carbon sources
A previous study (Tonukari et al. 2000) showed that the SNF1 homologue in Cochliobolus carbonum might also be involved in the uptake and/or utilization of certain simple sugars. To determine whether FoSNF has a similar role, we compared the growth rate of O-685 with that of the fosnf1 mutants in the presence of various carbon sources. When the growth rate of ectopic transformants was compared with that of O-685, there was no significant difference between them with respect to all carbon sources tested (data not shown). In the presence of glucose, the radial growth rate of the fosnf1 mutants, regardless of the allele used, did not differ from that of O-685 (Fig. 4). However, the growth rate of two independently isolated fosnf1-2 mutants (Fo9B1, Fo9C2) was reduced in the presence of arabinose, fructose, galactose, trehalose, xylan, or xylose, when compared with that of O-685, but on pectin the difference was minimal (Fig. 4). The fosn1-1 mutant 5B6 showed similar growth characteristics to Fo9B1 and Fo9C2 in the presence of arabinose, trehalose, xylan, and xylose, although it grew slightly better than fosnf1-2 mutants in the presence of fructose and pectin (data not shown). Since radial growth might not accurately reflect their mycelial biomass, we compared the mutant′s biomass with that of O-685 after 1 week of liquid-culturing in the presence of the simple sugars arabinose, glucose, and trehalose. The growth rate in the presence of glucose was indistinguishable among them, but was consistent with the radial growth rate. O-685 and its ectopic transformants grew better than the fosnf1 mutants in the presence of arabinose and trehalose (data not shown).

Comparison of the growth rates between F. oxysporum O-685 and the fosnf1-2 mutant Fo9B1 on various carbon sources. Growth conditions are described in the Materials and methods. Triangles and squares indicate the radial growth rate of O-685 and Fo9B1, respectively. A different scale was used for strains grown in the presence of galactose, because they grew significantly slower on this carbon source
The expression of three genes encoding cell wall-degrading enzymes is impaired in fosnf1 mutants
Three mutants, Fo9B1, Fo9C2, and Fo9D6, and three ectopic transformants, Fo9A3, Fo9B5, and Fo9C4, generated with fosnf1-2, were selected to analyze the expression of genes encoding exopolygalacturonase (PGX1), endopolygalacturonase (PGN1), and pectate lyase (PL). After 40 h of growth in the presence of 1% polygalacturonic acid as a sole carbon source (i.e., derepressive condition), transcript levels in the three fosnf1-2 mutants were substantially lower than those in O-685 and the ectopic transformants (Fig. 5). Although the transcript level of all three genes was increased in O-685 and the ectopic transformants under this condition, the amount of each transcript varied among the strains used. For instance, the level of PGX1 transcripts in the ectopic transformants Fo9A3 and Fo9C4 was higher than that in O-685 and Fo9B5. Furthermore, the PGN1 expression level in the ectopic transformant Fo9A3 was also higher than those of O-695, Fo9B5, or Fo9C4 (Fig. 5).

Expression of three genes encoding cell wall-degrading enzymes in F. oxysporum in a derepressible condition. Fungal growth and gene expression analysis are described in the Materials and methods. Blots were probed with pectate lyase (PL), exopolygalacturonase (PGX1), and endopolygalacturonase (PGN1) probes. WT Wild type, lanes 1–3 ectopic transformants, lanes 4–6 fosnf1-2 mutants
fosnf1 mutants exhibit reduced virulence on A. thaliana and cabbage
Strain O-685 was shown to be pathogenic on A. thaliana and various crucifer crops, including cabbage (Mullins et al., in preparation). Fusarium wilt symptoms in a susceptible cabbage variety infected by O-685 typically commenced with a chlorosis of the veins within the leaf structure. Over time, the symptoms expanded into the adjacent tissues, leading to a complete chlorosis of the leaf, with the frequent appearance of brown/black necrotic lesions within the bleached zones. This was soon followed by total plant collapse. In addition to these symptoms, infected plants were severely stunted. fosnf1 mutations significantly delayed the development of wilt symptoms in infected plants (Fig. 6). Plants of a susceptible cabbage variety (Grenadier) inoculated by SI with O-685 started to show vascular discoloration at 10 days post-inoculation (dpi) and total collapse by 15 dpi (Fig. 6D). In contrast, plants infected with the fosnf1-1 mutant displayed vascular discoloration at 12 dpi; and their total collapse was only observed after 21 dpi (Fig. 6E). Wilt symptom expression was further delayed (on average by 3 days with respect to that caused by the fosnf1-1 mutant) in plants inoculated with the fosnf1-2 mutant (Fig. 6F).

Response at 12 days post-inoculation (dpi) of Brassica oleracea plants to infection with F. oxysporum wild-type strain O-685 and fosnf1 mutants. Top row Plants inoculated by root-dipping with O-685 (A), fosnf1-1 (B), and fosnf1-2 (C). Bottom row Plants inoculated by soil-injection with O-685 (D), fosnf1-1 (E), and fosnf1-2 (F)
The RDI method accelerated the symptom development rate in cabbage relative to that achieved via SI (see Fig. 6 for a comparison). For example, cabbage plants inoculated with O-685 displayed vein-clearing at 5 dpi and moderate leaf chlorosis by 6 dpi. At 7 dpi, necrotic patches appeared at the leaf edges and plant collapse was observed shortly after 8 dpi. However, those infected by the fosnf1-2 mutant exhibited no disease symptoms at 7 dpi. They only started exhibiting initial symptoms at 10 dpi, showing moderate chlorosis at 11 dpi, necrosis by 12 dpi, and total collapse around 15 dpi.
In order to compare the advancement of the fosnf1-2 mutant and the wild type within cabbage plant tissues, we took advantage of the hygromycin B resistance trait of the ectopic transformants and the fosnf1-2 mutants. Plants inoculated with water, an ectopic transformant that is as virulent as O-685, and a fosnf1-2 mutant (Fo9B1) were sectioned into roots, stem, and leaves, following surface sterilization. These sections were placed on PDA plates supplemented with 50 µg hygromycin B ml−1. No fungal growth was observed in any of the sections from the water control, suggesting that the combination of hygromycin B and sterilization successfully prevented the growth of other microbes. Whereas the ectopic transformant could successfully colonize both the root and stem of inoculated plants, we found that the fosnf1-2 mutant was limited to the root system, suggesting that the mutant managed to colonize the root tissues but failed to advance through the vascular system in the stem. This observation is consistent with the delayed wilt symptom development caused by the fosnf1 mutant. Infection via the RDI method (in which the host roots were slightly trimmed using scissors, thus allowing the spores to directly enter into the vascular system) showed that, even when the fosnf1-2 mutants succeed in entering the vascular system, there is still a delay in causing wilt symptoms.
The delayed wilt symptom development caused by the fosnf1 mutants was also apparent in A. thaliana (Fig. 7). Three Arabidopsis ecotypes with different degrees of susceptibility to O-685, including Columbia (Col-0), Cape Verde (CV), and EST, were inoculated with O-685 and a fosnf1-1 mutant (5B6). At 8 dpi, only two out of 14 CV seedlings recorded any signs of vascular wilt (ADI of 2, 4, respectively) when infected with the fosnf1-1 mutant. In contrast, at 8 dpi, nine of the 14 CV seedlings infected by O-685 expressed severe wilt symptoms (ADI of 4), and the remaining seedlings also exhibited various degrees of wilt symptoms. A similar pattern was observed in Col-0 at 8 dpi. Only one out of 14 seedlings inoculated with the fosnf1-1 mutant displayed an ADI of 2; and three others appeared somewhat stunted in comparison with the remaining seedlings and the water controls. Among the Col-0 seedlings inoculated with O-685, six exhibited an ADI greater than 3 and two additional seedlings were severely stunted. At 8 dpi, the majority of EST seedlings (12/14) inoculated with O-685 were dead (ADI of 4), whereas four of the seedlings infected by the fosnf1-1 mutant exhibited wilt symptoms (ADI of 4).

Response from three different Arabidopsis ecotypes to infection with F. oxysporum wild-type strain O-685 and the fosnf1-1 mutant 5B6 at 8 dpi. Two plants per interaction are shown. A Cape Verde ecotype, B Columbia, C EST. Top row Water control, middle row inoculated with O-685, bottom row inoculated with 5B6
Discussion
The persistence of soil-borne fungal pathogens, compounded by the limited number of available control strategies, continuously threatens the sustainability of many crops. However, compared with foliar diseases, relatively few efforts have gone into studying the molecular and cellular basis of soil-borne fungal diseases. Therefore, we believe the A. thaliana–F. oxysporum system can be used as a model for generating such knowledge. In this work, we tested the hypothesis that the SNF1 homologue in F. oxysporum is important for its virulence on A. thaliana and cabbage. It was suggested that, in C. carbonum, the SNF1 homologue plays a role in pathogenicity (Tonukari et al. 2000). However, its function in soil-borne pathogens that colonize their hosts through the root had not been tested. Such a comparative analysis is important in order to understand how different fungal pathogens evolved distinct mechanisms enabling them to colonize specific host tissues and overcome the physical, physiological, and environmental modifications in multiple parts of the plant. A report (Dufresne and Osbourn 2001) showing that different factors condition the ability of M. grisea to colonize the root and leaf tissues supports this argument. Genes such as ALB1 and BUF1 and NPR1 are required only for leaf infection by M. grisea, while NUT1 is specifically required for its infection through the root. Genes such as PMK1, ABC1, IGD1, and NPR2 encode general factors required for infecting through both the leaf and root tissues.
The FoSNF1 gene product shows strong similarity to homologues from other filamentous ascomycete fungi, particularly in the serine/threonine protein kinase catalytic domain (Fig. 1). Phylogenetic analysis revealed that FoSNF1 is most closely related to HjSNF1 and a clear distinction can be made between the cluster containing these proteins and the group formed by ScSNF1 and Candida spp homologues (Fig. 2). However, in the FoSNF1 cluster, divergence along the evolutionary pathway appears to be significant, with several members showing an increased evolutionary distance from the earliest ancestor (Fig. 2). Bootstrap test values greater than 99% (after 1,000 replications) confirm such a phylogenetic distribution. Consistent with its degree of sequence similarity to CcSNF1, mutations in FoSNF1 also resulted in pleiotropic phenotypes similar to those observed in ccsnf1, including reduced virulence and impaired ability to uptake/utilize certain carbon sources. In addition, FoSNF1 is also important for proper derepression of the cell wall-degrading enzymes (Fig. 5). The seemingly conserved role of the SNF1 homologue in F. oxysporum and Cochliobolus carbonum suggests that it may function as a general virulence factor.
Although the ccsnf1 and fosnf1 mutants grew slower on both complex and simple carbon sources other than glucose, the degree of growth impairment appeared to vary significantly between F. oxysporum and C. carbonum, depending on the carbon source. The ccsnf1 mutant was unable to grow as fast as the wild type in complex carbon sources such as pectin and xylan (Tonukari et al. 2000) and, particularly in the presence of xylan, the growth of the ccsnf1 mutant was severely impaired relative to that of its wild-type strain (ca. 33% of the wild-type growth rate). Although the fosnf1 mutant also grew slower than O-685 in the presence of xylan and pectin (Fig. 3), the difference was not as pronounced as in C. carbonum. The growth rate difference on l-arabinose was also more pronounced in C. carbonum (ca. 17% of the wild-type growth rate) than in F. oxysporum (ca. 50% of the wild-type growth rate).
Since F. oxysporum does not produce a specialized infection structure, such as an appressorium, and appears to directly penetrate through the roots (Lagopodi et al. 2002), the most probable way of gaining access to and colonizing its hosts is by utilizing its cell wall-degrading enzymes. Indeed, the expression of genes encoding exo- and endo-polygalacturonase and pectate lyase in the fosnf1-2 mutants was significantly reduced (Fig. 5). These enzymes are believed to be involved in the depolymerization of pectin, a major component of the plant cell wall. Thus, the reduced (or impaired) production of such enzymes due to defects in FoSNF1 most likely impedes the initial penetration into its host. If the impaired ability to penetrate the vascular system is the main cause for reduced virulence in the fosnf1 mutants, the difference between O-685 and fosnf1-2 mutants in their virulence would be minimal when the mutants are allowed to directly enter the vascular system through artificial wound sites. When cabbage roots are cut to facilitate fungal entrance to the vascular system via RDI, the wilt symptom development by both the wild type and the fosnf1-2 mutants is accelerated in comparison with that achieved via SI. However, even when the fosnf1-2 mutants succeed in entering the vascular system, symptom development by fosnf1-2 mutants is clearly delayed, relative to that generated by O-685 and its ectopic transformant. These results suggest that both penetration into the vascular system and subsequent colonization of vascular tissues are important for the development of Fusarium wilt. In addition to assisting penetration into its host through the roots, cell wall-degrading enzymes are likely to help in the subsequent colonization of the xylem, by degrading the pit membrane of xylem vessels. Additionally, the impaired ability of the fosnf1-2 mutants to utilize certain carbon sources is likely to further impede their in planta growth, because they might not efficiently utilize sugars released from degraded cell walls as their carbon sources.
References
Agrios GN (1997) Plant pathology, 4th edn. Academic Press, San Diego
Altschul SF, et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Apel-Birkhold PC, Walton JD (1996) Cloning, disruption, and expression of two endo-1,4-B-xylanase genes, XYL2 and XYL2 from Cochliobolus carbonum. Appl Environ Microbiol 62:4129–4135
Armstrong GM, Armstrong JK (1981) Formae speciales and races of Fusarium oxysporum causing wilt diseases. Pennsylvania State University Press, University Park, Pa.
Carlson M, Osmond BC, Botstein D (1981) Mutants of yeast defective in sucrose utilization. Genetics 98:25–40
Carroll AM, Sweigard JA, Valent B (1994) Improved vectors for selecting resistance to hygromycin. Fungal Genet Newsl 41:22
Celenza JL, Carlson M (1984) Cloning and genetic mapping of SNF1, a gene required for expression of glucose-repressible genes in Saccharomyces cerevisiae. Mol Cell Biol 4:49–53
Church GM, Gilbert W (1984) Genomic sequencing. Proc Natl Acad Sci USA 81:1991–1995
Deese DC, Stahmann MA (1962) Pectic enzymes and cellulase formation by Fusarium oxysporum F. cubense on stem tissues from resistant and susceptible banana plants. Phytopathology 52:247–255
Dufresne M, Osbourn AE (2001) Definition of tissue-specific and general requirements for plant infection in a phytopathogenic fungus. Mol Plant-Microbe Interact 14:300–307
Escher D, Bodmer-Glavas M, Barberis A, Schaffner W (2000) Conservation of glutamine-rich transactivation function between yeast and humans. Mol Cell Biol 20:2774–2782
Fisher NL, Burgess LW, Toussoun TA, Nelson PE (1982) Carnation leaves as a substrate and for preserving cultures of Fusarium species. Phytopathology 72:151–153
Gao S, Choi GH, Shain L, Nuss DL (1996) Cloning and targeted disruption of enpg-1, the major in vitro extracellular endopolygalacturonase of the chestnut blight fungus, Cryphonectria parasitica. Appl Environ Microbiol 62:1984–1990
García-Maceira FI, Pietro AD, Roncero MIG (2000) Cloning and disruption of pgx4 encoding an in planta expressed exopolygalacturonase from Fusarium oxysporum. Mol Plant-Microbe Interact 13:359–365
Halila MH, Strange RN (1997) Screening of Kabuli chickpea germplasm for resistance to Fusarium wilt. Euphytica 96:273–279
Hardie DG, Carling D, Carlson M (1998) The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem 67:821–855
Johnston SA, Springer JK, Lewis GD (1979) Fusarium moniliforme as a cause of stem and crown rot asparagus and its association with asparagus decline. Phytopathology 69:778–780
Kanai T, Ogawa K, Ueda M, Tanaka A (1999) Expression of the SNF1 gene from Candida tropicalis is required for growth on various carbon sources, including glucose. Arch Microbiol 172:256–263
Kroes GMLW, Sommers E, Lange W (1998) Two in vitro assays to evaluate resistance in Linum usitatissimum to Fusarium wilt disease. Eur J Plant Pathol 104:561–568
Lagopodi AL, et al (2002) Novel aspects of tomato root colonization and infection by Fusarium oxysporum f. sp. radicis-lycopersici revealed by confocal laser scanning microscopic analysis using the green fluorescent protein as a marker. Mol Plant-Microbe Interact 15:172–179
Lo WS, et al (2001) Snf1—a histone kinase that works in concert with the histone acetyltransferase gcn5 to regulate transcription. Science 293:1142–1146
Moorman GW, Klemmer RA (1980) Fusarium oxysporum causes basal stem rot of Zygocactus truncatus. Plant Dis 64:1118–1119
Mullins E, Romaine CP, Chen X, Geiser D, Raina R, Kang S (2001) Agrobacterium tumefaciens-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology 91:173–180
Nakai K, Kanehisa M (1992) A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 14:897–911
O′Donnell K, Cigelnik E, Weber NS, Trappe JM (1997) Phylogenetic relationships among ascomycetous truffles and the true and false morels inferred from 18S and 28S ribosomal DNA sequence analysis. Mycologia 89:48–65
Palecek SP, Parikh AS, Huh JH, Kron SJ (2002) Depression of Saccharomyces cerevisiae invasive growth on non-glucose carbon sources requires the Snf1 kinase. Mol Microbiol 45:453-469
Petter R, Kwon-Chung KJ (1996) Disruption of the SNF1 gene abolishes trehalose utilization in the pathogenic yeast Candida glabrata. Infect Immun 64:5269–5273
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sambrook J, Russell DW (2001) Molecular cloning : a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Scott-Craig JS, Cheng Y, Cervone F, De Lorenzo G, Pitkin JW, Walton JD (1998) Targeted mutants of Cochliobolus carbonum lacking the two major extracellular polygalacturonases. Appl Environ Microbiol 64:1497–1503
Sink KC, Grey WE (1999) A root-injection method to assess verticillium wilt resistance of peppermint (Mentha x piperita L.) and its use in identifying resistant somaclones of cv. Black Mitcham. Euphytica 106:223–230
Sweigard JA, Chumley FG, Valent B (1992) Disruption of a Magnaporthe grisea cutinase gene. Mol Gen Genet 232:183–190
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tonukari NJ, Scott-Craig JS, Walton JD (2000) The Cochliobolus carbonum SNF1 gene is required for cell wall-degrading enzyme expression and virulence on maize. Plant Cell 12:237–247
Treitel MA, Kuchin S, Carlson M (1998) Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol Cell Biol 18:6273–6280
Walton JD (1994) Deconstructing the cell wall. Plant Physiol 104:1113–1118
Wu S-C, Ham K-S, Darvill AG, Albersheim P (1997) Deletion of two endo-β-1,4-xylanase genes reveals additional isozymes secreted by the rice blast fungus. Mol Plant-Microbe Interact 10:700–708
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Heitman
Rights and permissions
About this article
Cite this article
Ospina-Giraldo, M.D., Mullins, E. & Kang, S. Loss of function of the Fusarium oxysporum SNF1 gene reduces virulence on cabbage and Arabidopsis . Curr Genet 44, 49–57 (2003). https://doi.org/10.1007/s00294-003-0419-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-003-0419-y