
Abstract
The gene cluster catRABC, involved in catechol degradation, was isolated from Rhodococcus erythropolis CCM2595. The genes catA, catB, catC, and the divergently transcribed catR code for catechol 1,2-dioxygenase, cis,cis-muconate cycloisomerase, muconolactone isomerase, and an IclR-type transcriptional regulator, respectively. Measurements of catechol 1,2-dioxygenase activity showed that the expression of catA is induced by phenol but not by catechol or cis,cis-muconate. The activity of catechol 1,2-dioxygenase was repressed by succinate, but no repression by glucose was observed. The transcription start points of catA and catR were determined by primer extension analysis, and the respective promoters (P-catA and P-catR) were thus localized. Measurements of promoter activity during batch cultivation using transcriptional fusion with the gfpuv reporter gene showed that expression of the catR-catABC operon is regulated at the level of transcription. Both P-catR and P-catA are repressed by CatR, and the induction of P-catA by phenol is maintained in the absence of the repressor (in R. erythropolis ΔcatR). Two different potential binding sites for the IclR-type regulator and a recognition site for the cyclic AMP receptor protein (CRP) were identified within the intergenic region between catR and catA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A number of aerobic biodegradation pathways of aromatic compounds like phenol, benzoate, or naphthalene converge into catechol ring cleavage. The scission of the aromatic ring is catalyzed by dioxygenases in bacteria and occurs through ortho- or meta-cleavage. Ortho-cleavage is followed by further steps of the β-ketoadipate pathway, which finally converts the produced metabolites into intermediates of the citrate cycle. Other aromatic compounds, like p-cresol and p-hydroxybenzoate are converted to β-ketoadipate via protocatechuate (Harwood and Parales 1996). In various Gram-negative bacteria (e.g., in Pseudomonas putida or P. resinovorans), the genes catA, catB, and catC (coding for catechol 1,2-dioxygenase, cis,cis-muconate cycloisomerase, and muconolactone isomerase, respectively) involved in the degradation of catechol to β-ketoadipate form an operon transcribed divergently from the catR gene, coding for a LysR-type transcriptional regulator. In these bacteria, the CatR regulator acts as an activator of the adjacent catabolic genes by binding to the sites upstream of the cat operon in the presence of an inducer, which is usually a pathway intermediate like cis,cis-muconate (McFall et al. 1998; Nojiri et al. 2002). The gene coding for the LysR-type regulator was also found to be clustered to the genes catA, catB, and catC in the Gram-positive bacterium Arthrobacter sp. (Murakami et al. 2004). The regulatory catR gene transcribed divergently from the catabolic genes catABC was described in Rhodococcus opacus 1CP as well; however, the regulatory protein was a member of the IclR family of regulators (Eulberg and Schlömann 1998). IclR-type regulators are generally transcriptional repressors, although IclR proteins, which regulate the expression of catabolic genes, may also function as activators (Tropel and van der Meer 2004). The involvement of CatR in the regulation of catABC expression in R. opacus 1CP was proved by the binding of the protein within the intergenic catR-catA region (Eulberg and Schlömann 1998).
The Gram-positive actinobacterium Rhodococcus erythropolis CCM2595 was shown to utilize phenol, catechol, resorcinol, hydroxybenzoate, hydroquinone, and p-chlorophenol (Čejková et al. 2005). Its potential for biotechnological applications was studied due to the significance of the strain in bioremediation. Its cells were more resistant to toxic compounds in the form of a biofilm and in the presence of humic acids and were able to degrade higher concentrations of these substrates under these conditions (Čejková et al. 2005; Masak et al. 2005). We have developed a host-vector system for this strain (Veselý et al. 2003) as well as techniques for genetic manipulations within the chromosome (Čejková et al. 2005) and expressed the gfp gene coding for green fluorescent protein (GFP; Veselý et al. 2003). As catechol dioxygenases play a key role in the catabolism of various aromatic compounds, converting aromatics to aliphatics, we focused on the cloning and characterization of the genes involved in catechol degradation. In this paper, we describe the isolation of the catR-catABC operon from R. erythropolis CCM2595, induction of catechol 1,2-dioxygenase, and determination and analysis of the promoters driving transcription of the catR gene and catABC operon.
Materials and methods
Strains and vectors
The strain Escherichia coli DH5α (Hanahan 1985) was used for cloning, E. coli S17-1 (Simon et al. 1983) as a donor for conjugation and the strain R. erythropolis CCM2595 (Veselý et al. 2003) was the source of cat genes. The plasmids used are shown in Table 1.
Growth conditions
E. coli was grown in Luria–Bertani (LB) medium at 37°C. R. erythropolis CCM2529 was grown in LBP medium (van der Geize et al. 2001) with 0.5% glucose and 1.5% Tween 80 (used against cell clumping) at 30°C or in basal salt minimal medium (BSM; Ogawa and Miyashita 1995) with various carbon sources (phenol, catechol, benzoate, p-hydroxybenzoate, protocatechuate, cis,cis-muconate; each in concentration 1.7 mmol/l, glucose; 55 mmol/l, succinate; 37 mmol/l). The selection media contained kanamycin (30 μg/ml for E. coli and 200 μg/ml for R. erythropolis).
DNA manipulations, cloning, and sequencing
DNA isolation, polymerase chain reaction (PCR), transformation of E. coli, DNA cloning, and DNA analysis were done by standard methods (Sambrook and Russel 2001). Plasmid DNA was isolated from R. erythropolis by the same method (alkaline lysis) as from E. coli, with the following modifications. Before lysis, R. erythropolis cells were incubated for 2 h with lysozyme (5 mg/ml). For the lysis, a higher concentration of sodium dodecyl sulfate (5%) was used. Genomic DNA from R. erythropolis was isolated as described previously (Treadway et al. 1999). The primers for PCR amplification of the internal catA fragment were CDOF1 (CAGGATCCAGGGCCCGT/CACTAC) and CDOR1 (TCGGATCCGACCCAGTCACC). Total DNA from R. erythropolis CCM2595 was used as a template. The fragment was cloned into the BamHI site of pK18mobsacB, the resulting construct pCATA2 was introduced into the cells of R. erythropolis by conjugation, and after integration of the plasmid into the chromosome, regions flanking the cloned part of catA were isolated by the plasmid-rescue technique. The sequence of the complete cat cluster (3,650 bp) was determined by primer walking using the ABI Prism 2100 sequencer (Perkin Elmer). Deletion within the catR gene of R. erythropolis was constructed using the plasmid pDCATR consisting of pK18mobsacB and a fragment of the catRABC cluster (1,247 bp) with a deletion of 103 bp within catR. The plasmid was introduced into R. erythropolis by conjugation and clones in which the double recombination event occurred were identified by positive selection based on the conditional lethal effect of the sacB gene in R. erythropolis (Schäfer et al. 1994). The clones, free of the vector sequences and carrying the catR deletion, were identified by PCR and sequencing. Using the same gene replacement technique and an analogous procedure, the R. erythropolis ΔcatA clone with the 175-bp internal deletion was constructed.
RNA isolation and primer extension analysis
RNA from R. erythropolis cultures was isolated by phenol extraction as described by Eikmanns et al. (1994). Reverse transcription (RT) was performed with SuperScript II transcriptase (Invitrogen) and fluorescein-labeled primer GFP1 (CTAATTCAACAAGAATTGGGAC) complementary to the vector pEPR1, as described previously (Páteket al. 2003). The reverse transcript was run in an automatic ALF DNA Sequencer (Pharmacia Biotech) with the DNA sequencing reactions generated by using the same labeled primer.
RT-PCR analysis
The total RNA isolated from the cells of R. erythropolis cultivated on phenol was incubated with DNase (free of RNase; TopBio, Czech Republic) to remove traces of contaminating DNA. The template RNA (0.6 μg) was then transcribed into complementary DNA (cDNA) using SuperScript II reverse transcriptase and primer CATCR1 [CAGGGGCAGGTTCCAGAGA, nucleotides (nt) at positions 3,345 to 3,327 of the catRABC sequence, reverse]. Reverse transcription product (4 μl) was used in subsequent PCR reactions (in 50 μl) using Taq polymerase (TopBio). The primers CATA8 (CGAGAACGACGTGGCAACT, nt 1,851 to 1,869, forward) and CATBR1 (GATGTACTTCTCGATGATGACC, nt 2,213 to 2,192, reverse) were used to amplify the fragment AB (361 bp), whereas primers CATB3 (ATCGCCCTCAAGACCAC, nt 2,796 to 2,812, forward) and CATCR1 were used for the fragment BC (549 bp). All enzyme reactions were performed following the protocols recommended by the manufacturers. Products were analyzed by electrophoresis in a 1.2% agarose gel.
Transformation and conjugation of R. erythropolis
R. erythropolis cells were transformed by electroporation as described previously (Veselý et al. 2003). Conjugation was done essentially according to van der Geize et al. (2001), using the E. coli S17-1 (Simon et al. 1983) as a donor. The cells of R. erythropolis and E. coli were mixed and cultivated on LBP plates at 30°C for 24 h. Transconjugants were selected on LBP plates containing nalidixic acid (50 μg/ml) and kanamycin (200 μg/ml).
Enzyme assay
R. erythropolis cells were disrupted with an ultrasonic disintegrator (Ikasonic U50, IKA Labortechnik, Staufen, Germany), and after centrifugation (30 min; 18,000×g; 4°C), the activity of catechol 1,2-dioxygenase was measured spectrophotometrically in a cell-free extract using the method of Ngai et al. (1990). Specific activity was expressed as micromoles of cis,cis-muconate per minute per milligram of protein (U/mg protein).
GFP fluorescence intensity measurement
The cell suspension was washed twice with phosphate-buffered saline (PBS) (8.25 g Na2HPO4, 2.05 g NaH2PO4, 4 g NaCl, in 1,000 ml, pH 7.4), diluted with PBS to OD600 = 0.2 and its fluorescence measured with a Shimadzu RF-540 spectrofluorometer (excitation wavelength, 395 nm; emission wavelength, 465–600 nm; maximum at 509 nm). Fluorescence intensity was expressed in arbitrary units (AU) normalized to cell density.
Nucleotide sequence accession number
The nucleotide sequence of the catRABC gene cluster was deposited in GenBank under accession no. AJ605581.
Results
Cloning and sequencing of the internal fragment of catA
The genes coding for catechol-degrading enzymes were searched for in R. erythropolis CCM2595. As no activity of catechol 2,3-dioxygenase was detected in this strain using a simple biochemical test (Worsey et al. 1978), we designed primers for cloning the catA gene for catechol 1,2-dioxygenase (C1,2DO). The primers were based on the conserved regions of C1,2DO within the sequences from GenBank: Arthrobacter sp. (M94318), P. putida (AY029000), R. opacus (X99622), and Streptomyces setonii (AF277051). The amplified PCR fragment (genomic DNA of R. erythropolis CCM2595 was used as a template) of the appropriate size (about 400 bp) was cloned in the vector pK18mobsacB and sequenced. The nucleotide sequence revealed 68% identity with an internal section of catA from R. opacus 1CP according to database similarity searches. We concluded that part of the catA gene from R. erythropolis CCM2595 had been cloned.
Isolation of the complete catRABC gene cluster and its sequencing
The regions flanking the cloned part of catA were isolated by the plasmid-rescue technique as follows: The plasmid with the catA fragment (designated pCATA2) was introduced into the cells of R. erythropolis by conjugation, and KmR clones with the plasmid integrated (by homologous recombination) into the chromosome were identified using PCR. Total DNA of the resulting clone of R. erythropolis::pCATA2 was isolated, digested by EcoRI or PstI, and religated. After the transformation of E. coli, clones carrying the fragments flanking the original fragment of catA were isolated. Sequencing of the flanking fragments revealed the presence of four open reading frames. According to sequence similarity searches, these open reading frames correspond to the genes catA, catB, catC, and the divergently oriented catR (Fig. 1d) coding for catechol 1,2-dioxygenase, cis,cis-muconate cycloisomerase, muconolactone isomerase, and an IclR-type transcriptional regulator, respectively. The deduced amino acid sequences of the catA, catB, catC, and catR gene products from R. erythropolis CCM2595 showed the following degrees of identity with those from other strains of rhodococci: Rhodococcus sp. RHA1 (GenBank acc. no. CP000431; 73, 84, 81, and 72%, respectively), R. opacus 1CP (X99622; 72, 84, 80, and 71%, respectively), Rhodococcus sp. AN-22 (AB167712; 71, 72, and 87%, respectively; only the remnant of catR is present), R. erythropolis AN-13 (D83237; 71, 73, X, and 57%, respectively; X—the catC sequence is incomplete).

Analysis of the catR-catA intergenic region. a Determination of the catA transcription start point (TSP). The bottom peak (PEX) represents cDNA synthesized in reverse transcription (primer extension) using RNA from R. erythropolis. The peaks generated by the automatic sequencer represent the products of sequencing reactions (A, C, G, T) performed with the same fluorescein-labeled primer as that used for primer extension. A portion of the nucleotide sequence derived from the sequencing signals is shown below. Note that the sequence is complementary to that shown in Fig. 1c. TSP (underlined) was determined by a comparison of the position of the primer extension product and sequencing signal. b Determination of catR TSP. c Nucleotide sequence of the catR-catA intergenic region, regulatory features, and sequence elements. TSPs (+1) are underlined and transcription initiation indicated by the bent arrows. The proposed −35 and −10 hexamers and Shine–Dalgarno (SD) motifs are in bold letters. Database-deduced binding sites for the IclR-type regulator are boxed, and inverted and direct repeats are denoted by arrows within the boxes. The consensus-matching CRP-binding site is shaded. Translation initiation sites (in bold) are indicated by hollow arrows. d Scheme of the catRABC gene cluster
RT-PCR of the catABC gene cluster
To prove that the catA gene is cotranscribed with the genes catB and catC, a PCR involving an RT step was carried out with RNA isolated from phenol-grown R. erythropolis cells. The DNA fragments with expected sizes were amplified with the primers CATA8+CATBR1 spanning parts of the genes catA and catB and with the primers CATB3+CATCR1 spanning parts of the genes catB and catC when the same RT reaction product was used (data not shown). No PCR products were obtained when the RT step was omitted. These results indicate that a single transcript covers all three genes, which thus form an operon.
The catA gene codes for catechol 1,2-dioxygenase
To confirm the function of catA, an internal fragment (175 bp) of the chromosomal gene was deleted using the gene replacement technique. The resulting strain R. erythropolis ΔcatA grew in a minimal medium with glucose but did not grow in the medium with phenol (or catechol) as the only source of carbon. This result also showed that C1,2DO coded by catA codes for the only catechol-catabolizing enzyme present in phenol-degrading cells of in R. erythropolis CCM2595. Still another catechol dioxygenase might be present in the strain, which is induced with another substrate. C1,2DO activity was measured in cell extracts of R. erythropolis WT and R. erythropolis ΔcatA cultivated with both glucose and phenol present in the medium. The specific C1,2DO activity was 0.25 and ≤0.001 U/mg protein, respectively. The function of catA was confirmed by the observation that the specific C1,2DO activity in R. erythropolis carrying the catRABC cluster on the multicopy plasmid (pSRKcatRABC) was significantly higher than in the WT strain (0.71 U/mg protein). The plasmid pSRKcatRABC was also transferred to E. coli, and C1,2DO activity (0.2 U/mg protein) was found in transformants while no activity was detected in E. coli (pSRK21).
Activity of catechol 1,2-dioxygenase in R. erythropolis cells grown on various substrates
The specific activity of catechol 1,2-dioxygenase was measured in cell extracts of R. erythropolis CCM2595 grown in minimal medium with glucose, succinate, and phenol, both separately and in combination (Table 2). C1,2DO activity was apparently induced with phenol, whereas a very low activity was detected in extracts of cells grown with succinate or glucose. The strain grew poorly on catechol and cis,cis-muconate, and a very low activity of C1,2DO was also determined in the cell extracts from such cultures. When phenol and glucose were combined as substrates, C1,2DO activity was essentially the same as with phenol alone. Thus, no carbon catabolite repression was exerted by glucose. In contrast, when succinate was added to the medium with phenol, C1,2DO activity was only slightly above the basal level without induction (Table 2). This suggested that repression by succinate is involved in the control of catA expression.
Analysis of the promoters P-catA and P-catR
To analyze the transcriptional regulation of the catR-catABC gene cluster, the intergenic (catR-catA) DNA fragment (249 bp) also covering the 5′-ends (18 bp and 32 bp, respectively) of the divergently oriented coding regions was cloned in the promoter-probe vector pEPR1 in both orientations. The resulting constructs, pEPRPcatA and pEPRPcatR, were transferred to R. erythropolis by transformation. The colonies carrying pEPRPcatR exhibited clear green fluorescence on the plates with minimal or complete medium with or without phenol, whereas the colonies with pEPRPcatA exhibited fluorescence only in the presence of phenol in BSM medium. To localize the promoters of catA and catR, the transcription start points (TSPs) were determined by primer extension analyses (Fig. 1a,b) using RNA isolated from R. erythropolis (with the respective pEPR1 constructs) grown on BSM with phenol. The TSPs were found 117 and 84 nt upstream of the supposed translational starts of catA and catR, respectively (+1 in Fig. 1c). The −35- and −10-like elements of the promoter regions with appropriate spacing (TTGACA-17 nt-CACAGT-6 nt-TSP for catA and TTGTAC-17 nt-TACGCT-6 nt-TSP for catR) were found within the sequences upstream of the TSPs. A clear signal indicating the same TSP of catA was also determined with the RNA isolated from E. coli (pEPRPcatA) grown in LB medium (not shown), whereas no signal for the catR gene was detected. This confirmed that catA may be expressed from its own promoter in E. coli.
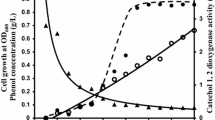
The activities of the promoters P-catA and P-catR (transcriptionally fused to gfpuv in pEPR1) in the cells of R. erythropolis WT cultivated on various carbon substrates were examined using the reporter green fluorescence emitted by GFP. The activity of the catA promoter in glucose or succinate-grown cells of the R. erythropolis WT was low over the whole cultivation period and showed little variation (Fig. 2a). As expected, the activity of P-catA markedly increased with cultivation on phenol (Fig 2a) but not on catechol or cis,cis-muconate (data not shown). Activity of P-catA was also very weakly induced by benzoate (less than 10% as compared to phenol). Protocatechuate and p-hydroxybenzoate, aromatic substrates degraded by the protocatechuate branch of β-ketoadipate pathway (Harwood and Parales 1996), did not induce the P-catA activity (data not shown). High activity of P-catA was also observed when the culture grew on a combination of phenol and glucose, whereas phenol in combination with succinate had almost no effect on P-catA activity in comparison with the growth on succinate alone. The results of these promoter studies were completely in agreement with the results of C1,2DO activity assays and demonstrated that both the induction of the enzyme activity by phenol and repression by succinate are controlled at the transcriptional level. Carbon catabolite repression is thus mediated by succinate but not by glucose. Repression of P-catA, similar to that caused by succinate, was also observed when the cells grew on a combination of phenol and protocatechuate or phenol and p-hydroxybenzoate. These substrates thus apparently also abolished the ability of phenol to induce expression of the operon.

Activity of the P-catA promoter on various substrates. Activity was determined as intensity of fluorescence of the GFP reporter. aR. erythropolis WT, bR. erythropolis ΔcatR
The same analysis of the catR promoter function showed that P-catR displayed only low basal activity (0.1 to 0.4 AU as measured by GFP fluorescence) independently of the carbon source over the whole cultivation period.
To elucidate the role of CatR in the expression of the catR gene and the catABC operon, the catR gene was inactivated by the internal deletion of 103 bp. The activity of P-catR and P-catA was then measured again using transcriptional fusion with gfpuv (in pEPR1) in the resulting strain R. erythropolis ΔcatR. The activity of the catR promoter in the ΔcatR strain was about 10 to 100-fold higher (1.5 to 10.3 AU) than the basal activity in the WT strain in all media tested. In the culture with succinate (the best carbon source of those tested), P-catR activity was (in comparison with the WT-strain) 60-fold higher after 12 h and even 100-fold higher after 24 h of growth. These results demonstrated that the expression of the catR gene is regulated by a strong autorepression.
The activity of the catA promoter in the absence of functional CatR was about three- to fourfold higher on glucose, succinate, and succinate+phenol than in the WT (catR+) strain (Fig. 2). The observed increase in the basal P-catA activity in the ΔcatR strain showed that CatR also acts as a repressor of P-catA. As in the WT strain, catA promoter activity in the ΔcatR strain was significantly elevated during growth on phenol alone or on a combination of phenol with glucose.
Inspection of the catR-catA intergenic DNA sequence (199 bp) and DNA similarity searches in the GenBank database indicated that two different motifs with a high degree of identity with various binding sites for the regulators of the IclR family from different bacteria (Fig. 3a,c) are located within this region. In addition, an inverted repeat closely similar to the CRP (cyclic AMP receptor protein) binding site from E. coli (Zheng et al. 2004) was detected within the sequence (Fig. 1c). As the IclR1 region and CRP motif overlap or are located immediately adjacent to the catA or catR promoters, we suppose that these potential binding sites are involved in the regulation of the expression of the catR-catABC operon.

Alignments of potential regulatory sequences from the intergenic region between catR and catA of R. erythropolis CCM2595. Nucleotides identical in all aligned sequences are shaded. R. erythropolis CCM2595 sequences are shown in bold. a IclR-box motif from R. erythropolis CCM2595 and experimentally proved IclR operator sequences from Acinetobacter sp. (PobR, PcaU; Gerischer et al. 1998) and P. putida (PcaR; pcaR, pcaIJ promoters; Guo and Houghton 1999). b Potential regulatory sequences from R. erythropolis CCM2595 (IclR1 and CRP) and sequences from catR−catA region from Rhodococcus and Nocardia strains. (1) R. erythropolis CCM2595, (2) R. opacus 1CP (GenBank acc. no X99622), (3) R. rhodochrous NCIMB 13259 (AF043741), (4) Rhodococcus sp. RHA1(CP000431), (5) Rhodococcus sp. AN-22 (AB167712), (6) Nocardia sp. C-14-1 (DQ267826). Inverted or direct repeats are indicated with arrows, the proposed −35 and −10 promoter elements are underlined, transcriptional start point (TSP) is denoted by a bent arrow and underlined. c Potential IclR2 box from R. erythropolis CCM2595 and consensus sequence of IclR binding sites from E. coli (Pan et al. 1996)
Discussion
The cloned gene cluster catR-catABC of R. erythropolis CCM2595 forms a regulatory unit (an operon, as proved by RT-PCR analysis) responsible for the degradation of catechol to β-ketoadipate enol-lactone that is further degraded via the β-ketoadipate pathway to intermediates of the citrate cycle. The organization of the catRABC genes from R. erythropolis CCM2595 is closely similar to that of homologous genes in R. opacus 1CP (Eulberg and Schlömann 1998), R. erythropolis AN-13 (GenBank acc. no. D83237), Rhodococcus sp. RHA1 (CP000431), and Nocardia sp. C-14-1 (DQ267826). In all these cases, the CatR proteins coded by the genes transcribed divergently from the adjacent catABC operons are members of the IclR family of transcriptional regulators. Genes responsible for catechol degradation in P. putida (McFall et al. 1998) and P. resinovorans (Nojiri et al. 2002) are organized in the catBCA operons with catR genes also located upstreamof catB in the opposite orientation. The operons catRBAC and catRBCA-II, in which all genes are transcribed in the same direction, were discovered in thermophilic S. setonii (An et al. 2001) and in benzamide-assimilating Arthrobacter sp., respectively (Murakami et al. 2004). In all these and most other cases, expression of these catabolic genes is controlled by LysR-type transcriptional regulators. These regulators involved in the degradation of aromatic compounds function as transcriptional activators (Tropel and van der Meer 2004). The involvement of an IclR-type CatR regulator in the regulation of catechol-degradation genes, which was first found in R. opacus 1CP (Eulberg and Schlömann 1998), seems to be less frequent. IclR-type regulators function prevalently in the gene clusters that code for the degradation pathways of p-hydroxybenzoate and protocatechuate. Such genes (pob and pca) were found in Acinetobacterbaylyi (DiMarco and Ornston 1994; Gerischer et al. 1998), in P. putida (Harwood and Parales 1996), in R. opacus (Eulberg et al. 1998), and in Corynebacterium glutamicum (Brinkrolf et al. 2006). Although the divergent arrangement of the regulatory genes (pobR, pcaU, pcaR, and catR) and degradative genes is similar to that found in operons under the control of LysR-type regulators, the position of the binding sites and their recognition sequences are different (Molina-Henares et al. 2006). In genes controlled by LysR-type regulators, intermediates like cis,cis-muconate or β-ketoadipate are usually the inducers, whereas gene clusters under the regulation of IclR-type proteins are mostly induced by aromatic substrates like protocatechuate (Gerischer et al. 1998), p-hydroxybenzoate (DiMarco and Ornston 1994), and gentisate (Brinkrolf et al. 2006). We have demonstrated that the activity of C1,2DO and the catA promoter are induced with phenol but not with cis, cis-muconate in R. erythropolis CCM2595. The activity of P-catR increased as much as 100-fold in the absence of functional CatR, which indicated that expression of the catR gene is autorepressed. This repression was independent of all the carbon sources tested and was not influenced by the inducer (phenol). No sensitivity of the autorepression to the inducers p-hydroxybenzoate and protocatechuate was observed in the A. baylyi ADP1 regulatory genes pobR (DiMarco and Ornston 1994) or pcaU (Gerischer et al. 1998). IclR-type proteins are generally repressors; however, some of the IclR-regulators that control degradative pathways have been shown to act as activators (Molina-Henares et al. 2006). According to our results from the comparison of P-catA activity in R. erythropolis ΔcatR and the WT strain, the P-catA promoter is also repressed by CatR. In C. glutamicum, deletion of the pcaR regulator gene also resulted in increased levels of transcription of pcaIJ genes (Brinkrolf et al. 2006). In A. baylyi ADP1, the PcaU regulator only repressed the target genes (pca operon for protocatechuate utilization) in the absence of the inducer (protocatechuate), whereas it functioned as an activator in its presence. The inactivation of pcaU reduced the induced expression of pca structural genes by about 90% (Gerischer et al. 1998). PcaU thus mainly acts as an activator. In contrast, we have found that the activity of P-catA in the presence of the inducer (phenol) increased three- to fourfold with the deletion of catR. However, the activity of P-catA increased approximately eightfold in the presence of phenol at its maximum (Fig. 2) in comparison with the basal activity (on succinate) in both the ΔcatR and catR+ strains. These results suggest that the main function of CatR is repression and that another mechanism of P-catA activation, which is not dependent on CatR, operates in R. erythropolis CCM2595. The genes catABC, similar in nucleotide sequence to catABC from R. erythropolis CCM2595, are constitutively expressed in Rhodococcus sp. AN-22 (Matsumura et al. 2006) during growth on both aromatic and non-aromatic substrates. The adjacent catR gene is disrupted by the IS204-like element in this strain, and the promoter within the IS element, which is therefore not regulated by original CatR, drives transcription of the operon. In R. opacus, NpdR, the IclR-type regulator, was found to repress the trinitrophenol degradation genes (Nga et al. 2004). In this case, deletion of the npdR gene resulted in an increase in hydrid transferase activity regardless of the presence of an inducer.
Transcriptional starts, promoter elements, and regulator binding sites (operators) were localized in the regions between the divergently oriented regulatory and target genes pobR-pobA (134 bp) and pcaU-pcaI (286 bp) from A. baylyi ADP1. In these cases, the IclR binding sites (IclR boxes) consist of three short repeats, two forming a palindrome, and a direct repeat separated by 10 nt. Although the relative positions of the promoters and IclR boxes in pobR-pobA and pcaU-pcaI intergenic regions are different, the repeats are very similar (18 out of 21 nt identical). Nearly identical operators for the regulator PcaR were also found to control the expression of the genes pcaI and pcaR in P. putida (Guo and Houghton 1999). The apparent sequence similarity of the inverted repeat found within the catR-catA intergenic region (199 bp) of R. erythropolis CCM2595 (IclR1 in Fig. 1) to the repeats forming operators of the genes responsible for the degradation of p-hydroxybenzoate and protocatechuate suggests that it also serves as an operator for the control of catR/catA expression (Fig. 3a). Moreover, we have identified several conserved sequences of catR-catA intergenic regions from various strains of Rhodococcus and Nocardia in the GenBank database. These sequences share completely identical inverted and direct repeats forming a typical IclR binding site and the proposed −35 and −10 hexamers of P-catA (Fig. 3b). The regulators PobR and PcaU from A. baylyi (DiMarco and Ornston 1994; Gerischer et al. 1998) and PcaR from P. putida (Guo and Houghton 1999) act as activators. The operators for these proteins are located upstream of the promoters of the target genes. In contrast, CatR of R. erythropolis CCM2595 was shown to act as a repressor, and its potential operator overlaps the catA promoter (and also the catR promoter, Fig. 1c). This is in agreement with the theory that the regulatory function of transcriptional regulators is determined by the position of the binding site for the regulator relative to the TSP and not merely by the family of the protein (Madan Babu and Teichmann 2003). In C. glutamicum, the PcaR regulator, whose operator also overlaps the potential promoter of the target gene pcaI, was also shown to serve as a repressor (Brinkrolf et al. 2006).
Another 12-bp palindromic sequence, highly similar to the consensus of the IclR binding site in E. coli (Pan et al. 1996) (Fig. 3c), was found upstream of the translation start site of catR in R. erythropolis CCM2565. In this case, no counterparts were recognized in the homologous sequences of rhodococci and nocardiae.
We found that induction of the catA promoter and C1,2DO activity with phenol was suppressed by succinate in the growth medium, whereas the presence of glucose did not influence it. This suggests that a kind of carbon catabolite repression affects the transcription of catA. In a number of cases, the preferred substrates prevent the induction of genes involved in the degradation of aromatic compounds (Collier et al. 1996; McFall et al. 1998). The repression of enzymes involved in the degradation of p-hydroxybenzoate and protocatechuate with succinate but not with glucose was described in A. baylyi ADP1 (Dal et al. 2002). It is worth mentioning that a conserved sequence, which only differs from the E. coli CRP consensus binding site TGTGA-N6-TCACA (Zheng et al. 2004) by a single nt, overlaps the catA promoter of R. erythropolis CCM2595 as well as the catA promoters within various Rhodococcus and NocardiacatR-catA sequences aligned in Fig. 3b. The gene coding for the regulatory protein of the CRP family (RHA1_ro04321) was found in the genome of Rhodococcus sp. RHA1 (the only Rhodococcus genome sequence available in the GenBank database).
R. erythropolis CCM 2595 grows well on p-hydroxybenzoate and protocatechuate, substrates degraded via the protocatechuate branch of β-ketoadipate pathway. The pathways for catechol and protocatechuate degradation represent metabolically parallel but distinct branches of degradation of aromatic compounds (Harwood and Parales 1996). Bacteria usually show some preference (hierarchy) in aromatic carbon source utilization when cultivated in two or more substrates. A. baylyi ADP1 was shown to consume benzoate before p-hydroxybenzoate in the presence of both carbon sources (Gaines et al. 1996). Repression of the catA promoter by protocatechuate and p-hydroxybenzoate in R. erythropolis CCM2595 suggests that these substrates are utilized preferentially to phenol.
The presence of two different IclR boxes and a potential CRP binding site within the catR-catA intergenic region seems unique among the regulatory regions of genes controlled by IclR transcriptional regulators. Adjacent to the catR gene, we have identified the genes coding for small and large subunits of phenol hydroxylase (pheA2, pheA1) and for an AraC-type transcriptional regulator (pheR). Future work will focus on the analysis of the R. erythropolis CCM2595 pheRA2A1 operon (GenBank AJ973228) and a study of the possible mechanisms of cross-regulation with catRABC genes.
References
An HR, Park HH, Kim ES (2001) Cloning and expression of thermophilic catechol 1,2-dioxygenase gene (catA) from Streptomyces setonii. FEMS Microbiol Lett 195:17–22
Brinkrolf K, Brune I, Tauch A (2006) Transcriptional regulation of catabolic pathways for aromatic compounds in Corynebacterium glutamicum. Genet Mol Res 5:773–789
Čejková A, Masák J, Jirků V, Veselý M, Pátek M, Nešvera J (2005) Potential of Rhodococcus erythropolis as a bioremediation organism. World J Microbiol Biotechnol 21:317–321
Collier DN, Hager PW, Phibbs PV Jr (1996) Catabolite repression control in the Pseudomonads. Res Microbiol 147:551–561
Dal S, Steiner I, Gerischer U (2002) Multiple operons connected with catabolism of aromatic compounds in Acinetobacter sp. strain ADP1 are under carbon catabolite repression. J Mol Microbiol Biotechnol 4:389–404
DiMarco AA, Ornston LN (1994) Regulation of p-hydroxybenzoate hydroxylase synthesis by PobR bound to an operator in Acinetobacter calcoaceticus. J Bacteriol 176:4277–4284
Eikmanns BJ, Thum-Schmitz N, Eggeling L, Ludtke KU, Sahm H (1994) Nucleotide sequence, expression and transcriptional analysis of the Corynebacterium glutamicumgltA gene encoding citrate synthase. Microbiology 140:1817–1828
Eulberg D, Schlömann M (1998) The putative regulator of catechol catabolism in Rhodococcus opacus 1CP-an IclR-type, not a LysR-type transcriptional regulator. Antonie Van Leeuwenhoek 74:71–82
Eulberg D, Lakner S, Golovleva LA, Schlömann M (1998) Characterization of a protocatechuate catabolic gene cluster from Rhodococcus opacus 1CP: evidence for a merged enzyme with 4-carboxymuconolactone-decarboxylating and 3-oxoadipate enol-lactone-hydrolyzing activity. J Bacteriol 180:1072–1081
Gaines G III, Smith L, Neidle EL (1996) Novel nuclear magnetic resonance spectroscopy methods demonstrate preferential carbon source utilization by Acinetobacter calcoaceticus. J Bacteriol 178:6833–6841
Gerischer U, Segura A, Ornston LN (1998) PcaU, a transcriptional activator of genes for protocatechuate utilization in Acinetobacter. J Bacteriol 180:1512–1524
Guo Z, Houghton JE (1999) PcaR-mediated activation and repression of pca genes from Pseudomonas putida are propagated by its binding to both the −35 and the −10 promoter elements. Mol Microbiol 32:253–263
Hanahan D (1985) Techniques for transformation of E. coli. In: Glover DM (ed) DNA cloning. A practical approach, vol 1. IRL, Oxford, pp 109–135
Harwood CS, Parales RE (1996) The β-ketoadipate pathway and the biology of self-identity. Annu Rev Microbiol 50:553–590
Madan Babu M, Teichmann SA (2003) Functional determinants of transcription factors in Escherichia coli: protein families and binding sites. Trends Genet 19:75–79
Masak J, Cejkova A, Jirku V, Kotrba D, Hron P, Siglova M (2005) Colonization of surfaces by phenolic compounds utilizing microorganisms. Environ Int 31:197–200
Matsumura E, Sakai M, Hayashi K, Murakami S, Takenaka S, Aoki K (2006) Constitutive expression of catABC genes in the aniline-assimilating bacterium Rhodococcus species AN-22: production, purification, characterization and gene analysis of CatA, CatB and CatC. Biochem J 393:219–226
McFall SM, Chugani SA, Chakrabarty AM (1998) Transcriptional activation of the catechol and chlorocatechol operons: variations on a theme. Gene 223:257–267
Molina-Henares AJ, Krell T, Eugenia Guazzaroni M, Segura A, Ramos JL (2006) Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol Rev 30:157–186
Murakami S, Kohsaka C, Okuno T, Takenaka S, Aoki K (2004) Purification, characterization, and gene cloning of cis,cis-muconate cycloisomerase from benzamide-assimilating Arthrobacter sp. BA-5-17. FEMS Microbiol Lett 231:119–124
Nga DP, Altenbuchner J, Heiss GS (2004) NpdR, a repressor involved in 2,4,6-trinitrophenol degradation in Rhodococcus opacus HL PM-1. J Bacteriol 186:98–103
Ngai KL, Neidle EL, Ornston LN (1990) Catechol and chlorocatechol 1,2-dioxygenases. Methods Enzymol 188:122–126
Nojiri H, Maeda K, Sekiguchi H, Urata M, Shintani M, Yoshida T, Habe H, Omori T (2002) Organization and transcriptional characterization of catechol degradation genes involved in carbazole degradation by Pseudomonas resinovorans strain CA10. Biosci Biotechnol Biochem 66:897–901
Ogawa N, Miyashita K (1995) Recombination of a 3-chlorobenzoate catabolic plasmid from Alcaligenes eutrophus NH9 mediated by direct repeat elements. Appl Environ Microbiol 61:3788–3795
Pan B, Unnikrishnan I, LaPorte DC (1996) The binding site of the IclR repressor protein overlaps the promoter of aceBAK.J Bacteriol 178:3982–3984
Pátek M, Muth G, Wohlleben W (2003) Function of Corynebacterium glutamicum promoters in Escherichia coli, Streptomyces lividans, and Bacillus subtilis. J Biotechnol 104:325–334
Sambrook J, Russel DV (2001) Molecular cloning. A laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73
Simon R, Priefer U, Pühler A (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/technology 1:784–791
Treadway SL, Yanagimachi KS, Lankenau E, Lessard PA, Stephanopoulos G, Sinskey AJ (1999) Isolation and characterization of indene bioconversion genes from Rhodococcus strain I24. Appl Microbiol Biotechnol 51:786–793
Tropel D, van der Meer JR (2004) Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol Mol Biol Rev 68:474–500
van der Geize R, Hessels GI, van Gerwen R, van der Meijden P, Dijkhuizen L (2001) Unmarked gene deletion mutagenesis of kstD, encoding 3-ketosteroid Delta1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counter-selectable marker. FEMS Microbiol Lett 205:197–202
Veselý M, Pátek M, Nešvera J, Čejková A, Masák J, Jirků V (2003) Host-vector system for phenol-degrading Rhodococcus erythropolis based on Corynebacterium plasmids. Appl Microbiol Biotechnol 61:523–527
Worsey MJ, Franklin FC, Williams PA (1978) Regulation of the degradative pathway enzymes coded for by the TOL plasmid (pWWO) from Pseudomonas putida mt-2. J Bacteriol 134:757–764
Zheng D, Constantinidou C, Hobman JL, Minchin SD (2004) Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res 32:5874–5893
Acknowledgments
This work was supported by grant 526/04/0542 from Czech Science Foundation (GACR) and by Institutional Research Concept no. AV0Z5020903. We thank D. Lukavská for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Veselý, M., Knoppová, M., Nešvera, J. et al. Analysis of catRABC operon for catechol degradation from phenol-degrading Rhodococcus erythropolis . Appl Microbiol Biotechnol 76, 159–168 (2007). https://doi.org/10.1007/s00253-007-0997-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-0997-6