
Abstract
The 454 pyrosequencing technique was applied to evaluate microbial community composition in sediment and water samples collected from the river receiving effluents from a swine farm and a farmhouse restaurant, respectively. For each sample, 4,600 effective sequences were selected and used to do the bacterial diversity and abundance analysis, respectively. Bacterial phylotype richness in the river sediment sample without effluent input was higher than the other samples, and the river water sample with addition of effluent from the swine farm had the least richness. Effluents from both the swine farm and the farmhouse restaurant have the potential to decrease the bacterial diversity and abundance in the river sediment and water, especially it is more significant in the river sediment. Effect of effluent from the swine farm on riverine bacterial communities was more significant than that from the farmhouse restaurant. Characterization of bacterial community composition in sediments from two tributaries of the downstream river showed that various effluents from the swine farm and the farmhouse restaurant have the similar potential to reduce the natural variability in riverine ecosystems, and contribute to the biotic homogenization in the river sediment.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Wastewater treatment plant (WWTP) effluent has the potential to influence the biological properties of the receiving ecosystem (Drury et al. 2013). The effluent can disrupt the community structure of algae, invertebrates, and fish in the effluent-receiving stream (Dyer et al. 2003; Spänhoff et al. 2007). Published studies have documented the potential ecosystem effects of the effluent, including increased nutrient loading (Waiser et al. 2011), eutrophication (Gücker et al. 2006), and oxygen deficits (Rueda et al. 2002). Researchers have evaluated the effects of the effluent on bacterial communities in the water column (Goñi-Urriza et al. 1999; Cébron et al. 2004). In contrast, only a few studies have presented evidence that WWTP effluent may affect sediment microbial communities. For instance, an increase in denitrification rates in sediments below WWTP effluent outfall was found by Lofton et al. (2007). Wakelin et al. (2008) reported that WWTP effluent altered the composition of the stream sediment microbial communities. Drury et al. (2013) demonstrated that WWTP effluent decreased the populations of benthic microbial communities inhabiting the river.
Along with the development of tourism, more and more farmhouse restaurants have been constructed and operated in Chinese rural areas during recent years. Effluents from the swine farm and the farmhouse restaurant along the riverside are most probably to influence riverine microbial ecosystems, but few published reports have considered this. Characterizing microbial communities in the river receiving sewage effluents would provide valuable information for evaluating effects of the effluents on the freshwater ecosystem health. As for the assessments of river health, microbes are informative of the status of aquatic ecosystems, owing to their ubiquitous presence and high abundance in the ecosystems (Lawrence et al. 2005).
The microbial community structure, which was previously investigated by cultural methods (Sandler and Kalff 1993), has recently been determined by molecular approaches (Wakelin et al. 2008; Ferrer et al. 2011; Drury et al. 2013). The microbial community has been studied for several decades by both isolation (Neilson 1978) and molecular methods (Erhart et al. 1997; Liu et al. 1997). The culturing approaches have been effective to determine microbial communities. However, most of bacteria cannot be cultured (Hugenholtz et al. 1998). The molecular approaches significantly improved our understanding of microbial communities. For complex environmental samples, however, these methods still cannot explore the panorama of microbial communities (Claesson et al. 2009). The 454 pyrosequencing is a high-throughput sequencing system that can create more than 400,000 reads with average quality of greater than 99.5 % accuracy (Glenn 2011). A certain number of DNA samples can be sequenced at the same time in a single run by incorporating barcode sequences on primers. This technique has been successfully used to explore the microbiota in a variety of environmental samples, such as swine manure (Lu et al. 2014), soil (Roesch et al. 2007), or wastewater (Ye and Zhang 2013).
In this study, in order to characterize bacterial community structures in the river impacted by sewage effluents, we evaluated 16S rRNA gene in water and sediment samples taken from the river receiving effluents from the swine farm and the farmhouse restaurant using 454 pyrosequencing. The conception of biotic homogenization suggests that anthropogenic modifications of the environment are reducing the biological variability in natural ecosystems. We demonstrate that although these effluents differed in physicochemical properties, they have the similar potential to decrease the natural variability in river ecosystems and show that these effluents contribute to the biotic homogenization in sediment ecosystems of the downstream river. This study describes the first effort to reveal the bacterial diversity and abundance in the river water and sediment impacted by effluents from the swine farm and the farmhouse restaurant using 454 pyrosequencing, and provides valuable data about the effects of these effluents on the biological integrity of river ecosystems.
Materials and methods
Nanxijiang river description and sample collection
Wenzhou (27°03′–28°36′ N, 119°37′–121°18′ E) is an industrialized city of Zhejiang Province of China. Nanxijiang River (27°58′–28°36′ N, 120°19′–120°59′ E) is located in Yongjia County adjacent to Wenzhou City, and they are separated by Oujiang River, which is a 142-km-long river that begins in the northern valleys of Yongjia County. It flows across the whole county from north to south, has an average slope of 6.0 ‰, and a drainage area of 2,436 km2. The source of the river is at an altitude of 1792 m. The upstream river winds through high mountains and deep valleys while downstream the watershed is flat. This river is a national water source area. A swine farm and a farmhouse restaurant were constructed and operated along the sides of the downstream river. In recent decades, with the rapid economy development and population increase, the river received more and more non-point-source runoff of the untreated rural domestic sewage from residents inhabiting the riverside, and point-source effluent of the treated wastewater from the swine farm and the farmhouse restaurant along the sides.
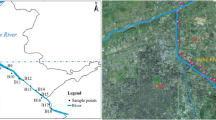
We collected water and sediment samples from two tributaries of the downstream Nanxijiang river to evaluate the effects of effluents from the swine farm and the farmhouse restaurant on the composition of riverine microbial communities. The NXS sediment sample and the NXW water sample were from the reference location in the midstream river. The SFW water sample and the SFS sediment sample were from the location, circa 500 m below the effluent outfall of the swine farm, in a downstream tributary (Fig. 1). The FRW water sample and the FRS sediment sample were from the location, circa 500 m below the effluent outfall of the farmhouse restaurant, in another downstream tributary (Fig. 1). The analyses of physicochemical properties of these samples were performed according to Standard Methods (APHA 1998). Cu and Zn of these samples were measured by inductively coupled plasma analysis.

The diagram of the layout of Nanxijiang River and the sampling locations. NXS and NXW sampling location is in the midstream river, undisturbed by effluents from the swine farm and the farmhouse restaurant; SFS and SFW, FRS and FRW sampling locations are in two downstream tributaries, and 5,00 m downstream of the effluents of the swine farm and the farmhouse restaurant, respectively
It was sunny and the temperature was 2–9 °C on January 10, 2013. Within each of the sampling places of NXW, NXS, SFW, SFS, FRW, and FRS in the river, three 7 × 5 m water areas were selected for use. Water and sediment samples were collected randomly from the six places in each of the three selected water areas. Each sample collected from the river sediment and water in each of the three water areas was mixed, respectively. In this way, three replicates of each sample were obtained. The average of the data about three replicates of each sample was then obtained for the downstream analysis. Sediment samples were collected using a Petite Ponar sampler (Wildlife Supply Company, Saginaw, MI, USA), and large debris was removed by hand. These sediments were stored in sterile 400-ml canning jars (Ball Corporation, Muncie, IN, USA). The sediments were then fixed on site by mixing with 100 % ethanol at a volume ratio of 1:1 and kept in an icebox for transportation and then stored in the laboratory at −20 °C before DNA extraction. Nine 500-ml plastic jars were made using polypropylene by Suzhou Lifeng Plastic Products Co., Ltd. (Suzhou, China). These river water samples were kept in the nine jars and delivered to our laboratory within 3 h. Immediately after its arrival at the laboratory, 400 ml river water was filtrated using a 0.45-μm glass fiber filter to collect the bacteria cells. The collected residue was used for DNA extraction.
DNA extraction, PCR amplification, and pyrosequencing
DNA of these samples was extracted using E.Z.N.A.® Bacterial DNA Kit for Soil (Omega Biotech, Norcross, GA, USA). A successful DNA isolation was confirmed by agarose gel electrophoresis. Before pyrosequencing, the above DNA was amplified with a set of primers targeting the hypervariable V1–V3 region of the 16S rRNA gene (RDP’s Pyrosequencing Pipeline: http://pyro.cme.msu.edu/pyro/help.jsp). The forward primer is 533R of 5′-TTACCGCGGCTGCTGGCAC-3′, and the reverse primer is 27 F of 5′-AGAGTTTGATCCTGGCTCAG-3′ (Shanghai Majorbio Bio-Pharm Tech, Shanghai, China). Barcodes that allow sample multiplexing during pyrosequencing were incorporated between the 454 adaptor and the forward primer. The PCR amplification was performed in a 20-μl reaction system using TransGen AP221-02: TransStart Fastpfu DNA Polymerase (TransGen Biotech, Beijing, China). The amplification was performed in an ABI GeneAmp® 9700 (ABI, Carlsbad, USA) under the following conditions: 95 °C for 2 min, 25 cycles at 95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s, and a final extension at 72 °C for 5 min, 10 °C until halted. The PCR products were purified using AxyPrep DNA Gel Extraction Kit (AXYGEN, Union City, CA, USA) and then mixed equally before pyrosequencing.
The PCR products of the V1–V3 region of 16S rRNA gene were sequenced using the Roche 454 FLX Titanium sequencer (Roche, Nutley, NJ, USA). These samples were individually barcoded to enable multiplex sequencing. The results are deposited into the NCBI short reads archive database (accession number: SRA096439).
Sequence processing and bacterial population analysis
After pyrosequencing, Python scripts were written to (1) filter out sequences containing more than one ambiguous base (‘N’), (2) check the completeness of the barcodes and the adaptor, and (3) remove sequences shorter than 200 bp. Sequencing noises were removed by Pre.cluster (Huse et al. 2010) tool in Mothur package. Chimeras introduced in the PCR process were detected using Chimera Slayer (Haas et al. 2011) in Mothur package. Because all sequences flagged as chimeras are not recommended to be discarded randomly (http://microbiomeutil.sourceforge.net/), so the reads flagged as chimeras were submitted to Ribosomal Database Project (RDP) Classifier (Wang et al. 2007). Those being assigned to any known genus with 50 % confidence threshold were merged with the non-chimera reads to create the “effective sequences” collection for the samples.
Although the used primers are bacteria specific primers, a few archaeal sequences might be obtained. In order to remove these archaeal sequences, the effective sequences were submitted to RDP Classifier again to identify the archaeal and bacterial sequences, and the archaeal sequences were filtered out using a self-written Python script. The “RDP Align” tool in RDP’s Pyrosequencing Pipeline was then used to align the effective sequences. A cluster file was created for each sample with “RDP Complete Linkage Clustering” tool. With the cluster file, the rarefaction curves were generated using the “RDP Rarefaction” tool.
The average length of the effective sequences without the primers was 462 bp. The effective sequences were compared with Greengenes 16S rRNA gene database (DeSantis et al. 2006) annotated with NCBI taxonomy using NCBI’s BLASTN tool and the default parameters except for the maximum hit number of 100 (Claesson et al. 2009). Subsequently, the sequences were assigned to NCBI taxonomies with MEGAN (Huson et al. 2007) using the lowest common ancestor algorithm and the default parameters except for the BLAST bitscore. The BLAST bitscore cutoff threshold 86 (Urich et al. 2008) was applied.
In this study, shared and unique species identified in 16S rRNA gene sequences recovered from barcoded pyrosequencing reads of the river sediment and water samples were represented by Venn diagram (at distance 0.03).
Statistical analysis
The biotic data about bacterial community structures was tested for significance using one-way analysis of variance based on location. P values of less than 0.05 were considered significant.
Results
Physicochemical properties of the samples
Related physicochemical information about the samples is summarized in Table 1, which shows that the physicochemical properties of the river sediment and water samples were distinct, owing to different wastewater effluents from the swine farm and the farmhouse restaurant. The effluent discharge altered the downstream chemistry of the river. The concentrations of ammonium, nitrate, and phosphate, and the contents of organic material and metals were higher downstream of the effluents.
Effectiveness check of the raw reads
We obtained 4,629, 5,421, 4,882, 5,693, 5,949, and 5,894 raw reads for the SFW, SFS, NXW, NXS, FRW, and FRS samples, respectively. After the initial quality check discussed above, the chimera and archaeal sequences were checked and removed. The percentages of 60.3–75.0 % of the raw reads met the quality and length criteria (Table 2). All the chimeras picked by Chimera Slayer were submitted to RDP classifier for further checking. Those sequences that cannot be assigned into a genus were excluded in the following analysis. In order to do the comparison at the same sequencing depth, 4,600 effective bacterial sequences were extracted from each sample for analysis.
Bacterial community structures of the river water and sediment
To compare the bacterial species richness among these samples, operational taxonomic units (OTUs) were determined for each sample at distance levels of 3 %, 5 % and 10 % (Table 2). The OTU amount of NXS was the largest, i.e., 2,174, 1,889 and 1,458 at distance cutoff levels of 3 %, 5 % and 10 %, respectively. The bacterial phylotype richness levels can be reflected using Shannon diversity index, which revealed that NXS had the highest bacterial diversity (Table 2). The Shannon diversity index of NXS was the highest, i.e., 7.03, 6.75 and 6.21 at distance cutoff levels of 3 %, 5 % and 10 %, respectively. Chao 1 richness estimation was also recorded highest in NXS followed by FRS, SFS, NXW, FRW and SFW at all calculated phylogenetic levels. The rarefaction curves of these samples at distance cutoff levels of 3 %, 5 % and 10 %, which demonstrated that the bacterial phylotype richness of NXS was higher than the other samples (Fig. 2). At the sequencing depth of 4,600, these rarefaction curves did not level off, indicating that this sequencing depth still could not cover the whole bacterial diversity.

Rarefaction curves of the river sediment and water samples at cutoff levels of 3 %, 5 %, and 10 % created using RDP’s pyrosequencing pipeline
The effective bacterial sequences were assigned into NCBI taxonomies using BLAST and MEGAN. From the phylum assignment result (Fig. 3), it was found that the bacterial diversity in NXS was significantly different from the other two sediments (P < 0.05). More than 37 % of the sequences in NXS were assigned into Cyanobacteria. In the other samples, Proteobacteria were the dominant phylum, accounting for approximately 54.3 %, 46.3 %, 60.4 %, 81.0 %, and 78.9 % in SFS, FRS, SFW, FRW, and NXW, respectively. There was no significant difference in the abundance of proteobacterial sequences between SFS and FRS (P > 0.05), but NXS showed a significantly higher abundance of Cyanobacteria (P < 0.01). Bacteroidetes were the secondary phylum in NXW, SFW, SFS, and FRW, corresponding to the percentages of approximately 8.0 %, 33.7 %, 11.8 %, and 9.1 %, respectively. The total phyla amount in NXS, SFS, FRS, NXW, SFW, and FRW were 17, 23, 25, 19, 13, and 13, respectively, indicating that the bacterial diversity in SFW and FRW was lower than the other samples at the phylum level. A total of 28 phyla were shared by these six samples. Detailed comparison of these phyla is shown in Table S1.

Relative abundances of different phyla in the river sediment and water samples (the results were obtained using BLASTN and MEGAN)
Bacterial diversity and abundance were also examined at the taxonomic units of class (Table S2), order (Table S3), and genus (Table S4). A high abundance of Burkholderiales in FRW was observed. According to the taxonomic analysis results of BLAST and MEGAN, there were 3,170 out of the totally 4,600 sequences assigned to Burkholderiales order, corresponding to a percentage of 68.9 % (Table S3). In the taxonomic units of class and order, the bacterial diversity of FRS was found higher than those of the other samples. At the genus level, however the bacterial diversity of NXS (144) was clearly higher than those of NXW (130), FRS (130), SFW (88), SFS (114), and FRW (100), respectively. The most dominant classes in NXW (64.2 %), FRS (20.5 %), SFW (52.4 %), SFS (21.3 %), and FRW (70.8 %) were the same class of Betaproteobacteria. At the order level, the top five dominant populations of SFS were Rhodocyclales (11.8 %), Xanthomonadales (10.1 %), Anaerolineales (10.0 %), Burkholderiales (5.5 %), and Desulfuromonadales (5.4 %), which were different from those of NXS, i.e., SubsectionIII (12.9 %), Burkholderiales (5.9 %), Micrococcales (5.8 %), Rhizobiales (4.1 %), and Sphingobacteriales (4.0 %). The differences could also be found between NXS and FRS, NXW and SFW, NXW and FRW (Table S3). The above data indicated that distinct effluents might differently impact riverine bacterial populations.
At the order level, it was found that the top five dominant populations in SFS sediment were Rhodocyclales (11.8 %), Xanthomonadales (10.1 %), Anaerolineales (10.0 %), Burkholderiales (5.5 %), and Desulfuromonadales (5.4 %), which were clearly different from those dominant populations of SFW water, i.e., Burkholderiales (45.0 %), Flavobacteriales (31.4 %), Rhodocyclales (5.4 %), Pseudomonadales (2.4 %), and Micrococcales (1.7 %). The differences could also be found between NXS and NXW, FRS and FRW. At the genus level, the diversity differences between the sediment samples and the water samples could be observed from Table S4. For example, Acidaminobacter, Algoriphagus, Anaerolinea, etc., were only detected in SFS sediment. Acinetobacter, Aeromonas, Albidiferax, etc., were found in SFW water but not in SFS sediment. The differences could also be found between NXS and NXW, FRS and FRW. The above data probably indicated that some bacterial populations in the river water may not proliferate in the river sediment and vice versa. This is an indication that the sedimentation process may change the abundance of bacteria. The bacteria in the sludge flocs were removed during the sedimentation process, while some free-swimming bacteria populations remained in the river water.
The dominant members of the bacterial community composition differed significantly between NXS and the samples of SFS (P < 0.01), and FRS (P < 0.01), respectively. The highest diversity was recorded from NXS taken from the midstream river, whereas a reduction in the diversity of the other two samples from the downstream branches was found. The lowest diversity was found in SFW. With sequence analysis and phylogenetic assignment of the bacterial communities of the samples, we found that the largest differences in bacterial community structures were between NXS and SFW. However, bacterial community structures between SFS and FRS were not significantly different (P > 0.05). Significant differences (P < 0.01) in phylogenetic structure between NXS and SFW were found at the class level. The 16 S rRNA gene sequences originating from NXS were mostly of Cyanobacteria (37.4 %), Proteobacteria (23.1 %), and Actinobacteria (12.3 %) origins, while sequences from SFW were of Proteobacteria (60.4 %), Bacteroidetes (33.7 %), and Actinobacteria (1.7 %) origins.
Shared species among the NXS (166), SFS (144), and FRS (158) sediments were represented by Venn diagram, and results clearly demonstrated that 18 species were common for all three sediment samples at 0.03 distances, while there were 32 species common for NXS and SFS, 49 species for NXS and FRS, and 69 for SFS and FRS (Fig. 4). Regarding the river water samples, shared species among the NXW (141), SFW (98), and FRW (123) water samples were also represented by Venn diagram, and results demonstrated that 24 species were common for all three water samples at 0.03 distances, while there were 51 species common for NXW and SFW, 49 species for NXW and FRW, and 31 for SFW and FRW (Fig. 4).

Venn diagram (at distance 0.03) showing shared and unique species identified in 16S rRNA gene sequences recovered from barcoded pyrosequencing reads of the river sediment and water samples
The above data indicated that phylogenetic shifts in the composition of riverine bacterial communities were promoted by effluent inputs. The effluents from the swine farm and the farmhouse restaurant decreased the riverine bacterial diversity and species richness at the downstream locations. There were 18 and 24 species were common for the sediment and water samples, respectively. However, the bacterial number of the sediment was greatly larger than that of the river water. This indicated that effects of the effluents on sediment bacterial communities were more significant than on the river water bacterial communities. Similarly, 32 species were common for NXS and SFS, while 49 for NXS and FRS, which indicated that the effluent from the swine farm impacted sediment bacterial communities more significantly than that from the farmhouse restaurant. However, there was no significant difference in the effects between the two effluents on the river water bacterial communities. The results showed that 32 species were common for NXS and SFS, 49 for NXS and FRS, whereas 69 for SFS and FRS. This indicated that effluents from the swine farm and the farmhouse restaurant contributed to biotic homogenization in sediment bacterial populations at the downstream locations. It was interesting that in spite of the higher concentrations of inorganic nutrients (Table 1), the numbers of water and sediment bacteria decreased below the effluents, which was in line with a previous study (Drury et al. 2013) on the shifts in benthic microbial communities caused by WWTP effluent. However, published studies also reported that increased concentrations of nitrogen and phosphorus associated with WWTPs stimulate planktonic bacterial growth (Goñi-Urriza et al. 1999) and benthic bacterial numbers (Wakelin et al. 2008). Toxic compounds may be present in the effluents, and these may have inhibited bacterial populations. Previous studies demonstrated that many of these compounds were not completely removed by wastewater treatment (Bartelt-Hunt et al. 2009; Akiyama and Savin 2010). Toxic compounds in the effluents could contribute to the decrease in bacterial diversity and species richness at the downstream locations, which conflicted with previous findings that demonstrated an increase in bacterial diversity below a WWTP effluent (Wakelin et al. 2008). Moreover, Wakelin et al. (2008) found that WWTP effluent led to an increase in carbon of the sediment in the downstream creek, which was in line with the findings in this study (Table 1) but disagreed with the findings of Drury et al. (2013) in the sediment downstream the rivers. However, besides the study of Drury et al. (2013), biotic homogenization and decreases in the population of sediment bacterial communities were also found in this study. This indicated that toxic chemicals from effluents probably played major roles in biotic homogenization of sediment microbial ecosystems. However, biotic homogenization was not clearly found in water microbial communities at the downstream locations, which probably associated with effects of the water flow on the water bacterial communities at the downstream locations.
Discussion
Differences in riverine bacterial communities between this and previous studies
The bacterial diversity in the river water and sediment samples was evaluated in this study. In NXS, at a distance cutoff of 3 %, 2,174 OTUs were obtained from 4,600 sequences (Table 2), indicating that NXS is a highly species-rich ecosystem. Most of the sequences (45 %) in SFW concentrated in Burkholderiales order. The OTU amount of the river water samples (NXW, FRW and SFW) was much smaller than a previous study conducted on WWTP wastewater (McLellan et al. 2010), in which more than 3,000 OTUs were identified for each sample. The results of McLellan et al. (2010) on the bacterial diversity in WWTP wastewater showed that Actinobacteria, Bacteroidetes, and Firmicutes were the most dominant three bacterial groups, adding up to 37.5 % of the total bacteria. In this study, the top three bacterial phyla of NXW and FRW were Proteobacteria, Bacteroidetes, and Cyanobacteria (Table S1). At the phylum level, the top three dominant groups of SFW were Proteobacteria, Bacteroidetes, and Actinobacteria (Table S1). Therefore, the dominant bacterial groups in this study were different from those of McLellan et al. (2010).
A few studies have been conducted on the bacterial community of the river sediment receiving effluent discharge from WWTP. Wakelin et al. (2008) examined the bacterial community in the sediment using PCR-denaturing gradient gel electrophoresis (DGGE) of 16S rRNA genes and clone library analysis. Proteobacteria was found to be the dominant phylum, which was in line with this study. Their cloning results showed that Betaproteobacteria was the most abundant class in Proteobacteria phylum, followed by Gammaproteobacteria. However, the results of this study suggested that Betaproteobacteria (20.5 %) and Anaerolineae (16.8 %) were the top two classes in FRS (Table S2). Another published study evaluated the bacterial diversity of the lake sediment (Laguna de Carrizo, Spain) based on the analyses of the diversity of 16S rRNA amplicons and a 3.1 Mb of consensus metagenome sequence (Ferrer et al. 2011). Their results showed that Beta- and Deltaproteobacteria were the most abundant in the sediment. Deltaproteobacteria is a class which comprises the major group of sulfate-reducing bacteria (SRB). In this study, the most represented SRB sequences of the samples affiliated with Desulfobulbaceae, Myxococcales, Syntrophobacterales, and Geobacteraceae. This inconsistence between the present study and previous studies may result from the different physicochemical properties of the sediments. FRS sample in this study was collected from the river receiving the treated effluent from the farmhouse restaurant, whereas in previous studies sediment samples were taken from the river receiving effluent from WWTP or from the Ca2+-rich anoxic and sub-saline lake. Wakelin et al. (2008) reported that differences in sediment bacterial communities had a high correlation with the physicochemical properties of the sediments. Phylogenetic and biochemical analyses by Ferrer et al. (2011) demonstrated that the inherent physicochemical properties of the Ca2+-rich anoxic sediment of the sub-saline lake, coupled with adaptation to anthropogenic activities, resulted in an efficient microbial community.
Consistent with the high abundance of Betaproteobacteria (21.3 %), Bacteroidetes (11.8 %), and Alphaproteobacteria (1.6 %) bacteria in SFS, some researchers also found that there were Betaproteobacteria (19.2–38.6 %), Bacteroidetes (3.6–20.0 %), and Alphaproteobacteria (11.5–19.2 %) groups in the eutrophic Taihu Lake based on cloning results (Tang et al. 2009). In SFS, most of the Bacteroidales bacteria were affiliated with the genera of Bacteroides, Alkaliflexus, Paludibacter, Parabacteroides, and Prevotella. The Bacteroidales order comprises of many different members. The dominant Bacteroidetes group was organic-aggregate-associated bacteria, which play major roles in microbial food webs and in the cycling of major elements, especially in freshwater systems (Tang et al. 2009).
The results of Wakelin et al. (2008) on the bacterial diversity in the sediment below the WWTP outfall, showed that Beta-, Gamma-, and Deltaproteobacteria were the most dominant three bacterial classes in the sediment sample. In this study, however the top three bacterial classes in NXS were Cyanobacteria (no_rank), Actinobacteria (class), and Alphaproteobacteria (Table S2). At the order level, the top three dominant groups were SubsectionIII, Burkholderiales, and Micrococcales (Table S3). In this way, we could also reveal the differences between the reports by Wakelin et al. (2008) and the other two sediments of FRS and SFS at the class and order levels, respectively. Therefore, the dominant bacterial groups in the sediments were different from those of Wakelin et al. (2008).
Significant bacterial diversity differences between NXS and the other two sediments of FRS and SFS could be observed in Table S4. For instance, Acidovorax, Actinomycetospora, Adhaeribacter, etc., were only detected in NXS. Afipia, Alicycliphilus, Anaeromyxobacter, etc., were found in FRS but not in NXS. Another notable difference in sediment bacterial communities was a 128-fold difference in the abundance of Cyanobacteria sequences, which accounted for more than 37 % of the sequences in NXS but less than 0.3 % of the sequences in FRS (Table S1). This indicates that various effluents may differently alter the components of the river sediments (Table 1). The difference in the compositions of sediments (Table 1) probably impacts the abundance of bacteria in sediments. Microbial community structures in sediments had a high correlation with physicochemical properties of the sediments (Ferrer et al. 2011).
Effects of effluent addition to riverine bacterial communities
In this study, effluent discharge altered the chemistry and bacterial communities of the downstream river. The concentrations of nitrogen and phosphorus became higher below the effluents, similar to what has been observed for a variety of ecosystems (Gücker et al. 2006; Spänhoff et al. 2007; Waiser et al. 2011). These effluents influenced sediment ammonium, nitrate, phosphate concentrations, and organic material content. The effluents significantly (P < 0.01) changed bacterial community composition in the sediments of FRS and SFS, compared to that of NXS. This was correlated with changes in chemical properties of sediments at the sampling locations. These effluents also decreased the bacterial community diversity and richness (Table 2). We found no significant differences (P > 0.05) in bacterial community composition between FRS and SFS. The differences in bacterial community structures between NXS and the samples of FRS and SFS were evident at the phyla level (Table S1), indicating a large phylogenetic shift. The effluents significantly decreased the abundance of Cyanobacteria in sediment bacterial communities (Table S1). The effluents also reduced the abundance of Actinobacteria, Armatimonadetes, Gemmatimonadetes, and Planctomycetes (Table S1). In contrast, there was an increase in the abundance of Chlorobi, Chloroflexi, Firmicutes, Fusobacteria, Lentisphaerae, Nitrospirae, Proteobacteria, Spirochaetes, and TM6 in FRS and SFS (Table S1). Comparison of the used sediments provides insight into the effects of different effluents on sediment bacterial communities. Similar spatial partitioning in microbial community structure as estimated using DNA sequence analyses has been reported for ammonia oxidizers (Cébron et al. 2003) and for total bacterial communities in reservoir sediments (Wobus et al. 2003). In the above cases, the chemical properties were the major determinants of microbial composition.
Bacterial communities in FRS and SFS were dominated by Proteobacteria, a metabolically diverse group of Gram-negative bacteria often found in freshwater ecosystems (Fazi et al. 2005). Bacteroidetes and Chloroflexi were the secondary phylum in SFS and FRS, respectively. Bacteroidetes are Gram-negative heterotrophic bacteria that are ubiquitous in freshwater ecosystems and are known to degrade high-molecular-weight organic compounds (Drury et al. 2013). Therefore, the higher abundance of Bacteroidetes in SFS (11.8 %) and FRS (5.2 %) might result from higher concentrations of organic compounds (Table 1).
Sequences from the midstream sample of NXS included those belonging to representatives of Caulobacterales and Sphingomonadales. These bacterial orders are considered to be oligotrophic-adapted to conditions of low availability of metabolic substrate (Pang and Liu 2006). Sphingomonads are able to transform metals and nutrients (Vilchez et al. 2007), indicating that they have physiological attributes of functional relevance to maintain ecosystem health. Their presence in this section of the river may indicate the higher habitat quality at the midstream location. There was a significant increase in Nitrospirae abundance in the downstream samples of FRS and SFS, which was in agreement with the reports of previous studies (Wakelin et al. 2008; Drury et al. 2013). Nitrospira species are Gram-negative bacteria that catalyze the second step in nitrification and are the dominant nitrite oxidizers in freshwater sediments (Altmann et al. 2003). The downstream sediments had a higher ammonium concentration (Table 1), which represents more substrate for nitrification and could elucidate the higher Nitrospira abundance in FRS (0.29 %) and SFS (0.05 %).
The biotic homogenization has been demonstrated by numerous studies performed on plant and animal communities but has been less well explored for microbial communities (McKinney 2006). In this study, sewage effluents caused shifts in sediment bacterial composition. The results suggest that the effluent input may be a driver of the biotic homogenization in sediment bacterial communities. Specific changes in bacterial composition resulted from the effluents. For instance, there was a significant decrease in Cyanobacteria abundance below the effluents. These effluents also caused a reduction in Actinobacteria abundance. In contrast, the Nitrospirae abundance increased below the effluents. Nitrospirae catalyze the second step in nitrification, so their increased abundance may result from the increased ammonium concentrations at the downstream locations.
In general, significant differences in the bacterial abundances between NXS and the samples of FRS and SFS, may be resulted from distinct physicochemical properties of the effluents from the farmhouse restaurant and the swine farm. These effluents impacted the sediment chemistry and bacterial communities in the downstream river. Despite the higher concentrations of inorganic nutrients, the numbers of sediment bacteria decreased below the effluents. These effluents probably contained compounds toxic to microorganisms. Toxic compounds in the effluents could lead to a decrease in bacterial diversity and species richness at the downstream locations.
Differences in microbial community structures provide useful information about the influence of disturbance on the biological integrity of ecosystems. In this study, although the effluents from the wastewater treatment systems of the swine farm and the farmhouse restaurant discharged into the river may meet water quality criteria, these effluents can impact downstream ecosystems. These effluents caused increased nutrients in the waterways. The downstream sediments showed decreased bacterial diversity, shifting to bacterial communities contained more nitrate-oxidizing bacteria. Although the two downstream tributaries of the river have received effluents with distinct properties, sediment bacterial communities in these tributaries homogenized downstream.
This study provides an insight into the phylogenetic structure of bacterial communities of the river water and sediment, showing that although effluents from the farmhouse restaurant and the swine farm are distinct in their physicochemical properties, they have the similar potential to decrease the bacterial diversity and abundance in the sediment and water below the effluents. The effect of these effluents on sediment bacterial communities was more significant than that on the river water bacterial communities. The effect of the swine farm effluent on sediment bacterial communities was more significant than that of the farmhouse restaurant effluent. The numbers of bacterial genera and species in NXS exceeded those in FRS and SFS, respectively. The amounts of microbial genera and species in NXW exceeded those in FRW and SFW, respectively. Moreover, the amount of the shared species between FRS and SFS was clearly larger than that between NXS and FRS, and SFS, respectively. This indicated that the addition of various effluents contributed to biotic homogenization of river sediment microbial ecosystems. Results suggest that anthropogenic activities beside the river may have exerted selective pressure on the riverine microbial community to adapt it to toxic chemicals. Differences in riverine microbial communities provide useful information on the influence of disturbance and stress on the biological integrity of river ecosystems. In order to evaluate the effect of effluent discharge on the functional ecology of riverine microbial communities, further investigations will be required to characterize functional genes and metabolic pathways of the river microbial community.
References
Akiyama T, Savin MC (2010) Populations of antibiotic-resistant coliform bacteria change rapidly in a wastewater effluent dominated stream. Sci Total Environ 408(24):6192–6201
Altmann D, Stief P, Amann R, De Beer D, Schramm A (2003) In situ distribution and activity of nitrifying bacteria in freshwater sediment. Environ Microbiol 5(9):798–803
APHA (1998) Standard methods for the examination of water and wastewater, 20th edn. American Public Health Association, American Water Works Association, Water Environment Federation, Washington, DC (USA)
Bartelt-Hunt SL, Snow DD, Damon T, Shockley J, Hoagland K (2009) The occurrence of illicit and therapeutic pharmaceuticals in wastewater effluent and surface waters in Nebraska. Environ Pollut 157(3):786–791
Cébron A, Berthe T, Garnier J (2003) Nitrification and nitrifying bacteria in the lower Seine river and estuary (France). Appl Environ Microbiol 69(12):7091–7100
Cébron A, Coci M, Garnier J, Laanbroek HJ (2004) Denaturing gradient gel electrophoretic analysis of ammonia-oxidizing bacterial community structure in the lower Seine River: impact of Paris wastewater effluents. Appl Environ Microbiol 70(11):6726–6737
Claesson M, óSullivan O, Wang Q, Nikkila J, Marchesi J, Smidt H, De Vos W, Ross R, óToole P (2009) Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 4(8):e6669
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Drury B, Rosi-Marshall E, Kelly JJ (2013) Wastewater treatment effluent reduces the abundance and diversity of benthic bacterial communities in urban and suburban rivers. Appl Environ Microbiol 79(6):1897–1905
Dyer SD, Peng C, McAvoy DC, Fendinger NJ, Masscheleyn P, Castillo LV, Lim JMU (2003) The influence of untreated wastewater to aquatic communities in the Balatuin River, The Philippines. Chemosphere 52(1):43–53
Erhart R, Bradford D, Seviour R, Amann R, Blackall L (1997) Development and use of fluorescent in situ hybridization probes for the detection and identification of Microthrix parvicella in activated sludge. Syst Appl Microbiol 20(2):310–318
Fazi S, Amalfitano S, Pernthaler J, Puddu A (2005) Bacterial communities associated with benthic organic matter in headwater stream microhabitats. Environ Microbiol 7(10):1633–1640
Ferrer M, Guazzaroni ME, Richter M, García-Salamanca A, Yarza P, Suárez-Suárez A, Solano J, Alcaide M, van Dillewijn P, Molina-Henares MA, López-Cortés N, Al-Ramahi Y, Guerrero C, Acosta A, de Eugenio LI, Martínez V, Marques S, Rojo F, Santero E, Genilloud O, Pérez-Pérez J, Rosselló-Móra R, Ramos JL (2011) Taxonomic and functional metagenomic profiling of the microbial community in the anoxic sediment of a sub-saline shallow lake (Laguna de Carrizo, Central Spain). Microb Ecol 62(4):824–837
Glenn TC (2011) Field guide to next generation DNA sequencers. Mol Ecol Resour 11(5):759–769
Goñi-Urriza M, Capdepuy M, Raymond N, Quentin C, Caumette P (1999) Impact of an urban effluent on the bacterial community structure in the Arga River, Spain, with special reference to culturable Gram-negative rods. Can J Microbiol 45(10):826–832
Gücker B, Brauns M, Pusch MT (2006) Effects of wastewater treatment plant discharge on ecosystem structure and function of lowland streams. J N Am Benthol Soc 25(2):313–329
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E (2011) Chimeric 16 S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21(3):494–504
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180(18):4765–4774
Huse SM, Welch DM, Morrison HG, Sogin ML (2010) Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12(7):1889–1898
Huson D, Auch A, Qi J, Schuster S (2007) MEGAN analysis of metagenomic data. Genome Res 17(3):377–386
Lawrence JR, Swerhone GDW, Wassenaar LI, Neu TR (2005) Effects of selected pharmaceuticals on riverine biofilm communities. Can J Microbiol 51(8):655–669
Liu WT, Marsh TL, Cheng H, Forney LJ (1997) Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol 63(11):4516–4522
Lofton DD, Hershey AE, Whalen SC (2007) Evaluation of denitrification in an urban stream receiving wastewater effluent. Biogeochemistry 86(1):77–90
Lu XM, Lu PZ, Zhang H (2014) Bacterial communities in manures of piglets and adult pigs bred with different feeds revealed by 16S rDNA 454 pyrosequencing. Appl Microbiol Biotechnol 98(6):2657–2665
McKinney ML (2006) Urbanization as a major cause of biotic homogenization. Biol Conserv 127(3):247–260
McLellan S, Huse S, Mueller Spitz S, Andreishcheva E, Sogin M (2010) Diversity and population structure of sewage derived microorganisms in wastewater treatment plant influent. Environ Microbiol 12(2):378–392
Neilson A (1978) The occurrence of aeromonads in activated sludge: isolation of Aeromonas sobria and its possible confusion with Escherichia coli. J Appl Microbiol 44(2):259–264
Pang CM, Liu WT (2006) Biological filtration limits carbon availability and affects downstream biofilm formation and community structure. Appl Environ Microbiol 72(9):5702–5712
Roesch L, Fulthorpe R, Riva A, Casella G, Hadwin A, Kent A, Daroub S, Camargo F, Farmerie W, Triplett E (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1(4):283–290
Rueda J, Camacho A, Mezquita F, Hernandez R, Roca JR (2002) Effects of episodic and regular sewage discharges on the water chemistry and macroinvertebrate fauna of a Mediterranean stream. Water Air Soil Pollut 140(1–4):425–444
Sandler BC, Kalff J (1993) Factors controlling bacterial production in marine and freshwater sediments. Microb Ecol 26(2):79–99
Spänhoff B, Bischof R, Böhme A, Lorenz S, Neumeister K, Nöthlich A, Küsel K (2007) Assessing the impact of effluents from a modern wastewater treatment plant on breakdown of coarse particulate organic matter and benthic macroinvertebrates in a lowland river. Water Air Soil Pollut 180(1):119–129
Tang X, Gao G, Qin B, Zhu L, Chao J, Wang J, Yang G (2009) Characterization of bacterial communities associated with organic aggregates in a large, shallow, eutrophic freshwater lake (Lake Taihu, China). Microb Ecol 58(2):307–322
Urich T, Lanzén A, Qi J, Huson D, Schleper C, Schuster S (2008) Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 3(6):e2527
Vilchez R, Pozo C, Gómez MA, Rodelas B, González-López J (2007) Dominance of sphingomonads in a copper-exposed biofilm community for groundwater treatment. Microbiology 153(2):325–337
Waiser MJ, Tumber V, Holm J (2011) Effluent-dominated streams: Part 1. Presence and effects of excess nitrogen and phosphorus in Wascana Creek, Saskatchewan, Canada. Environ Toxicol Chem 30(2):496–507
Wakelin SA, Colloff MJ, Kookana RS (2008) Effect of wastewater treatment plant effluent on microbial function and community structure in the sediment of a freshwater stream with variable seasonal flow. Appl Environ Microbiol 74(9):2659–2668
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267
Wobus A, Bleul C, Maasen S, Scheerer C, Schuppler M, Jacobbs E, Röske I (2003) Microbial diversity and functional characterization of sediments from reservoirs of different trophic states. FEMS Microbiol Ecol 46(3):331–347
Ye L, Zhang T (2013) Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 pyrosequencing. Appl Microbiol Biotechnol 97(6):2681–2690
Acknowledgments
This work was supported by the Foundation of Zhejiang Educational Committee (No. Y201224611), the China Postdoctoral Science Foundation (No. 2013 M541647), and the Jiangsu Planned Projects for Postdoctoral Research Funds (No. 1302130C).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 988 kb)
Rights and permissions
About this article

Cite this article
Lu, XM., Lu, PZ. Diversity, abundance, and spatial distribution of riverine microbial communities response to effluents from swine farm versus farmhouse restaurant. Appl Microbiol Biotechnol 98, 7597–7608 (2014). https://doi.org/10.1007/s00253-014-5772-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5772-x