
Abstract
A highly active amide hydrolase (DamH) was purified from Delftia sp. T3-6 using ammonium sulfate precipitation, diethylaminoethyl anion exchange, hydrophobic interaction chromatography, and Sephadex G-200 gel filtration. The molecular mass of the purified enzyme was estimated to be 32 kDa by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis. The sequence of the N-terminal 15 amino acid residues was determined to be Gly-Thr-Ser-Pro-Gln-Ser-Asp-Phe-Leu-Arg-Ala-Leu-Phe-Gln-Ser. Based on the N-terminal sequence and results of peptide mass fingerprints, the gene (damH) was cloned by PCR amplification and expressed in Escherichia coli BL21(DE3). DamH was a bifunctional hydrolase showing activity to amide and ester bonds. The specific activities of recombinant DamH were 5,036 U/mg for 2′-methyl-6′-ethyl-2- chloroacetanilide (CMEPA) (amide hydrolase function) and 612 U/mg for 4-nitrophenyl acetate (esterase function). The optimum substrate of DamH was CMEPA, with K m and k cat values of 0.197 mM and 2,804.32 s−1, respectively. DamH could also hydrolyze esters such as 4-nitrophenyl acetate, glycerol tributyrate, and caprolactone. The optimal pH and temperature for recombinant DamH were 6.5 and 35 °C, respectively; the enzyme was activated by Mn2+ and inhibited by Cu2+, Zn2+, Ni2+, and Fe2+. DamH was inhibited strongly by phenylmethylsulfonyl and SDS and weakly by ethylenediaminetetraacetic acid and dimethyl sulfoxide.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chloroacetanilide herbicides are one of the most widely used herbicide families. Members of this family share a molecular core consisting of 2-chloroacetanilide and differ only in the type and arrangement of substitutions (Fig. 1) (Saha et al. 2012). Acetochlor (2-chloro-N-(ethoxymethyl)-N-(2-ethyl-6-methylphenyl)-acetamide) is an important chloroacetanilide herbicide. Chloroacetanilide herbicides are selective pre-emergent herbicides that are used to control broadleaf weeds and annual grasses in corn fields (Xu et al. 2006). Acetochlor has been used worldwide for more than 40 years; the estimated annual consumption is 35,000,000 lbs in the USA and >100,000,000 lbs in China (Grube et al. 2011; Hua 2011), mostly for corn cultivation. Previous studies have revealed that acetochlor negatively impacts both the environment and agricultural ecosystems. Acetochlor exerts inhibitory effects on soil microorganisms, which could affect the growth and reproduction of earthworms (Zhou et al. 2006; Yasmin and D'Souza 2010). It also exhibits certain toxic effects in humans and animals (Crump et al. 2002; Liu et al. 2006; Xiao et al. 2006; Zafeiridou et al. 2006). As such, acetochlor has been identified as a class B-2 carcinogen by the US Environmental Protection Agency (Coleman et al. 2000; Xiao et al. 2006).

Structure of chloroacetanilide herbicides. R1-ethyl, R2-methyl—alachlor. R2-butyl—butachlor. R1-methyl, R2-ethyl—acetochlor. R2-isopropyl—propisochlor
Microorganisms play major roles in the degradation of acetochlor (Liu et al. 2002). Acetochlor biodegradation pathways include dechlorination, hydroxylation, N-dealkylation, C-dealkylation, and dehydrogenation (Xu et al. 2006), resulting in breakdown products, including 2-ethyl-6-methylaniline (MEA), chloroacetyl-indoline, and 2′-methyl-6′-ethyl-2-chloroacetanilide (CMEPA) (Jablonkai 2000; Dagnac et al. 2002; Ye et al. 2002).
The hydrolysis of an amide bond in the structure is an important reaction during the microbial degradation of acetanilide herbicides. In recent years, several arylamidases that play roles in the degradation of acetanilide herbicides have been cloned and characterized (Zhang et al. 2011, 2012; Shen et al. 2012). These enzymes belong to the amidase family and commonly contain a GGSS motif and Ser-Ser-Lys catalytic triads (Mayaux et al. 1990; Shin et al. 2002).
We previously isolated an efficient CMEPA-hydrolyzing strain T3-6 from acetochlor-polluted soil. This strain was identified as Delftia sp. T3-6 and isolated from a microbial consortium that could mineralize acetochlor completely. One member of this consortium could transform acetochlor into CMEPA, whereas another could grow using MEA as its sole carbon source and supply carbon sources for other consortium members. In this study, an amide hydrolase that hydrolyzed CMEPA into MEA was purified from Delftia sp. T3-6, and its encoding gene was cloned and expressed in a heterologous manner in Escherichia coli.
Materials and methods
Chemicals
CMEPA (96.5 % purity) and 2′,6′-diethyl-2-chloroacetanilide (CDEPA; 96.7 % purity) were purchased from Qingdao Vochem Co. Ltd. (Shandong, China). Acetochlor, butachlor, metamifop, propanil, and MEA (96.7 % purity) were purchased from Shanghai Jingchun Co. Ltd. All other chemicals used in this study were purchased from Sigma (St. Louis, MO, USA) and were analytical grade or higher purity.
Bacterial cultivation and crude extract preparation
Delftia sp. T3-6 (CCTCC No. M 2012526), which exerted strong hydrolase activity on CMEPA, was isolated from soil samples. The strain was precultured in Luria–Bertani medium (LB; containing 10.0 g/L tryptone, 5.0 g/L yeast extract, and 10.0 g/L NaCl, pH 7.0), harvested by centrifugation at 6,000 × g at 4 °C for 5 min, washed, and resuspended in 20 mM sodium phosphate buffer (PBS; pH 7.0). The cells were disrupted by sonication for 20 min (Sonicator 201 M, Kubota, Japan); cell suspensions were then centrifuged at 12,000 rpm for 20 min at 4 °C. The supernatants obtained from this step were referred to as crude extracts. Protein concentrations were determined using the Bradford (1976) method.
Purification of CMEPA hydrolase DamH
All purification steps were performed at temperatures ≤4 °C to avoid any possible enzyme denaturation. The cell extracts were fractionated using ammonium sulfate, and the precipitate (60–80 % saturation) was harvested by centrifugation at 12,000 rpm for 25 min. Precipitates were dissolved in ~50 mL of 20 mM PBS (pH 8.0) and dialyzed. The dialyzed enzyme fraction was then applied to a DEAE-Toyopearl 650 M (Toyopearl, Japan) column. The active fractions were pooled for hydrophobic interaction chromatography. The fractions containing CMEPA hydrolase activity were collected and concentrated. The enzyme was then subjected to gel filtration on a Sephadex G-200 column (2.6 × 150 cm, GE Healthcare, USA). Fractions containing CMEPA hydrolase activity were collected (Mayaux et al. 1990).
Zymogram analysis of CMEPA hydrolase
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was performed according to the method of Laemmli (1970). After electrophoresis, the gel was cut into two parts. One part was stained with 0.1 % Coomassie brilliant blue R-250 and used to analyze the size of DamH; the other was washed twice with 2.5 % (v/v) Triton X-100 for 30 min to remove the SDS. After washing, the gel was incubated with 0.1 M Tris-HCl buffer (pH 8.0) for 1 h at 4 °C to renature DamH and then stored in 20 mM Tris-HCl buffer (pH 8.0) at 4 °C for zymogram analysis. Gel sections with renatured DamH were incubated on 20 mM Tris-HCl agar plates containing 200 mg/L CMEPA. After incubation at 37 °C for 30 min, the gel was stained with 0.04 M potassium hexacyanoferrate(III) and 0.02 M 4-aminoantipyrene solution (El-Dib et al. 1975; Norwitz and Keliher 1980); the location where DamH existed became visible as a purple band. The stained gels were then compared with zymogram, and the molecular weight of the CMEPA hydrolase was determined (Bano et al. 2011).
Standard assay for the enzyme activities with cell extracts
DamH activity was determined by measuring the release of MEA formed during CMEPA hydrolysis. A reaction mixture containing 2 mM CMEPA and 10 μL enzyme diluted in 3 mL 20 mM Tris-HCl (pH 7.0) was incubated at 30 °C for 20 min. After incubation, 30 μL 0.2 M potassium hexacyanoferrate(III) and 30 μL 0.1 M 4-aminoantipyrene were added to the reaction mixture. MEA reacted with 4-aminoantipyrene to form a purple compound with a maximum absorption at 545 nm (Kotresha and Vidyasagar 2008). One unit of CMEPA hydrolase activity was defined as the amount of enzyme needed to release 1 μmol of MEA in 1 min.
Protein assay
Proteins were quantified using the Bradford (1976) method using bovine serum albumin as the standard; protein concentrations during purification studies were then calculated from the standard curve. The eluted fractions from the chromatographic separations were monitored at 280 nm.
Protein sequencing and mass spectroscopy analysis
N-terminal amino acid sequencing and peptide mass fingerprint analysis of the DamH sequence were performed using the Edman degradation method on an ABI Precise 494 protein sequencer (Shanghai GeneCore Bio Technologies Co. Ltd.). The stained gel band corresponding to the position of the purple band in the renatured gel was excised and used for peptide mass fingerprint analysis (Bo-Yuan Biological Technology Co. Ltd.). The resulting peptide fragments were analyzed by searches in Mascot website databases (http://www.matrixscience.com) to determine the sequences of peptide fragments.
Gene cloning, expression, and purification of the recombinant DamH
Genomic DNA was extracted from Delftia sp. T3-6 cells and purified as described previously to serve as the PCR template (Sambrook and Russell 2001). Oligonucleotide primers were designed to amplified the damH gene (Table 1). Primer DamPF (forward) and DamPR (reverse) was applied to amplify the internal 214 bp of damH. The 5′-nucleotide sequence ( 1–876 bp of damH) was amplified using primers DamNF and DamPR. The 3′-nucleotide sequence was amplified by thermal asymmetric interlaced (TAIL)-PCR using the specific primes F-SP2 and F-SP1, arbitrary primers ARB1, ARB2, and ARB3 (Liu and Whittier 1995; Terauchi and Kahl 2000). The complete gene was PCR-amplified using the following primers: Dam-F (forward), containing an NdeI site (underlined) at the initiation site of the damH gene; and Dam-R (reverse), containing a SalI site (underlined) after the stop codon and a 6× His tag before the stop codon.
The PCR products were digested using NdeI and SalI and inserted into the NdeI-SalI sites of pET29a(+) to obtain the plasmid pET29a-damH. The plasmid was then transformed into E. coli BL21(DE3). The transformants were grown in LB medium containing 50 μg/mL kanamycin at 37 °C until they reached mid-log phase; they were then induced with 0.2 mM IPTG at 18 °C for 24 h.
Crude enzyme extracts of E. coli BL21(DE3) were prepared using ultrasonic disruption, as described previously. The recombinant DamH was purified with Ni2+-NTA resin (Qiagen, Valencia, CA, USA) (Janknecht et al. 1991). The fractions containing recombinant DamH were collected and dialyzed against 20 mM Tris-HCl (pH 7.0) overnight at 4 °C to remove imidazole.
Biochemical properties of the purified recombinant DamH
Substrate specificity
The substrate specificity of DamH was determined using propanil, acetochlor, butachlor, metamifop, formamide, acetamide, propanamide, acrylamide, benzamide, benzeneacetamide, nicotinamide, pyrazinamide, anthranilamide, CMEPA, CDEPA, formanilide, acetanilide, 4-nitroacetanilide, 4-acetamido phenol, glyceryl triacetate, glycerol tributyrate, glycerol tricaproate, 4-nitrophenyl acetate, 4-nitrophenyl butyrate, 4-nitrophenyl caprylate, and ε-caprolactone. Substrate solutions were prepared in 20 mM Tris-HCl (pH 7.0) at a concentration of 1 mM. Amidase activity was determined by the release of ammonia using the phenol-hypochlorite ammonia detection method (Weatherburn 1967), esterase activity was assessed using the release of fatty acids (Ihara et al. 1991; Wei et al. 2003; Kaiser et al. 2006; Rashamuse et al. 2009), and arylamidase activity was determined under standard assay conditions, as described previously.
DamH activity on different concentrations (0.025–2 mg/mL) of the test substrates was determined, as described above. The kinetic rate constants K m and V max were calculated using a Lineweaver–Burk plot (Dowd and Riggs 1965) at a concentration range of 0.025–4 mg/mL.
Effects of pH and temperature on activity and stability
The optimal reaction pH was assessed in several buffers of varying pH at 30 °C. The following buffers were used: 20 mM citrate buffer, pH 3.0–6.0; 20 mM PBS, pH 6–8.0; 20 mM Tris-HCl buffer, pH 7.5–8.8; and 0.05 M glycine NaOH buffer, pH 8.8–11.0. The optimal reaction temperatures were determined using the optimal pH at temperatures from 4 to 50 °C.
To measure pH stability, the enzyme was incubated at 4 °C for 24 h in different buffers, and the residual activity was determined using the enzyme assay conditions described above. The thermal stability of DamH was assessed by incubating the enzyme preparations at different temperatures. Aliquots were removed at specific time intervals, and the activity remaining was measured using the enzyme assay conditions described above. Non-heated enzyme was used as the control (100 %).
Effect of metal ions and chemical agents on enzyme activity
The effects of potential inhibitors or activators on the enzymatic activity of DamH were determined by adding 1 mM of various metal salts (Cu2+, K+, Fe2+, Co2+, Ni2+, Fe3+, Zn2+, Ca2+, Mg2+, Cr3+, Ba2+, and Mn2+), chemical agents (Triton X-100, Tween-80, SDS, dimethyl formamide (DMF), and PMSF), and organic reagents (methanol, ethanol, acetone, acetonitrile, isopropanol, and DMSO) to the reaction mixture that had been pre-incubated for 10 min at 30 °C. Enzyme activity without any additive was used as the control and defined as 100 %.
Nucleotide sequence accession number
The sequence for the amide hydrolase gene damH has been deposited in the GenBank database under the accession number KF638270.
Results
Purification of DamH from Delftia sp. T3-6
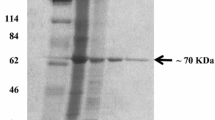
The CMEPA hydrolase DamH is a constitutive enzyme in Delftia sp. T3-6. When cultivated in LB media, T3-6 showed high DamH activity (up to 233 U/mg) without the addition of any inductive compounds. A summary of the purification process for DamH is shown in Table 2. After the four-step purification process, DamH was purified by 17.7-fold, with a 0.3 % recovery. SDS–PAGE was then used to analyze the purified DamH to verify its purity and determine its molecular weight. As shown in Fig. 2, the enzyme with CMEPA hydrolysis activity formed a single band on SDS–PAGE with a molecular weight of ~32 kDa.

Zymogram analysis and molecular weight measurement of the DamH by SDS–PAGE. Lane 1 protein molecular weight markers; lane 2 purified CMEPA; lane 3 potassium hexacyanoferrate(III) and 4-aminoantipyrene solution staining on CMEPA plate
Cloning and expression of the damH gene
The N-terminal 15-residue amino acid sequence of DamH was determined to be NH2-GTSPQSDFLRALFQS. Peptide fingerprint analysis also identified two additional peptide fragments of DamH as NH2-APEADDELRR and NH2-TNPLANPLKASYQGFPR. These amino acid sequences were used to design primers to amplify the damH gene from the genome of strain T3-6. The primers DamPF and DamPR were used to amplify the internal 214 bp of damH, the 3′ terminal nucleotide sequence of damH was amplified by TAIL-PCR, and the 5′ terminal nucleotide sequence of damH was amplified by DamNF and DamPR. Based on the 5′ and 3′ information obtained from these reactions, oligonucleotides for the complete nucleotide sequence were designed, which was then PCR-amplified using genomic DNA as the template. The cloned damH gene was 903 bp in length, encoding a 300-amino acid protein with a calculated molecular mass of 32 kDa. The corresponding protein was searched against the GenBank database using the BLASTP program (http://blast.ncbi.nlm.nih.gov). The searches revealed that the most related proteins were an arylamidase (98 % identity) and acetyl-hydrolase from Acinetobacter sp. SE19 (67 % identity) (Fig. 3). DamH contained the sequence (G-X-S-X-G) (Bornscheuer 2002), which is highly conserved and characteristic of the esterase family. A catalytic triad that is also highly conserved in this family, Ser-Glu-His (Ser149, Glu244, and His274), was also identified.

Phylogenetic tree of DamH and related esterase/arylamidase constructed by the neighbor-joining method. DamH was aligned with the following proteins, with their preferred structure indicated: Sphingobium quisquiliarum arylamidase/esterase, Acinetobacter sp. SE19 acetyl-hydrolase (Cheng et al. 2000), Xanthobacter flavus 6-hexanolactone hydrolase (Van Beilen et al. 2003), Rhodococcus sp. HI-31 caprolactone hydrolase (Mirza et al. 2009), Arthrobacter sp. Rue61a N-acetylanthranilate amidase (Kolkenbrock et al. 2006), Sphingomonas sp. Y57 propanil hydrolase (Zhang et al. 2011), Alcaligenaceae bacterium FB188 histone deacetylase (Nielsen et al. 2005), Rhodococcus sp. N771 amidase (Ohtaki et al. 2010), Paracoccus sp. M1-1 aryl acylamidase (Shen et al. 2012), Paracoccus sp. FLN-7 aryl-amidase (Zhang et al. 2012)
The overexpression of damH in E. coli BL21 was successful. The expressed and purified recombinant DamH was analyzed using SDS–PAGE (Fig. 4).

Analysis of the expression and purification of recombinant DamH. Lane 1 protein molecular weight marker; lane 2 extract of a 300-mM imidazole-eluted sample; lane 3 extract of a 200-mM imidazole eluted sample; lane 4 extract of a 100-mM imidazole-eluted sample; lane 5 extract of a 50-mM imidazole-eluted sample; lane 6 extract of a 0-mM imidazole eluted sample; lane 7 total protein of uninduced BL21(DE3) harboring recombinant damH in pET-29a(+)
Substrate specificity
The substrate specificity of the purified enzyme was examined using various amide and ester compounds. Data revealed that DamH catalyzed anilide compounds such as CMEPA, CDEPA, formanilide, acetanilide, 4-nitroacetanilide, and 4-acetamido phenol but could not hydrolyze propanil, acetochlor, butachlor, metamifop, formamide, acetamide, propanamide, acrylamide, benzamide, benzeneacetamide, nicotinamide, pyrazinamide, or 2-aminobenzamide. Although it shares homology and a similar catalytic triad like typical carboxylesterases, its amidase activity was much higher than its esterase activity. The substrate specificity and K m and k cat values of DamH were determined using various amide substrates (Table 3). The specific activities of DamH were 5,036.28 U/mg (CMEPA) and 611.94 U/mg (4-nitrophenyl acetate). The most suitable substrate was CMEPA, with K m and k cat values of 0.197 mM and 2,804.32 s−1, respectively.
Effects of pH and temperature on enzyme activity and stability of recombinant DamH
The effects of temperature and pH on the CMEPA hydrolase activity of DamH were assayed using purified enzyme. Recombinant DamH exerted high levels of activity at pH 6.5–7.0, with an optimum pH of 6.5 (Fig. 5a). Little activity was detected at a pH below 3.0 or above 8.0, but DamH retained >50 % activity after storage at pH 4.0–8.0 for 24 h (Fig. 5b). The enzyme was active at 5–50 °C, with an optimum temperature of 35 °C (Fig. 5c). The thermal stability of the purified enzyme was assessed by incubating DamH for 11 h in the absence of substrate at 0–55 °C (Fig. 5d). DamH was stable and retained >60 % residual activity for 11 h at temperatures <40 °C but was unstable at temperatures >45 °C. This suggests that DamH is a mesophilic enzyme.

Effects of pH and temperature on enzyme activity and stability of the recombinant DamH. a Determination of the optimal pH. Assays were carried at 35 °C for 15 min in buffers of varying pH. b pH stability. Activity was measured under optimal conditions (20 mM PBS, pH 6.5, 35 °C, 15 min) after incubation of the purified enzyme with buffers of varying pH at 4 °C for 24 h. c Determination of the optimal temperature. Activity was measured in 20 mM PBS, pH 6.5, at 5–50 °C for 15 min. d Thermal stability. Activity was measured under optimal conditions after incubation of the enzyme at indicated temperatures for 11 h
Effect of metal ions and chemical agents on the enzymatic activity of DamH
Metal ions play an important role in the activity of enzymes. As shown in Table 4, 1 mM Mn2+ stimulated DamH enzyme activity strongly, whereas Mg2+, Ba2+, K+, and Co2+ all reduced activity slightly, and Cu2+, Fe2+, Ni2+, and Zn2+ inhibited enzyme activity strongly. Ca2+ and Cr3+ ions had little effect on the enzyme activity at a concentration of 1 mM. The effect of various chemical agents on enzyme activity was also assessed (Table 5). Ethylenediaminetetraacetic acid (EDTA) did not inhibit the enzyme activity at a concentration of 10 mM. In contrast, detergents and most organic solvents inhibited the activity of DamH. Taken together, these data suggest that DamH is a metal ion-independent hydrolase, and EDTA did not chelate a possible metal ion required for the activity of the enzyme.
Discussion
Acetochlor is an important herbicide widely used for weeds controlling due to its high efficacy and low cost. Long-term application of acetochlor caused serious residue in soil. Microbial degradation is an important removal pathway of such compounds from the environment (Dictor et al. 2008). Many studies have focused on the microbial degradation of acetochlor, and several biodegradation intermediates have been identified (Coleman et al. 2000; Dagnac et al. 2002; Ye et al. 2002; Xu et al. 2006, 2008; Dictor et al. 2008). CMEPA is the key metabolic intermediate during the biodegradation of acetochlor; similar structures are also formed during the degradation of alachlor, butachlor, metolachlor, pretilachlor, and propisochlor. Till now, no strain that could mineralize acetochlor had been isolated (Xu et al. 2006, 2008). Delftia sp. T3-6 is a member of an acetochlor-degrading consortium which can completely mineralize acetochlor. Strain T3-6 can transform CMEPA to MEA, but enzyme and gene involved in the transformation process are unknown.
Propanil is an acetanilide herbicide with similar core structure with CMEPA and can be hydrolyzed by amidases (Shen et al. 2012; Zhang et al. 2012) and histone deacetylase-like propanil hydrolase PrpH (Zhang et al. 2011). The amidases AmpA (Zhang et al. 2012) and PamH (Shen et al. 2012) possess fatty acyl amidase and aryl arylamidase activities and can act on various short-chain aliphatic amines and aryl acylamides. Both amidases could not hydrolyze chloroacetamide herbicides such as acetochlor or butachlor, and their activity on CMEPA and CDEPA was not reported. PrpH is a histone deacetylase-like propanil hydrolase PrpH which could hydrolyze propanil only (Zhang et al. 2011). However, DamH showed different substrates specificity compared with PrpH, AmpA, and PamH. DamH could catalyze the hydrolysis of aryl acylamides and various ester substrates. DamH showed no activity on acetochlor or butachlor but could hydrolyze their degradation intermediates CMEPA and CDEPA; we deduced that steric hindrance effects of the ethoxymethyl/butoxymethyl groups of the parental compounds inhibited the approach of the these substrates to the active sites of enzyme. Meanwhile, DamH could not hydrolyze fatty acid amides and propanil, indicating that its catalytic mechanism is different from PamH and AmpA.
Sequence alignment analysis of hydrolases that act on amides or arylamides revealed that they could be clustered into three groups (Fig. 3). PamH and AmpA formed a group of amidases, whereas PrpH is a histone deacetylase homolog that formed an independent group. PamH and AmpA possess the typical amidase GGSSXG signature sequence and the Lys-Ser-Ser catalytic triad, suggesting that they belong to typical amidase family (Capel et al. 1995; Patricelli et al. 1999; Shen et al. 2012; Zhang et al. 2012). In contrast, DamH possessed the common G-X-S-X-G (amino acid residues 147 to 151) esterase motif (Bornscheuer 2002) and a S149, E244, H274 catalytic triad. Like many “GX class” esterases and lipases, DamH contains an HGX motif (79HGG81), indicating that it belongs to the “GX-type hydrolases (Kolkenbrock et al. 2006). Although it shares homology and a similar catalytic triad like typical carboxylesterases (Arpigny and Jaeger 1999), the enzyme clearly shows the highest activity in the hydrolysis of the model compounds CMEPA, which is, however, a tertiary amine. Hence, the enzyme is a special esterase with amide hydrolase activity. DamH showed much higher catalytic efficacy compared with other amidases. It worked five times faster in catalytic efficacy and 50 times more specific than AmpA on 4-acetamido phenol.
Of the enzymes with reported amides and ester hydrolytic activity, most were classified predominantly as amidases, with weaker esterase activity (Schmidt et al. 2007; Kourist et al. 2008; Wang et al. 2009; Syrén and Hult 2011). Biochemical characterization revealed that DamH was able to hydrolyze aryl arylamides and various esters. Similar phenomenon was observed in several aryl arylamide hydrolases (Pohlenz et al. 1992; Akutsu-Shigeno et al. 2006; Kolkenbrock et al. 2006). Amq from Arthrobacter nitroguajacolicus Rü61a is a bi-functional hydrolase that also acts on aryl-acylamides and esters (Kolkenbrock et al. 2006). Amp is predominantly an esterase with phenylacetate as the optimal substrate, whereas DamH tended to function as an amidase, using CMEPA as the optimal substrate. Pcp from Arthrobacter oxydans P52 is also an amide hydrolase classified as carboxyesterase which could hydrolyze p-nitrophenylbutyrate (Pohlenz et al. 1992). In recent years, many enzymes have been identified that catalyze more than one chemical transformation, which is known as catalytic promiscuity. This process plays an important role in the evolution of enzymes and in enzyme engineering (Khersonsky et al. 2006). The unique catalytic activity of DamH makes it an interesting research entity for the evolution and mechanisms of catalytic promiscuity.
References
Akutsu-Shigeno Y, Adachi Y, Yamada C, Toyoshima K, Nomura N, Uchiyama H, Nakajima-Kambe T (2006) Isolation of a bacterium that degrades urethane compounds and characterization of its urethane hydrolase. Appl Microbiol Biotechnol 70(4):422–429
Arpigny J, Jaeger K (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Bano S, Qader SAU, Aman A, Syed MN, Azhar A (2011) Purification and characterization of novel α-amylase from Bacillus subtilis KIBGE HAS. AAPS PharmSciTech 12(1):255–261
Bornscheuer UT (2002) Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol Rev 26(1):73–81
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(1):248–254
Capel PD, Ma L, Schroyer BR, Larson SJ, Gilchrist TA (1995) Analysis and detection of the new corn herbicide acetochlor in river water and rain. Environ Sci Technol 29(6):1702–1705
Cheng Q, Thomas SM, Kostichka K, Valentine JR, Nagarajan V (2000) Genetic analysis of a gene cluster for cyclohexanol oxidation in Acinetobacter sp. strain SE19 by in vitro transposition. J Bacteriol 182(17):4744–4751
Coleman S, Linderman R, Hodgson E, Rose RL (2000) Comparative metabolism of chloroacetamide herbicides and selected metabolites in human and rat liver microsomes. Environ Health Perspect 108(12):1151–1157
Crump D, Werry K, Veldhoen N, Van Aggelen G, Helbing CC (2002) Exposure to the herbicide acetochlor alters thyroid hormone-dependent gene expression and metamorphosis in Xenopus Laevis. Environ Health Perspect 110(12):1199–1205
Dagnac T, Jeannot R, Mouvet C, Baran N (2002) Determination of oxanilic and sulfonic acid metabolites of acetochlor in soils by liquid chromatography–electrospray ionisation mass spectrometry. J Chromatogr A 957(1):69–77
Dictor M-C, Baran N, Gautier A, Mouvet C (2008) Acetochlor mineralization and fate of its two major metabolites in two soils under laboratory conditions. Chemosphere 71(4):663–670
Dowd JE, Riggs DS (1965) A comparison of estimates of Michaelis–Menten kinetic constants from various linear transformations. J Biol Chem 240(2):863–869
El-Dib MA, Abdel-Rahman MO, Aly OA (1975) 4-Aminoantipyrine as a chromogenic agent for aromatic amine determination in natural water. Water Res 9(5):513–516
Grube A, Donaldson D, Kiely T, Wu L (2011) Pesticides industry sales and usage: 2006 and 2007 market estimates. In: Office of Chemical Safety and Pollution Prevention. US EPA, Washington
Hua N (2011) Amide herbicides formulations and their progress of R&D. Mod Agrochem 10(1):8–15
Ihara F, Kageyama Y, Hirata M, Nihira T, Yamada Y (1991) Purification, characterization, and molecular cloning of lactonizing lipase from Pseudomonas species. J Biol Chem 266(27):18135–18140
Jablonkai I (2000) Microbial and photolytic degradation of the herbicide acetochlor. Int J Environ Anal Chem 78(1):1–8
Janknecht R, de Martynoff G, Lou J, Hipskind RA, Nordheim A, Stunnenberg HG (1991) Rapid and efficient purification of native histidine-tagged protein expressed by recombinant vaccinia virus. Proc Natl Acad Sci U S A 88(20):8972–8976
Kaiser P, Raina C, Parshad R, Johri S, Verma V, Andrabi KI, Qazi GN (2006) A novel esterase from Bacillus subtilis (RRL 1789): purification and characterization of the enzyme. Protein Expr Purif 45(2):262–268
Khersonsky O, Roodveldt C, Tawfik DS (2006) Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol 10(5):498–508
Kolkenbrock S, Parschat K, Beermann B, Hinz H-J, Fetzner S (2006) N-Acetylanthranilate amidase from Arthrobacter nitroguajacolicus Rü61a, an α/β-hydrolase-fold protein active towards aryl-acylamides and-esters, and properties of its cysteine-deficient variant. J Bacteriol 188(24):8430–8440
Kotresha D, Vidyasagar G (2008) Isolation and characterisation of phenol-degrading Pseudomonas aeruginosa MTCC 4996. World J Microbiol Biotechnol 24(4):541–547
Kourist R, Bartsch S, Fransson L, Hult K, Bornscheuer UT (2008) Understanding promiscuous amidase activity of an esterase from Bacillus subtilis. ChemBioChem 9(1):67–69
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Liu Y, Whittier RF (1995) Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 25(3):674–681
Liu Y, Zhang Y, Liu J, Huang D (2006) The role of reactive oxygen species in the herbicide acetochlor-induced DNA damage on Bufo raddei tadpole liver. Aquat Toxicol 78(1):21–26
Liu Z, Yang H, Huang Z, Zhou P, Liu S-J (2002) Degradation of aniline by newly isolated, extremely aniline-tolerant Delftia sp. AN3. Appl Microbiol Biotechnol 58(5):679–682
Mayaux JF, Cerebelaud E, Soubrier F, Faucher D, Pétré D (1990) Purification, cloning, and primary structure of an enantiomer-selective amidase from Brevibacterium sp. strain R312: structural evidence for genetic coupling with nitrile hydratase. J Bacteriol 172(12):6764–6773
Mirza IA, Yachnin BJ, Wang S, Grosse S, Bergeron H, Imura A, Iwaki H, Hasegawa Y, Lau PCK, Berghuis AM (2009) Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J Am Chem Soc 131(25):8848–8854
Nielsen TK, Hildmann C, Dickmanns A, Schwienhorst A, Ficner R (2005) Crystal structure of a bacterial class 2 histone deacetylase homologue. J Mol Biol 354(1):107–120
Norwitz G, Keliher PN (1980) Effect of acidity and alkalinity on the distillation of phenol: interferences of aromatic amines and formaldehyde with the 4-aminoantipyrine spectro-photometric method for phenol. Anal Chim Acta 119(1):99–111
Ohtaki A, Murata K, Sato Y, Noguchi K, Miyatake H, Dohmae N, Yamada K, Yohda M, Odaka M (2010) Structure and characterization of amidase from Rhodococcus sp. N-771: insight into the molecular mechanism of substrate recognition. BBA-Proteins Proteomics 1804(1):184–192
Patricelli MP, Lovato MA, Cravatt BF (1999) Chemical and mutagenic investigations of fatty acid amide hydrolase: evidence for a family of serine hydrolases with distinct catalytic properties. Biochemistry 38(31):9804–9812
Pohlenz H-D, Boidol W, Schüttke I, Streber WR (1992) Purification and properties of an Arthrobacter oxydans P52 carbamate hydrolase specific for the herbicide phenmedipham and nucleotide sequence of the corresponding gene. J Bacteriol 174(20):6600–6607
Rashamuse K, Magomani V, Ronneburg T, Brady D (2009) A novel family VIII carboxylesterase derived from a leachate metagenome library exhibits promiscuous β-lactamase activity on nitrocefin. Appl Microbiol Biotechnol 83(3):491–500
Saha S, Dutta D, Karmakar R, Ray DP (2012) Structure–toxicity relationship of chloroacetanilide herbicides: relative impact on soil microorganisms. Environ Toxicol Pharmacol 34(2):307–314
Sambrook J, Russell D (2001) Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Schmidt M, Henke E, Heinze B, Kourist R, Hidalgo A, Bornscheuer UT (2007) A versatile esterase from Bacillus subtilis: cloning, expression, characterization, and its application in biocatalysis. Biotechnol J 2(2):249–253
Shen W, Chen H, Jia K, Ni J, Yan X, Li S (2012) Cloning and characterization of a novel amidase from Paracoccus sp. M-1, showing aryl acylamidase and acyl transferase activities. Appl Microbiol Biotechnol 94(4):1007–1018
Shin S, Lee T-H, Ha N-C, Koo HM, Kim S-Y, Lee H-S, Kim YS, Oh B-H (2002) Structure of malonamidase E2 reveals a novel Ser-cisSer-Lys catalytic triad in a new serine hydrolase fold that is prevalent in nature. EMBO J 21(11):2509–2516
Syrén P-O, Hult K (2011) Amidases have a hydrogen bond that facilitates nitrogen inversion, but esterases have not. Chemcatchem 3(5):853–860
Terauchi R, Kahl G (2000) Rapid isolation of promoter sequences by TAIL-PCR: the 5′-flanking regions of Pal and Pgi genes from yams (Dioscorea). Mol Gen Genet 263(3):554–560
Van Beilen JB, Mourlane F, Seeger MA, Kovac J, Li Z, Smits TH, Fritsche U, Witholt B (2003) Cloning of Baeyer–Villiger monooxygenases from Comamonas, Xanthobacter and Rhodococcus using polymerase chain reaction with highly degenerate primers. Environ Microbiol 5(3):174–182
Wang J-L, Xu J-M, Wu Q, Lv D-S, Lin X-F (2009) Promiscuous enzyme-catalyzed regioselective Michael addition of purine derivatives to α, β-unsaturated carbonyl compounds in organic solvent. Tetrahedron 65(12):2531–2536
Weatherburn M (1967) Phenol-hypochlorite reaction for determination of ammonia. Anal Chem 39(8):971–974
Wei Y-L, Kurihara T, Suzuki T, Esaki N (2003) A novel esterase from a psychrotrophic bacterium, Acinetobacter sp. strain no. 6, that belongs to the amidase signature family. J Mol Catal B Enzym 23(2):357–365
Xiao N, Jing B, Ge F, Liu X (2006) The fate of herbicide acetochlor and its toxicity to Eisenia fetida under laboratory conditions. Chemosphere 62(8):1366–1373
Xu J, Qiu X, Dai J, Cao H, Yang M, Zhang J, Xu M (2006) Isolation and characterization of a Pseudomonas oleovorans degrading the chloroacetamide herbicide acetochlor. Biodegradation 17(3):219–225
Xu J, Yang M, Dai J, Cao H, Pan C, Qiu X, Xu M (2008) Degradation of acetochlor by four microbial communities. Bioresour Technol 99(16):7797–7802
Yasmin S, D'Souza D (2010) Effects of pesticides on the growth and reproduction of earthworm: a review. Appl Environ Soil Sci 2010(2010):1–9
Ye C, Wang X, Zheng H (2002) Biodegradation of acetanilide herbicides acetochlor and butachlor in soil. J Environ Sci China 14(4):524–529
Zafeiridou G, Geronikaki A, Papaefthimiou C, Tryfonos M, Kosmidis EK, Theophilidis G (2006) Assessing the effects of the three herbicides acetochlor, 2, 4, 5-trichlorophenoxyacetic acid (2, 4, 5-T) and 2, 4-dichlorophenoxyacetic acid on the compound action potential of the sciatic nerve of the frog (Rana ridibunda). Chemosphere 65(6):1040–1048
Zhang J, Sun J-Q, Yuan Q-Y, Li C, Yan X, Hong Q, Li S-P (2011) Characterization of the propanil biodegradation pathway in Sphingomonas sp. Y57 and cloning of the propanil hydrolase gene prpH. J Hazard Mater 196:412–419
Zhang J, Yin J-G, Hang B-J, Cai S, He J, Zhou S-G, Li S-P (2012) Cloning of a novel arylamidase gene from Paracoccus sp. strain FLN-7 that hydrolyzes amide pesticides. Appl Environ Microbiol 78(14):4848–4855
Zhou Q-X, Zhang Q-R, Liang J-D (2006) Toxic effects of acetochlor and methamidophos on earthworm Eisenia fetida in phaiozem, northeast China. J Environ Sci-China 18(4):741–745
Acknowledgments
Grants from the Natural Science Foundation of Jiangsu Province, China (No. BK2012029), the Natural Science Foundation of China (31270095), and the National Science and Technology Support Program (2012BAD14B02) supported this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, F., Hou, Y., Zhou, J. et al. Purification of an amide hydrolase DamH from Delftia sp. T3-6 and its gene cloning, expression, and biochemical characterization. Appl Microbiol Biotechnol 98, 7491–7499 (2014). https://doi.org/10.1007/s00253-014-5710-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5710-y