
Abstract
The realization that majority of microbes are not amenable to cultivation as isolates under laboratory conditions has led to the culture-independent metagenomic approach as a novel technique for novel biocatalyst discovery. A leachate fosmid shotgun metagenome library was constructed and subsequently screened for esterolytic activities on a tributyrin agar medium. Nucleotide sequencing and translational analysis of an esterase-positive fosmid clone led to the identification of a 1,281 bp esterase gene (estC) encoding a protein (EstC) of 427 aa with translated molecular weight of 46.3 kDa. The EstC primary structure contained a signal leader peptide (29 aa), which could be cleaved to form a mature protein of 398 aa with molecular weight 43.3 kDa. Homology searches revealed that EstC belonged to the family VIII esterases, which exploit a serine residue within the S-x-x-K motif as a catalytic nucleophile. Substrate specificity studies showed that EstC prefers short to medium acyl chain length of p-nitrophenyl esters, a characteristic typical of “true” carboxylesterases. Moreover, EstC represents the first member of the family VIII esterases with a leader peptide and a detectable promiscuous β-lactam hydrolytic activity. Site-directed mutagenesis studies also revealed that in addition to Ser103 and Lys106 residues, the Tyr219 residue also plays a catalytic role in EstC. The organic solvent stability and the specificity towards esters of tertiary alcohols linalyl acetate (3,7-dimethyl-1,6-octadien-3-yl acetate) make EstC potentially useful in biocatalysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Culture enrichment techniques involving the cultivation of micro-organisms and subsequent screening of isolated strains for desired catalytic activities have conventionally been used to discover novel microbial enzymes (Wahler and Reymond 2001). However, due to the limitations in microbial cultivation, there is a widely held notion that these enrichment techniques fail to capture the majority of the total environmental genetic pool (Gans et al. 2005).
Recent advances in the technologies for novel enzyme discovery have included the culture-independent metagenomic approach (Lorenz et al. 2002; Rondon et al. 2000; Rashamuse et al. 2009). In principle, the metagenomic technique provides access to the total genetic pool in a given environment (Cowan et al. 2005; Gilliespie et al. 2002). A number of metagenome libraries have successfully been constructed and screened, providing novel enzymes and antibiotics with potential industrial and medical applications (Lammle et al. 2007; Gilliespie et al. 2005).
Microbial carboxylesterases are classified in eight families (families I–VIII) based on a comparison of primary structures (Arpigny and Jaeger 1999). Family VIII esterases represent a poorly characterized esterase family, with high sequence identity to class C β-lactamases and penicillin binding protein (Bornscheuer 2002). Despite high sequence identity to class C β-lactamases, members of family VIII esterases reported to date lack activity against standard β-lactam substrates (Elend et al. 2006; Rashamuse et al. 2007a; Ogino et al. 2004; Petersen et al. 2001; Sakai et al. 1999; Nishizawa et al. 1995). Moreover, unlike other microbial esterase families where the active-site serine residue is often located within the G-x-S-x-G or G-D-S-L motif (Bornscheuer 2002), family VIII active-site serine residue is situated within the S-x-x-K motif.
Domestic landfills provide a unique and varied environment in which a wide range of natural and synthetic compounds are found, potentially providing a basis for broad microbial diversity and evolution. Hence, they are of interest as sources of novel biomolecules discovery. In this study, we report on the construction and screening of carboxylesterase encoding gene(s) from a leachate metagenome shotgun fosmid library. One of the esterase-positive fosmid clones was sequenced, and the identified gene encoding carboxylesterase was expressed in Escherichia coli and the encoded enzyme was biochemically characterized.
Materials and methods
Strains, plasmids, and culturing conditions
E. coli EPI100-T1R (F- mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ-rpsL nupG trfA dhfr) was used as the host for cloned environmental DNA (Epicentre Biotechnologies, USA). E. coli JM109 was used as the host for routine subcloning, while E. coli BL21 (DE3) was used as the expression host. The cloning vectors, pSmart (Lucigen) and pTZ57R/T (Fermentas), were used for subcloning of the PCR products and enzyme-restricted DNA fragments. The vector pET28a (Novagen) was used for expression. All the restriction and modification enzymes were used as recommended by manufacturers. Primers were synthesized and purchased from Inqaba Biotech (Pretoria, South Africa). The standard molecular biology protocols followed were as described by Sambrook and Russell (2001).
Fosmid library construction and screening
A sample for environmental DNA (eDNA) isolation was an aqueous acidic leachate (pH 4.2, with brownish appearance) collected from the Chloorkop landfill site East of Johannesburg South Africa (26°03′17.50″S). A sample (20 ml) was immediately (without storage) subjected to environmental DNA extraction using the direct-lysis protocol described by Gilliespie et al. (2002).
The purified eDNA was subjected to metagenome library construction using the EpiFOS™ Fosmid Library Production Kit (Epicentre Biotechnologies, USA). Primary screening of recombinant clones in E. coli EPI100-T1R was performed on LB agar plates supplemented with isopropyl-β-d-thiogalactoside (IPTG, 0.1 mM), chloramphenicol (12.5 μg ml-1), tributyrin 1% (v/v), and gum arabic 0.1% (w/v), followed by incubation at 37°C. Esterase-positive clones were identified by the presence of zone of clearance around the colony margins.
DNA sequencing and sequence analysis
Nucleotide sequences of the esterase-positive fosmid clone were determined by random shotgun sequencing using an automated high throughput GS 20 DNA sequencer (Roche applied science). The open reading frame (ORF) identification and annotation were performed using CLC Combine Workbench software (CLCBIO, Denmark) with an aid of BLASTP search (Altschul et al. 1997). Sequence analysis and manipulation were performed using the BioEdit sequence alignment editor (Hall 1990).
Analytical methods
Esterase assays:
Unless otherwise stated, all enzyme assays were performed in triplicate. Routine esterase activity assays were performed by a standard colorimetric method measuring the release of p-nitrophenol from p-nitrophenyl esters at 410 nm (Rashamuse et al. 2007b), using a Beckman DU850 UV/visible spectrophotometer with a peltier temperature controller. Unless otherwise described, enzyme activity was measured at 30°C in 20 mM Tris–HCl, pH 7.5 with 1 mM p-nitrophenyl butyrate (dissolved in isopropanol) as the substrate. The extinction coefficient of p-nitrophenol under these conditions was 13,800 M-1 cm-1.
Determination of β-lactamase activity:
The β-lactam hydrolytic activity of EstC was determined spectrophotometrically using Nitrocefin [3-(2, 4 dinitrostyrl)-(6R, 7R-7-(2-thienylacetamido)-ceph-3-em-4-carboxylic acid, E-isomer)] as a substrate (Oxoid kit manual, 6th edition 1990, Unipath, Basingstoke, UK). The enzyme was incubated with Nitrocefin (1 mM) solution in (0.1 M phosphate; 1 mM EDTA, pH 7.0) at 30°C, and the rate change at 486 nm was recorded. The molar extinction coefficient of Nitrocefin under these conditions was 20,500. A β-lactamase from Bacillus cereus (Sigma) and the esterase (EstBL) from Burkholderia multivorans (Rashamuse et al. 2007a) were used as positive and negative control, respectively. The activity of EstC against non-chromogenic β-lactam substrates (ampicillin, carbenicilin, cephalosporin C, and Cephalotin) were measured as described by Avison et al. (2000).
Peptidase/amidase assay:
The amide hydrolytic activity of EstC was assayed using casein and labeled with N-(resorufin-4-carbonyl)-piperidine-4-carbonic acid based on the universal protease substrate kit (Roche Applied Science) and p-nitrobutyranilide as described by Kourist et al. (2008a).
Expression constructs
The estC gene was PCR-amplified from the plasmid (pSmart-EstC) using the pEst2CF/pEst2CR primer pair (Table 1), which introduces the NcoI and XhoI sites at the 5′- and 3′-end of the gene, respectively. The primer pair (pET2CF/pET2CR) targeted the truncated esterase gene without the leader peptide coding sequence and allowed the recombinant gene to be expressed in-frame with the 6x-His tag sequence at the 3′-end of the gene. The amplified PCR product was digested with NcoI/XhoI, followed by ligation into pET28a linearized with the same enzymes. The resultant pEstC expression construct was placed under the control of the T7 inducible promoter (IPTG inducible).
Site-directed mutagenesis
The pEstC template plasmid and the complementary mutagenic oligonucleotide pairs (Table 1) designed to introduce the S103A, K106A, Y219A, and S373A substitutions were used for PCR amplification using the HiFi™ DNA polymerase (Kapa Biosystems, South Africa). The PCR mixtures were treated with DpnI to digest the methylated parental template DNA. The resultant mutant plasmid constructs were used to transform E. coli BL21 cells, and the substitutions were confirmed by DNA sequencing.
Protein expression and purification
The E. coli BL21 cells harboring pEstC and the mutant expression constructs were used to inoculate 250 ml LB broth/kanamycin (30 µg ml-1). The growing cultures were incubated with shaking at 37°C to an OD600nm of 0.5, followed by induction with 1 mM IPTG for 4 h. Harvested cells (8,000×g, 10 min, 4°C; ~2 g wet weight) were resuspended in 10 ml B-PER bacterial protein extraction reagent (Pierce, USA) and incubated at room temperature for 10 min. The suspension was centrifuged (13,000×g, 10 min, 4°C), and the supernatant was used as a source for intracellular soluble proteins.
The intracellular soluble fraction was loaded onto HisPrepTM FF (16/10) IMAC column. The tagged protein was eluted with 250 mM imidazole in 50 mM phosphate buffer containing 0.3 M NaCl (pH 8.0). The eluted fraction was passed through VIVASPIN 10 kDa cut-off spin column (Vivascience, UK) to remove imidazole. The protein concentration of the purified sample was determined by the method of Bradford (1976), using bovine serum albumin as standard, while the purity of the sample was analyzed on denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) according to the method of Laemmli (1970).
Thin layer chromatography
Lyophilized purified enzyme (35 mg) was rehydrated in 1 ml phosphate buffer (0.1 M, pH 7.5) for 30 min. The rac-linalyl acetate substrate (15 mg) was added, and the reaction mixture was shaken (150 rpm) at 30°C. Samples were withdrawn and analyzed by TLC using Merck Silica gel 60 F and hexane/ethyl acetate (5:1) as an eluent. Compounds were visualized by spraying anisaldehyde solution [anisaldehyde (15 g)/ ethanol (250 ml)/conc. sulfuric acid (2.5 ml)] and heat treatment.
Biochemical characterization
The optimum temperature of the EstC was determined by measuring the rate of p-nitrophenyl butyrate hydrolysis over a temperature range (10–60°C). Substrate, 1 mM in 50 mM Tris–HCl buffer (pH 7.5), was incubated at desired temperatures and followed by addition of enzyme. Where necessary, the pH adjustments at set temperatures were performed to take into account the effect that changing temperature has on altering pH. The thermostability profile of EstC was measured by incubating the enzyme at various temperatures (20°C to 50°C), followed by measuring residual activities after every 30 min using the standard esterase assay.
To investigate the substrate specificity of EstC, the enzyme activity was determined using the standard assay in the presence of 1 mM of the specified p-nitrophenyl esters of various chain lengths: p-nitrophenyl acetate (C2), p-nitrophenyl butyrate (C4), p-nitrophenyl caprylate (C8), and p-nitrophenyl laurate (C12). Experimental data of initial velocity versus substrate concentration were fitted to the Michaelis–Menten equation.
Accession number
The gene nucleotide sequence data of the esterase gene (estC) has been deposited in the GenBank under the accession number FJ025785.
Results
Environmental DNA extraction
A recovery of a high molecular weight eDNA in a substantial concentration is an essential key step in the successful construction of a diverse gene library from environmental sources (Gabor et al. 2004). A modified environmental DNA protocol described by Gilliespie et al. (2005) was adapted for the eDNA extraction from aqueous leachate samples, yielding a pure eDNA (3.8 μg) with the A 260/230 nm and A 260/A280nm ratios of 1.8 and 2.3, respectively.
Library construction and screening
A fosmid library was constructed using a copy-controlled pEpiFOS-5 vector resulted in a library size of approximately 1.4 × 106 colony forming units. The restriction fragment length polymorphisms of 45 randomly selected clones using BamHI restriction enzymes showed non-redundant patterns and the average insert size of 35 kb.
A library screening for esterase-positive hits was carried out by identifying the halo-formation around the colony margins on the tributyrin agar plates. Approximately 10,000 chloramphenicol-resistant recombinants were screened and 87 halo-forming clones identified (hit rate 1:115). Out of the 87 esterase-positive clones, one fosmid clone designed Est01 (harboring recombinant fosmid designed pEst01) showed consistent high esterase activity and was chosen for further studies. This clone formed a halo in the absence of the IPTG within a 12 h period, indicating that the gene encoding esterolytic activity was being expressed in E. coli under the control of the intrinsic regulator elements. A secondary screening on the olive oil–Rhodamine B lipase specific agar assay (Kouker and Jaeger 1987) showed that Est01 clone lacks lipase activity.
Sequence analysis of the cloned pEST01 fosmid DNA insert
In order to locate the gene(s) encoding esterolytic activity within the pEst01 fosmid, a random shotgun sequencing of a complete insert DNA was performed. Nucleotide sequencing of the insert DNA revealed a 52.1 kb insert size, with an average GC content of 52.5%. Translational analysis of the nucleotide sequence of the 52 kb insert fragment revealed 32 complete open reading frames. One of the open reading frames (ORF7) was identified as putative carboxylesterase based on sequence similarity search using BLASTP (Altschul et al. 1997).
Nucleotide sequence and primary structure analyses of ORF7
ORF7 consisted of 1,281 bp, commencing with an ATG start codon and terminating with a TAA stop codon. The nucleotide sequence of ORF7 had a GC content of 56.3%. ORF7 encodes a 46.3 kDa deduced protein of 427 amino acids in length, with a predicted pI value of 4.77. The SignalP 3.0 signal peptide prediction program (Bendten et al. 2004) revealed that ORF7 encoded a pre-protein containing a putative 29 aa N-terminal signal peptide (with a maximum cleavage site probability of 0.9 between Ala29 and Gln30), which could be cleaved to form a mature protein of 398 aa with predicted molecular weight and pI values of 43.3 kDa and 4.38, respectively.
Analysis of the deduced protein sequence of ORF7 revealed the S-M-T-K sequence (amino acid positions 103–106), compatible with the S-x-x-K motif that is conserved in class C β-lactamases (Knox et al. 1996), penicillin binding proteins (PBPs) (Joris et al. 1988), and family VIII carboxylesterases (Arpigny and Jaeger 1999). The G-M-S-E-G sequence (amino acid positions 371–375), corresponding with a classical pentapeptide signature (G-x-S-x-G) motif, which harbors the catalytic serine in other esterase families (Bornscheuer 2002), was observed at the C terminus of the EstC primary structure.
Homology searches
The amino acid sequence of ORF7 showed 61% similarity and 44% identity to Hyphomonas neptunium family VIII carboxylesterase, 59% similarity and 43% identity to Solibacter usitatus class C β-lactamase, and 43% similarity and 36% identity to Caulobacter crescentus low molecular weight peptidase. Based on the high sequence similarity to family VIII carboxylesterases, the gene encoded by ORF7 was designated estC and the corresponding protein EstC. The multiple sequence alignment of EstC and closely related homologues showed that the S-x-x-K motif is well conserved (Fig. 1a). A corresponding phylogenetic tree revealed that EstC clusters with esterases from family VIII (Fig. 1b).

a Multiple sequence alignment of the S-x-x-K region of the mature EstC with other related proteins from family VIII carboxylesterases, class C β-lactamases, and penicillin binding proteins. Family VIII carboxylesterases represented by Est-Ag (Arthrobacter globiformis, accession no. AAA99492), Est-An (Arthrobacter nitroguajacolicus, accession no. CAD61039), EstB (B. gladioli, accession no. AAF59826), EstBL (B. multivorans, accession no. AAV97951), and Est-Sc (Streptomyces chrysomallus, accession no. CAA78842). Class C β-lactamases are represented by Lac-1 (Enterobacter cloacae, accession no. D4479) and Lac-2 (E. coli, accession no. X74512), and penicillin binding proteins are represented by PBP-1 (Streptomyces R61, accession no. P15555) and PBP-2 (B. cereus, accession no. CAA09676). b The corresponding evolutionary distance phylogram showing the position of EstC in relation to family VIII carboxylesterases, class C β-lactamase, and penicillin binding proteins. The phylogenetic tree is based on neighbor-joining analysis using PHYLOWIN package. The scale represents 0.1 amino acid substitution per position. The numbers on the tree show the levels of bootstrap support based on 500 resamplings. Sequences were obtained from GenBank
Protein expression and purification
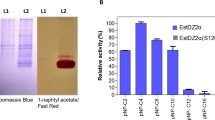
In order to study the biochemical properties of the recombinant enzyme, the estC gene (lacking the N-terminal coding sequence) was directionally ligated into pET28a to allow the expression of the gene under a strong T7 promoter. Following induction with IPTG for 6 h, the recombinant EstC was produced in a biologically active form in the soluble cytoplasmic fraction of E. coli cells. A one-step IMAC purification procedure was used to purify the enzyme since the expression construct was designed to allow the recombinant protein to be fused with the C-terminal 6x histidine tag. This purification procedure resulted in 5 mg of EstC (specific activity of 1043 U mg, using p-nitrobutyrate as a substrate) with a sevenfold enrichment and a yield of 55%. The SDS-PAGE analysis of the purified sample yielded a single protein band corresponding to a molecular weight of 43 kDa (Fig. 2a). A zymogram assay involving the staining of PAGE gel α-naphthol acetate substrate solution in the presence of fast blue B dye (Rashamuse et al. 2007b) exhibited single dark-red protein band (Fig. 2b), indicating that the purified enzyme was biologically active.

a SDS-PAGE (12%) electrophoregram showing different fractions from IMAC column: lane M molecular weight markers, lane 1 crude cell free fraction, lane 2 flow through fraction, lane 3 concentrated wash fraction, and lane 4 concentrated eluted fractions. b A zymogram showing EstC activity stain under native PAGE conditions: lane 1 marker, lane 2 α-naphthol acetate/fast blue B-stained EstC
Biochemical characterization
Temperature profile:
Activity of EstC was investigated over a temperature range of 10–60°C (Fig. 3a). The derived results indicate that EstC has a temperature optimum at 40°C. No activity was found at 60°C. Thermostability data (Fig. 3b) showed that EstC was thermo-labile with an approximate half-life of 30 min at 50°C.

a Effect of temperature on the recombinant EstC. b Thermostability profile of EstC
Effect of additives:
The effect of various additives was investigated using the standard assay by performing a 60 min incubation of the enzyme with 1 mM of additives (PMSF, DTT, and EDTA). The addition of PMSF resulted in 89% loss of activity (indicating a serine hydrolase mechanism), while in the presence of DTT and EDTA, EstC retained 100% activity (suggesting that neither cysteine residues nor metal co-factors are required for activity in this enzyme).
Solvent stability:
The solvent stability of EstC was examined by standard assay techniques with the enzyme being incubated in various buffer–solvent mixtures for 1 h. EstC was quite stable in the presence of a number of hydrophilic (low logP) solvents at 30% (v/v). For instance, EstC exhibited a sixfold higher activity in the presence of methanol (logP of −0.24), relative to activity in the control buffer, while activity was not affected by the presence of DMSO (logP of −1.3) and isopropanol (logP of −0.28). In the presence of N,N-dimethylformamide (logP of −1.0), acetone (logP of −0.23), and acetonitrile (logP of −0.33), EstC retained 89%, 44%, and 20% activity, respectively. No activity was detected when the buffer contained hydrophobic (high logP) solvents such as toluene (logP of 2.5) and hexane (logP of 3.5) at either 5% (v/v) or 30% (v/v).
Substrate selectivity and specificity:
The hydrolytic activity of EstC against different fatty acid esters was investigated using a range of p-nitrophenyl (p-NP) esters; (acetate, C2; butyrate, C4; caprylate, C8; laurate, C12; palmitate, C16). The EstC hydrolytic pattern against p-NP esters (Table 2) showed a strong preference towards short to medium length acyl chains of C2 to C8, with p-NP-C4 being the most easily hydrolyzed substrate. The hydrolytic activity of EstC against long chain esters decreased drastically (i.e. 97% lower for p-NP-C12 and no activity against p-NP-C16). The kinetic parameters of EstC were determined for short to medium chain length substrates (p-NP-C2, p-NP-C4, p-NP-C8). The K M value suggested that EstC has high affinity for p-NP-C8, while the catalytic efficiency constant (k cat/K M) showed that p-NP-C8 was the preferred substrate (Table 2).
The high sequence similarity of EstC to a number of class C β-lactamases and PBPs sequences prompted the investigation of the activity of this enzyme on β-lactam and amide-based substrates. While EstC did not show detectable amide hydrolytic activity against resorufin-labeled casein and p-nitrobutryranilide, it however exhibited β-lactam hydrolytic activity against Nitrocefin, with a specific activity of 13.51 U mg-1, which was 11% relative to the β-lactamase from B. cereus.
However, the activity of EstC against non-chromogenic β-lactam substrates (ampicillin, carbenicilin, cephalosporin C, and Cephalotin) was not detected.
A number of esterases have been tested for their ability to hydrolyze esters of tertiary alcohols (Kourist et al. 2008b). We have qualitatively examined the ability of EstC to hydrolyze esters of tertiary alcohol using 3,7-dimethyl-1,6-octadien-3-yl acetate (linalyl acetate) as a substrate. TLC analysis (data not shown) revealed the evidence of linalyl acetate hydrolysis to linalool by EstC. Although the hydrolytic activity of EstC against linalyl acetate was low, the ability of esterase EstC to cleave sterically hindered esters of tertiary alcohol is remarkable, particularly in view of the fact that a large number of carboxylesterases reported to date lacks the activity against similar substrates (Kourist et al. 2008b).
Site-directed mutagenesis:
In order to identify catalytic important residues within the EstC primary structure, we used a site-directed mutagenesis approach. Complementary mutagenic oligonucleotides were used to direct replication of the EstC encoding gene. These oligonucleotides were designed to direct the synthesis of both strands of the pEstC DNA template in a linear (non-PCR-based approach) manner and generate the proposed mutations: S103A, K106A, Y219A, and S373A. The alanine residue was chosen as a substitute residue because it lacks a bulky side chain and therefore would not likely impose any steric and electrostatic effects or disrupt main-chain conformation as may occur with other residues such the proline and glycine residues, respectively.
Multiple sequence alignment revealed that three of the targeted residues (Ser103, Lys106, Tyr219) were highly conserved in all of the three different enzyme groups (β-lactmases, PBPs, and family VIII esterases) harboring the S-x-x-K motif. Heterologous expression of the mutant constructs revealed the same level of protein expression as the wild type (data not shown). Activity analysis of the mutants relative to wild type showed that S103A and K106A substitutions resulted in complete loss of activity, while the Y219A substitution resulted in 90% loss of activity. The catalytic efficiency (k cat/K M) of EstC and S373A were essentially similar, while that of Y219A substitution was fivefold lower (Table 3).
Discussion
Activity-based screening of metagenomic libraries has a large potential to discover new classes of genes with novel and useful functions (Lorenz et al. 2002). In this study, the application of the culture-independent metagenomic approach coupled with functional screening was demonstrated and led to the discovery and characterization of a novel family VIII carboxylesterase (EstC).
The EstC primary structure revealed the presence of the S-M-T-K sequence corresponding to the catalytic S-x-x-K motif, which is found in a number of hydrolase families namely: family VIII esterases (Arpigny and Jaeger 1999), class C β-lactamases (Knox et al. 1996), and penicillin binding proteins (Joris et al. 1988). However, the other two highly conserved class C β-lactamases (-Y-A-N-) and (K-T/S-G) motifs (Joris et al. 1988) located at the middle (position 209–211) and C terminus (426–428), respectively, were not observed within the primary structure of EstC. The G-M-S-E-G sequence corresponding with the G-x-S-x-G motif was present at the C-terminal of the EstC primary structure. The corresponding C terminus located G-x-S-x-G sequences have previous been reported in other members of family VIII esterases (Berger et al. 1988; Schutte and Fetzner 2007).
The EstC primary structure analysis also showed the presence of a putative amino acid N-terminal leader peptide characterized by a positively charged N-terminus, a hydrophobic core region, and a polar C-terminal region (Bendten et al. 2004). This observation suggests that EstC could be a periplasmic or extracellular enzyme, which is directly exported through the membrane with the aid of the leader sequence via the two step Xcp-dependent secretion pathways, which are normally mediated by the N-terminal signal peptide (Arpigny and Jaeger 1999).
Literature searches revealed that all primary structures reported to date from family VIII esterases lack the N-terminal signal peptide (Bornscheuer 2002; Arpigny and Jaeger 1999). Based on this information, the expression construct (pEstC) was designed, excluding the leader peptide encoding sequence but in-frame with the C-terminal His-tag encoding sequence. The estC gene was functionally expressed in E. coli in a biologically active form without the leader peptide coding sequence. The molecular mass of the purified EstC was estimated to be 43 kDa, consistent with the estimated molecular weight calculated from the translated nucleotide sequence of the mature protein. The monomeric 43 kDa molecular state of EstC was within the 42–45 kDa range, which has been reported for other family VIII esterases (Elend et al. 2006; Schutte and Fetzner 2007; Rashamuse et al. 2007a; Ogino et al. 2004; Petersen et al. 2001; Sakai et al. 1999; Nishizawa et al. 1995)
The EstC showed no lipase activity on olive oil/Rhodamine B lipase specific assay (Kouker and Jaeger 1987), suggesting that the enzyme was a “true” carboxylesterase. The observation was further confirmed by substrate specific profiling (using p-nitrophenyl esters of different chain length), which revealed that EstC prefers short to medium chain length, typical true carboxylesterases (Jaeger et al. 1999). EstC lost approximately 90% of the activity in the presence of 1 mM PMSF, consistent with the reports from other members of family VIII, which also showed significant loss of activity in the presence of PMSF (Ogino et al. 2004; Petersen et al. 2001; Nishizawa et al. 1995). The inhibition of these enzymes confirms the involvement of a serine residue in the catalytic mechanism of EstC. It is well established that PMSF inhibits many serine hydrolyses by covalent linkage to the activated serine hydroxyl group, thereby mimicking the first transition state in ester hydrolysis (Jaeger et al. 1999).
Substrate profiling revealed that, in addition to ester bond hydrolysis, EstC hydrolyzes the amide bond of the β-lactam ring of the Nitrocefin substrate. To our knowledge, all members of family VIII esterases reported to date lack of detectable β-lactam ring hydrolysing activity. This suggests that the recognition of β-lactam substrates by EstC could have been maintained as the enzyme evolves from class C β-lactamases (Petersen et al. 2001). Preliminary data derived from Burkholderia gladioli EstB suggested that the lack β-lactam-hydrolyzing activity in family VIII esterases could be due to steric reasons (Wagner et al. 2002). The observed β-lactam-hydrolyzing activity of EstC could imply that the EstC substrate binding pocket is designed to overcome steric hindrances observed in other family members. Moreover, the ability of EstC to hydrolyze Nitrocefin provides an indication of the possible function of this enzyme in antibiotic resistance (Elend et al. 2006).
Enantio-pure alcohols are important building block in many pharmaceutical applications (Kourist et al. 2008b). Sequence similarity (31% identity) between EstC and EstB from B. gladioli, which hydrolyze ester of tertiary alcohol, linalyl acetate (Wagner et al. 2002), promoted the investigation of EstC activity against the same substrate. Based on TLC analysis, it was shown that EstC hydrolyzes linalyl acetate but with low efficiency. Further analysis of EstC showed the GGGL sequence, which corresponds to GGGX (X-denoting hydrophobic residue) motif. Henke and co-workers have shown that the presence of the GGGX motif is linked with the specificity for tertiary alcohols (Kourist et al. 2008b; Henke et al. 2002). All enzymes investigated bearing this motif were found to be active towards acetates of tertiary alcohols, while enzymes bearing the more common GX motif did not catalyze the model substrates (Henke et al. 2002). It is noted however that the GGGX motif in these esterases was located adjacent to the oxyanion region (Kourist et al. 2008b; Henke et al. 2002) unlike in EstC, where this motif was located towards the C terminus of the protein. While the significance of the locality of the GGGX motif is unclear, the ability of EstC to hydrolyze linalyl acetate supports the importance of this motif.
The EstB 3-D structural data (Wagner et al. 2002) suggested a number of active-site residues (Ser103, Lys106, Tyr219 and Ser318), which are potentially involved in a hydrogen bonding network with either the catalytic Ser103 residue or with substrate and associated water molecules. The corresponding putative active-site residues (Ser74, Lys77, and Tyr 181) were also identified in EstBL model structure (Rashamuse et al. 2007a). We have selected a number of corresponding residues (Ser103 Lys106, Tyr219, and Ser373) from the EstC primary structure for mutagenesis studies. With an exception of S373A mutation, all of other mutations were shown to be catalytically important in EstC reaction mechanism (Table 3). Although the roles of serine and Lys residues in the consensus S-x-x-K motif as catalytic nucleophile and base, respectively, are well established (Sakai et al. 1999; Wagner et al. 2002), it was the Y219A substitution that was intriguing. The loss of activity by the Tyr219 substitution seem to suggest that Tyr219 plays a role analogous to that proposed for Tyr150 from the X-ray crystal structure of the Citrobacter freundii class C β-lactamase (Knox et al. 1996; Oefner et al. 1990; Zawadzke et al. 1996). It could be suggested that the Tyr219 residue plays a role in the catalytic mechanism of EstC as a result of its optimally positioning to act as a proton acceptor for the hydrogen atom of Ser103, thereby activating Ser103 for nucleophilic attack of the ester-carbonyl of the substrate molecule. At the same time, such reaction consequently position Tyr219 favorably to act as a proton donor to the main-chain NH groups of residues Ser103 during the release of the tetrahedral intermediate.
The application of the activity-based screening of large insert metagenomic libraries has led to the discovery of a new member of family VIII esterases. The EstC primary structure analysis revealed only moderate identity (44%) to any known carboxylesterases in the databases. The unique primary structure of EstC and the promiscuous β-lactamase activity of the enzyme demonstrate the usefulness of applied metagenome technology in the discovery of novel enzyme genes.
References
Altschul SF, Madden TS, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402
Arpigny KL, Jaeger KE (1999) Bacterial lipolytic enzymes: classification and properties. J Biochem 343:177–183
Avison BM, Niumsup P, Walsh TR, Bennett PM (2000) Aeromonas hydrophila AmpH and CepH ß-lactamases: depressed expression in mutants of Escherichia coli lacking creB. J Antimicro A Chemother 46:695–702
Bendten JD, Nielsen H, von Heijnie G, Brunak S (2004) Improved prediction of signal peptide: SignalP 3.0. J Mol Biol 340:783–795
Berger R, Hoffmann M, Keller U (1998) Molecular analysis of a gene encoding a cell bound esterase from Streptomyces chrysomallus. J Bacteriol 180:6396–6399
Bornscheuer UT (2002) Microbial carboxylesterases: classification, properties and application in biocatalysis. FEMS Microbiol Rev 26:73–81
Bradford MM (1976) A rapid and sensitive method for quantification of microgram quantities of protein utilizing the principle of protein-dye binding. J Anal Biochem 72:248–254
Cowan D, Meyer Q, Stafford W, Muyanga S, Cameron R, Wittwer P (2005) Metagenomic gene discovery: past, present and future. Trends Biotech 23:321–329
Elend C, Scheisser C, Leggewie C, Babiak P, Carballeira D, Steele L, Reymond L, Jaeger K, Streit W (2006) Isolation and biochemical characterization of two novel metagenome-derived esterases. Appl Environ Microbiol 72:3637–2645
Gabor EM, Alkema WB, Janssen DB (2004) Quantifying the accessibility of the metagenome by random expression cloning technique. Environ Microbiol 6:948–958
Gans J, Wolinsky M, Dunbar J (2005) Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309:1387–1390
Gilliespie DE, Brady SF, Bettermann AD, Cianciotto NP, Liles MR, Rondon MR, Goodman RM, Handelsman J (2002) Isolation of antibiotics turbomycinA and B from metagenomic library of soil microbial DNA. Appl Environ Microbiol 68:4301–4306
Gilliespie DE, Rondon MR, Goodman RM, Handelsman J, Williamson LL (2005) Metagenomic library from uncultured microorganisms. In: Osborn AM, Smith CJ (eds) Molecular microbial ecology. Taylor and Francis group, New York, pp 261–279 Ch1
Hall TA (1990) BioEdit: a user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl acids Symp Ser 41:95–98
Henke E, Pleiss J, Bornscheuer UT (2002) Activity of lipases and esterases towards tertiary alcohols: insights into structure-function relationships. Angew Chem Int Ed 41:3211–3213
Jaeger KE, Dijkstra BW, Reetz MT (1999) Bacterial biocatalysis: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Ann Rev Microbiol 53:315–351
Joris B, Ghuysens JM, Dive G, Renard A, Dideberg O, Charlier P, Frere JM, Kelly JA, Boyington JC, Moews PC (1988) The active-site-serine penicillin-recognizing enzymes as members of the Streptomyces R61 DD-peptidase family. J Biochem 250:313–324
Knox JR, Moews PC, Frere JM (1996) Molecular evolution of bacterial β-lactam resistance. Chem Bio 3:937–947
Kouker G, Jaeger KE (1987) Specific and sensitive plate assay for bacterial lipases. Appl Environ Microbiol 53:211–213
Kourist R, Bartsch S, Fransson L, Hult K, Bornscheuer U (2008a) Understanding promiscuous amidase activity of an esterase from Bacillus subtilis. Chem BioChem 9:67–69
Kourist R, de Maria P, Bornscheuer U (2008b) Enzyme synthesis of optically active tertiary alcohols: expanding the bioclaysis toolbox. Chem BioChem 9:491–498
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lammle K, Zipper H, Breuer M, Hauer B, Buta C, Brunner H, Rupp S (2007) Identification of novel enzymes with different hydrolytic activities by metagenome expression cloning. J Biotechnol 127:575–592
Lorenz P, Liebeton K, Niehaus F, Eck K (2002) Screening novel enzymes for biocatalytic processes: accessing the metagenome as a resource of novel functional sequences space. Curr Opin Biotechnol 13:572–577
Nishizawa M, Shimizu M, Ohkawa H, Kanaoka M (1995) Stereoselective production of (+)-trans-chrysantemic acid by microbial esterase: cloning, nucleotide sequence, and overexpression of the esterase gene of Arthrobacter globiformis in Escherichia coli. Appl Environ Microbiol 61:3208–3215
Oefner C, D’Arcy A, Daly JJ, Gubernator K, Charnas RL, Heinze I, Hubschwerlen C, Winkler FK (1990) Refined crystal structure of beta-lactamase from Citrobacter freundii indicates a mechanism for beta-lactam hydrolysis. Nature 343:284–288
Ogino H, Mimitsuka T, Muto T, Matsumura M, Yasuda M, Ishimi K, Ishikawa H (2004) Cloning, expression, and characterization of a lipolytic enzyme gene (lip8) from Pseudomonas aeruginosa LST-03. J Mol Microbiol Biotechnol 7:212–223
Petersen EI, Valinger G, Solkner B, Stubenrauch G, Schwab H (2001) A novel esterase from Burkholderia gladioli shows high deacetylation activity on cephalosporins is related to β-lactamases and DD-peptidases. J Biotechnol 89:11–25
Rashamuse K, Ronneburg F, Hennessy F, Visser D, van Heerden E, Piater L, Litthauer D, Moller C, Brady D (2009) Discovery of a novel carboxylesterase through functional screening of pre-enriched environmental library. J Appl Microbiol. doi:https://doi.org/10.1111/j.1365-2672.2008.04114.x
Rashamuse K, Burton S, Stafford W, Cowan D (2007a) Molecular characterization of a novel family VIII esterase from Burkholderia multivorans UWC10. J Mol Microbiol Biotechnol 13:181–188
Rashamuse K, Burton S, Cowan D (2007b) A novel recombinant ethyl ferulate esterase from Burkholderia multivorans. J Appl Microbiol 103:1610–1620
Rondon MR, August PR, Bettermann AD, Brady SF, Grossman TH, Liles MR, Loiacone KA, Lynch BA, MacNeil IA, Minor C (2000) Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 66:2541–2547
Sakai Y, Ishikawa J, Fukasaka S, Yumiroto H, Mitsui R, Yanaese H, Kato N (1999) A new carboxylesterase from Brevibacterium lines IFO 12171 responsible for the conversion of 1,4-butanediol diacrylate to 4-hydroxybutyl acrylate: purification, characterization, gene cloning and gene expression in Escherichia coli. Biosci Biotechnol Biochem 63:688–697
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Schutte M, Fetzner S (2007) EstA from Arthrobacter nitroguajacolicus R_61a, a thermo- and solvent-tolerant carboxylesterase related to class C β-lactamases. Curr Microbiol 54:230–236
Wagner GU, Petersen EI, Schwab H, Kratky C (2002) EstB from Burkholderia gladioli: A novel esterase with a β-lactamase fold reveals steric factors discriminate between esterolytic and β-lactam cleaving activity. Protein Sci 11:467–478
Wahler D, Reymond JL (2001) Novel methods for biocatalyst screening. Curr Opin Chem Biol 5:152–158
Zawadzke LE, Chen CCH, Banjeree S, Li Z, Wasch S, Kapadia G, Moult J, Herzberg O (1996) Elimination of the hydrolytic water molecule in a class A β-lactamase mutant—crystal structure and kinetics. J Biochem 35:16475–16482
Acknowledgments
The work was supported by the CSIR-YREF (Young Researcher Establishment Fund). The authors would also like to thank Mr Harris Tshwane Manchidi for the help with sample collection, Dr Edwin Mutlane for synthesizing the p-nitrobutyranilide substrate, and the members of the CSIR (Enzyme technologies group) for their useful comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rashamuse, K., Magomani, V., Ronneburg, T. et al. A novel family VIII carboxylesterase derived from a leachate metagenome library exhibits promiscuous β-lactamase activity on nitrocefin. Appl Microbiol Biotechnol 83, 491–500 (2009). https://doi.org/10.1007/s00253-009-1895-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-1895-x