
Abstract
Transposons are developing molecular tools commonly used for several applications: one of these is the delivery of genes into microorganisms. These mobile genetic elements are characterised by two repeated insertion sequences that flank a sequence encoding one or more orfs for a specific transposase that moves these sequences to other DNA sites. In the present paper, the IS2 transposon of Escherichia coli K4 was modified in vitro by replacing the sequence coding for the transposase with that of the kfoC gene that codes for chondroitin polymerase. KfoC is responsible for the polymerisation of the bacterial capsular polysaccharide whose structure is analogous to that of chondroitin sulphate, a glycosaminoglycan with established and emerging biomedical applications. The recombinant construct was stably integrated into the genome of E. coli K4 by exploiting the transposase from endogenous copies of IS2 in the E. coli chromosome. A significant improvement of the polysaccharide production was observed, resulting in 80 % higher titres in 2.5-L fed-batch cultivations and up to 3.5 g/L in 22-L fed-batch cultures.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Different classes of active transposons have been observed in all genomes studied so far, including microorganims, plants and animals (also humans), with just few exceptions. Although their level of activity varies in the different species, they play key roles in the evolution of genomes generating variability by modifying genes and genomes from both structural and functional points of view.
Transposons are mobile genetic elements characterised by the presence of two terminal, repeated and inverted insertion sequences that flank one or more genes coding for a transposase, the enzyme that is specifically responsible for their mobility. IS2 is a 1.3-kb transposable element normally found in both the genome and in the F-plasmid of Escherichia coli strains (Ghosal et al. 1978). It is a member of the IS3 family that is characterised by an internal sequence coding for two orfs indicated as OrfA and OrfB flanked by imperfect inverted repeats, namely inverted left repeat (IRL) and inverted right repeat (IRR). OrfA codes for InsA, a 14-kDa protein that was shown to negatively regulate transposition (Hu et al. 1994). OrfA and OrfB code for a 46-kDa protein indicated as InsAB, via a −1 translational frameshift mechanism. InsAB is the transposase and, therefore, is responsible for the expression and transposition of IS2 (Hu et al. 1996). Lewis and Grindley (1997) demonstrated, in fact, that the overexpression of InsAB increases the frequency of transpositional recombination and the formation of two transpositional products and figure-eight molecules that are processed into IS2 minicircles.
An IS2 insertion sequence is located within region 2 of the gene cluster responsible for capsular polysaccharide (CPS) biosynthesis of E. coli K4 (Ninomiya et al. 2002). The latter belongs to the serological group II of K antigens organised into three regions. Regions 1 and 3 include the kps genes, common to all group II members. They are primarily involved in the transport of the polymer into the periplasm and outside the cell wall (Smith et al. 1990). The genes coding for enzymes that direct the synthesis and assembly of the final polysaccharide are found in region 2, the serotype-specific region. Besides three genes of unknown function (kfoB, kfoG, kfoD), region 2 is comprised of kfoE that is responsible for the addition of fructose residues (Trilli et al. 2011), kfoA coding for a UDP-glucose 4-epimerase, kfoF coding for a UDP-glucose dehydrogenase and the well-characterised kfoC gene coding for chondroitin polymerase (Ninomiya et al. 2002; Suzuki 2008). The insertion sequence, IS2, is located between the kfoC and kfoD genes.
E. coli K4 is a natural source of a capsular polysaccharide whose structure is closely related to that of the glycosaminoglycan (GAG) chondroitin sulphate (CS). As a matter of fact, they share a common carbon backbone, chondroitin, formed by repeated disaccharide units of N-acetyl-d-galactosamine (GalNAc-β 1:4 linked) and glucuronic acid (GlcA-β 1:3 linked) and differ in the presence of fructose and sulphate residues. CS has a central role in the extracellular matrix of vertebrates since it boosts the biosynthesis of connective tissue components and it inhibits enzymes that degrade cartilage (Imada et al. 2010). For this reason, CS is widely used for the treatment of osteoarthrithis. However, recent literature has also investigated attractive potential applications of this GAG in the formulation of skin substitutes, cancer prevention and vaccine development (Achur et al. 2008; Wang et al. 2006; Pothacharoen et al. 2006).
In order to guarantee an unlimited source of CS and bypass the current animal-based extraction procedures, numerous attempts to increase the levels of CPS produced by wild-type E. coli K4 have focused on the improvement of fermentation and purification strategies from simple batch processes on a 12-L scale (Manzoni et al. 1996) to the development of high cell density cultivation followed by purification of the polysaccharide through membrane ultrafiltration strategies and ethanol precipitation (Cimini et al. 2012; Schiraldi et al. 2012). Furthermore, a natural CS equivalent was obtained by chemical sulphation of the purified chondroitin as described by Bedini et al. (2011). Yields were further improved by single overexpressions of two genes involved in the biosynthesis and regulation of CPS production, namely kfoC and rfaH. In the first case, even though KfoC was shown to be crucial in the improved production of polysaccharide, the use of a vector expression system was found to be unsuitable for fermentation processes due to its instability in the unconventional genetic background of E. coli K4 (Cimini et al. 2010a, b). In the second case, the employment of an integration cassette for rfaH overexpression resulted in high yields (5.3 g/L) of CPS in DO-stat 2-L fed-batch processes (Cimini et al. 2013). Recently, the kfoE gene was deleted from the genome of E. coli K4 in order to produce a defructosylated polymer that is more similar to the target active principle (Trilli et al. 2011).
The presence of transposons clearly impacts the host genome where they can exert deleterious effects by disrupting genes or by negatively influencing gene regulation or even promoting damaging recombinational events (Hedges and Deininger 2007; Callinan and Batzer 2006). However, besides the established primary role in genome evolution, they represent an important tool for cell engineering. They have long been used in molecular biology for several purposes. For instance, transposon-based vectors are useful for integrating genes into different mammalian cell types. In fact, the sleeping beauty transposition element was used for transfecting hematopoietic cells in a stable and effective manner (Izsvak et al. 2009) and also for generating induced pluripotent stem cells (Muenthaisong et al. 2012). Several applications have also been reported which regard the use of transposons for the random mutagenesis of both prokaryotes and eukaryotes (Hayes 2003) or for the identification of functional genes (Lewenza et al. 2005; Shin et al. 2006; Wright et al. 2001). Tn5-based mutagenesis was in fact used as a tool to identify essential genes in E. coli for the construction of a minimal bacterial genome (Yu and Kim 2008). Several authors have also described the use of transposons for the creation of transcriptional or translational fusions for the study of differentially regulated genes (Lewenza et al. 2005; Buchan et al. 2008).
Transposons therefore represent a novel and potentially interesting approach for the engineering of E. coli K4. In the present paper, we describe the modification of an IS2 element of E. coli K4 and its use for the generation of a recombinant strain overexpressing the endogenous gene kfoC that is involved in capsular polysaccharide biosynthesis. A significant enhancement of CPS production was obtained, demonstrating the efficacy of the engineering method and confirming the critical role of KfoC, as previously demonstrated (Cimini et al. 2010a, b). The stability of the engineered strain was verified not only by repeated growth cycles in shake-flasks but also through batch and fed-batch experiments in 2.5- and 22-L fermenters. These results are central towards pilot-scale and large-scale production processes.
Materials and methods
Medium
The standard medium used for all shake-flask and fermenter production studies consisted of a defined salt medium (KH2PO4 2 g/L, K2HPO4 9.7 g/L, Na3C6H5O7 0.5 g/L, (NH4)2SO4 1 g/L, MgCl2 0.1 g/L) supplemented with glycerol (10 g/L) as the main carbon source and neutralised soy peptone (1 g/L) as an additional nitrogen source (Oxoid). Luria-Bertani (LB) medium was used during transformation experiments. Transformed cells were grown on LB medium supplemented with kanamycin (50 μg/mL). The wild-type strain used in all experiments was E. coli K4 serotype O5:K4:H4 (CCUG 11307), purchased from the Culture Collection of the University of Gothenburg.
Materials
Genomic DNA, plasmid DNA and RNA were isolated using the Qiagen DNeasy kit and the QIAamp DNA tot, the Qiagen miniprep kit and the Qiagen RNeasy kit (Qiagen, Valencia, CA), respectively, according to the manufacturer’s instructions. Restriction endonuclease digestions, DNA ligations, SDS-PAGE and agarose gel electrophoresis were performed using standard techniques (Sambrook and Russel 2001).
Construction of the cassette for kfoC overexpression: IRL-kfoC-IRR
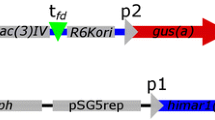
The strategy used for the construction of the transposon containing kfoC is shown in Fig. 1. The kfoC gene and the IRL region belonging to the IS2 insertion sequence were amplified from E. coli K4 chromosomal DNA. kfoC was amplified with primers 5′-kfoC (Cgg gAT CCC gAT gAg TAT TCT TAA TCA AgC) and 3′-kfoC (CgA gCT Cgg CCA gTC TAC ATg TTT ATC) containing the BamHI and SacI linker sites, respectively. Primers irla (5′-TAg ACT ggC CCC CTg AAT CTC C-3′) and irlb (5′-Cgg gAT CCT CCA ATg ACT AgT CTA AAA ACT Ag-3′) were used for the amplification of the IRL sequence from IS2. Irlb contains the linker site for BamHI. Polymerase chain reaction (PCR) was carried out with Expand High fidelity PCR System (Roche, Monza, Italy) according to the manufacturer’s protocol.

Schematic representation of the engineering strategy used for the construction of BK4071
The complementary fragments, containing the SacI linker site, IRR (5′-Tgg ATT TgC CCC TAT gTT TCC AgA TAC CTg TTA TCA CTT AAA gCT-3′) and anti-IRR (5′-TTA AgT gAT AAC Agg TAT CTg gAA ACA TAg ggg CAA ATC CA-3′) that correspond to the IRR region of the IS2 transposon, were directly synthesised by Roche.
DNA fragments were recovered from agarose gels using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). Restriction endonucleases were purchased from New England Biolabs, and ligases were purchased from Invitrogen (Carlsband, CA). Nucleotide sequencing of all cloned PCR fragments was carried out at BMR Genomics (Padova, Italy) to verify that the sequences were correct.
The assembled fragment containing the kfoC gene flanked by the ends of IS2 rensponsible for transposition was amplified with primers irla and irrb (5′-Tgg ATT TgC CCC TAT gTT TCC Ag-3′) and cloned into the XL cloning vector (Invitrogen, CA) following the manufacturer’s instructions. This generated plasmid pCP04.
Construction of BK4071
Transformation
For the construction of all strains, E. coli K4 electrocompetent cells were prepared according to the instrument instructions and transformed through electroporation using a Bio-Rad Gene Pulser (2-mm cuvettes, 2.5 kV, 200 Ω, 25 μF). E. coli K4 was transformed with the vector pCP04 and plated on LB plates supplemented with 50 μg/mL kanamycin to select for positive clones. Three clones were analysed in shake flask, as described in the following sections, for the production of K4 polysaccharide. The best performer identified here as E. coli K4-pCP04_1 was selected for the following experiments.
Transposition assay
Strain E. coli K4-pCP04_1 was grown in a shake flask in order to induce the transposition event causing integration of the modified transposon (IRL-kfoC-IRR) into the genome of E. coli K4. Flasks with 200 mL of medium in the presence and absence of kanamycin (50 μg/mL) were inoculated with a starting concentration of 0.04 gcdw/L of cells and incubated at 37 °C at 200 rpm in a rotary shaker. Three consecutive 24 h experiments were performed for a total of 72 h of growth. After 8, 24, 32, 48, 56 and 72 h, samples of biomass were collected for the extraction of genomic and plasmid DNA. Samples of broth were plated on LB with and without antibiotic at all time points to follow the loss of recombinant plasmid over time. Three of the colonies grown after 72 h on plates without antibiotic were randomly selected to perform the following shake-flask experiments.
Shake-flask experiments
Shake-flask experiments were performed in order to evaluate the effect of kfoC gene overexpression on the production of K4 polysaccharide. Before each experiment, cells from 20 % (w/v) glycerol stock preparations were streaked on agar plates and grown overnight (o/n) at 37 °C. Single colonies were then used to inoculate pre-cultures that were incubated o/n at 37 °C in shaking conditions.
The medium used for all shake-flask production studies is described in the “Medium” section.
For all experiments, 200-mL cultures of wild-type and recombinant E. coli K4 were grown in 1-L baffled flasks, keeping a 1:5 medium/air volume ratio, at 37 °C and 200 rpm in a rotary shaker incubator (model Minitron, Infors, Bottmingen, Switzerland). Samples were withdrawn during the course of the experiment to analyse polysaccharide production. Every shake-flask experiment was repeated at least four times.
Evaluation of strain stability
In order to test the stability of the recombinant strain, repeated shake-flask experiments consisting of a total of 150 h of growth were performed in the same conditions described above. The final titre of polysaccharide produced was analysed every 24 h before restarting the culture. Experiments were performed in triplicate.
Analysis of mRNA expression level
Semi-quantitative reverse transcriptase PCR was performed on E. coli K4 and on the BK4071 recombinant strain to evaluate the increase of the kfoC mRNA pool in time during growth. For this purpose, the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase was used (Invitrogen Carlsband, CA). RNA was treated with DNase using the DNA-free kit following the supplied protocol (Ambion Inc Austin, TX) before reverse transcription and amplification. The 16S rRNA gene was used as a control to normalise data.
Fermentation experiments
Batch (2.5 L)
Fermentation experiments were carried out as biological triplicates in a Biostat CT reactor (Sartorius Stedim; Melsungen, Germany) with a 2-L working volume. A constant pH of 7.5 was maintained via automated addition of 30 % v/v NH4OH and 30 % v/v H2SO4.
Seed cultures were prepared by inoculating a single colony into 200 mL of medium in 1-L baffled shake flasks. The flasks were incubated o/n at 37 °C and 200 rpm. During incubation, 5-mL samples were withdrawn from the reactors at regular time intervals for the determination of substrates and extracellular metabolites and for polysaccharide quantification.
For batch experiments, the concentration of dissolved oxygen (DO) was maintained above 20 % with varying airflow rate (1–1.5 L/min) and stirring rate (200–800 rpm) according to oxygen demand. The basic medium used for all experiments was the one described in the “Medium” section; however, experiments in which the medium was modified by the addition of 9.5 mg/L of arginine and 45 mg/L of glutamic acid, or by the addition of 0.1 g/L of sucrose, or by the use of 1.5 g/L of soy peptone were also performed.
Fed batch (2.5–22 L)
For fed-batch experiments, cells were grown in the semi-defined medium containing glycerol and soy peptone. After the batch phase, when the DO spiked indicating C source exhaustion, a concentrated nutrient solution (360 g/L glycerol and 36 g/L soy peptone for experiments in the 2.5-L fermenter and 480 g/L glycerol and 48 g/L soy peptone for experiments in the 22-L fermenter, plus inorganic salts 20-fold concentrated) was fed to the culture in the following growth phase. The feeding rate was between 0.5 and 3 g/L h.
Quantification of CPS
Broth samples were collected from shake-flask and fermentation experiments and centrifuged at 1,700 × g. Supernatants were then ultrafiltered on 10-kDa centrifugal filter devices (YM-10 Centricon, Millipore, Bedford, MA, USA) at 5,000 × g and concentrated to 1/10th of their initial volume. The retentate was twice diafiltered and then analysed for the content of K4 polysaccharide.
Capillary electrophoresis analysis was performed on a Beckman-Coulter HPCE instrument (P/ACE MDQ, Palo Alto, CA) equipped with a diode array detector and a UV lamp, using an uncoated fused-silica tube (70 cm in total length, 60 cm in effective length, 50 μm in I.D.) at 25 °C. Separation and quantification of the K4 and K4 defructosylated polysaccharides were performed according to the previously described method (Restaino et al. 2009).
kfoC gene copy number determination
The number of copies of kfoC present in the genome of BK4071 was established by real-time PCR. The total DNA of the wild type (wt) E. coli K4 and of the recombinant strain was extracted by using the QIAamp DNA Mini kit (Qiagen). The dsx gene was used as an internal control since only one copy of the gene was present in the genome of E. coli strains (Hahn et al. 2001). The primers used for the amplification of kfoC and dsx are reported in Table 1. PCR amplification was performed in an iQ5 instrument (Bio-Rad, CA). Amplification was carried out in 25-μL volume containing 5 μL DNA, 12.5 μL of iQ Syber Green Supermix (Bio-Rad, CA) and 0.5 μL of each primer at a concentration of 0.4 μM. After incubation at 95 °C for 3 min, amplification proceeded with 40 cycles of 95 °C for 10 s and 62 °C for 1 min. Tenfold serial dilutions of the purified kfoC and dsx fragments were used for the construction of calibration curves in which the copy n°/n° of molecules was related to Ct values according to Whelan et al. (2003) and Lee et al. (2006). The ratio between the number of amplified kfoC and dsx molecules in the wt and in the recombinant strain indicated the number of kfoC gene copies in BK4071. Two biological samples each were analysed in triplicate.
Results
Strain construction
E. coli K4 was initially modified by the addition of the pCP04 plasmid containing a modified IS2 element in which OrfA and OrfB were replaced by kfoC (Fig. 1). The gene was therefore flanked by IRL and IRR, required for the transposition, and it was located downstream of the PIRL promoter. Repeated shake-flask experiments on the standard medium without antibiotic supplementation led, after 72 h, to the complete loss of the recombinant plasmid and to the integration of the modified transposon into the genome of E. coli K4, yielding a strain that does not require antibiotic selection. An overview of the engineering strategy used is given in Fig. 1.
Loss of the XL vector was verified by plating the cells on the medium supplemented with kanamycin and by extraction of plasmid DNA verifying the absence of the recombinant vector. Although no plasmid bands were visible on agarose gels, specific primers (M13 Forward and M13 Reverse, furnished by Invitrogen) were used as a further control for the amplification of eventual plasmid residues; however, no band was obtained (data not shown).
In order to verify that the integration of the modified IS2 element occurred by transposition and not by homologous recombination, two primer pairs, namely M13 Forward/irla and M13 Reverse/irrb, were used to amplify the total DNA of BK4071 and no band was obtained, indicating the absence of plasmid fragments from the genome of BK4071 and, therefore, confirming the transposition event (data not shown).
Shake-flask experiments
E. coli K4 –pCP04 was grown in shake-flasks on the standard medium containing glycerol and soy as main carbon and nitrogen sources, respectively, and the titre of capsular polysaccharide was analysed to determine whether it was affected by the presence of additional pIRL-controlled kfoC copies (Table 2). Following integration of the modified insertion sequence (IRL-kfoC-IRR) and elimination of the XL vector, the resulting strains were also analysed in shake flasks in the same conditions. All of the three randomly selected colonies grown in parallel showed an increase of CPS production compared to the wild type after 24 h of growth; however, the best clone that demonstrated a significant 1.8-fold increase was chosen for following experiments and designated as BK4071 (Table 2). By comparing the titre of CPS pre (strain E. coli K4 –pCP04)- and post (strain BK4071)-integration, a decrease was observed, probably due to a lower number of additional copies of the cassette present in the integrative strain. In order to verify the genetic stability of BK4071, repeated growth experiments in shake-flasks were also performed showing that after three cycles, each lasting 24 h, the production of biomass and associated CPS remained unaltered (data not shown).
In comparison to the wild type, BK4071 also showed a 1.7-fold higher Y K4/X of 94 ± 7 mgK4/gcdw, thereby demonstrating that the additional kfoC copy enhances CPS production without affecting the yield of biomass (Table 2).
The abundance of kfoC transcripts in the wt and recombinant strain was determined at three time points during growth. Results demonstrate an increased expression of the gene by 20 and 60 % after 6 and 24 h of growth, respectively, whereas expression is similar to that observed in the wt strain after 8 h of growth (Fig. 2).

Analysis of kfoC gene expression in E. coli K4 and BK4071 in shake-flask experiments. Histogram representing the fold overexpression of the gene under investigation in the recombinant strain BK4071 compared to the wt E. coli K4
Batch experiments in 2.5-L bioreactors
The physiology of BK4071 was studied in 24 h batch experiments in the standard medium (SM), in order to compare the strain performance to that obtained by the wt E. coli K4, as well as in SM supplemented with either glutamic acid and arginine or sucrose in order to stimulate growth. Results are summarised in Table 3. The addition of the two sources of amino acids unexpectedly inhibited growth, whereas the final titre of polysaccharide was only slightly reduced; therefore, a high Y K4/X was obtained. Both the concentration of biomass and CPS were slightly lower when sucrose was added to the SM, whereas the increased initial concentration of soy peptone in the broth (1.5 vs 1) mainly stimulated biomass production without affecting the synthesis of CPS. Compared to experiments performed with the wt E. coli K4, the best results were obtained in the SM without any supplementation. BK4071 showed a 40 % increase in the final titre of CPS over wild-type levels. Moreover, the maximum concentration of product (425 mg/L) was reached after only 8 h of growth, greatly improving process productivity.
Fed-batch experiments on 2.5- and 22-L scales
BK4071 was analysed in fed-batch fermentations on 2.5- and 22-L scales. In both fermentations, agitation speed and oxygen supply were modified according to the strain’s metabolic demand in order to keep the pO2 always above 20 % of saturation. The final production of biomass and K4 polysaccharide was higher in the process performed on the higher scale culminating in 3.47 g/L of K4 CPS after 32 h of growth (Fig. 3). However, the ratio of K4 CPS produced to biomass did not vary in the two processes, as indicated by a Y K4/X value of 0.22 g/g. This discrepancy might be attributed to small differences in the feeding strategy; in fact, a residue (1–2 g/L) of carbon source was always present in the broth for experiments performed in the 22-L bioreactor, whereas no accumulation of carbon source was observed during processes performed in the 2.5-L fermenter. A summary of the results obtained with the wt and recombinant strains during fed-batch experiments on the 2.5- and 22-L scales is reported in Table 4.

Time course of fed-batch experiments in 2.5- and 22-L fermenters with strain BK4071. a Production of biomass and K4 CPS. b Consumption of glycerol and production of total acids
kfoC gene copy number determination
Real-time PCR amplifications of the total DNA from samples of cultures of BK4071 and of the wt E. coli K4 were performed to compare the number of copies of kfoC present in each strain. The dsx gene was used as an internal single-copy control gene. Standard curves for each gene were constructed in order to relate the Ct cycle to the n° of copies/molecules of the target gene as described in the “Materials and methods” section. The ratio between the number of kfoC and dsx molecules in the wild-type strain was used as a calibrator. The number of copies of kfoC in BK4071 was obtained by dividing the kfoC-to-dsx ratio in BK4071 by that of the calibrator. As reported in Table 1, the obtained value is equal to 1.76 ± 0.06, probably indicating the presence of one additional gene copy, introduced with the modified insertion sequence, compared to the wt E. coli K4.
Discussion
The goal of obtaining microbially derived chondroitin sulphate was addressed in our recent studies of the development of fermentation and engineering strategies to maximise the capacity of E. coli K4 to produce CS-like capsular polysaccharide (Cimini et al. 2012, 2013; Restaino et al. 2013). A plasmid-based expression system was recently used for the homologous overexpression of the kfoC gene in E. coli K4 (Cimini et al. 2010a). kfoC codes for a glycosyltransferase that is responsible for the polymerisation of the nascent chains of capsular polysaccharide by the alternate addition of glucuronic acid and N-acetyl-galactosamine to the growing end of the polymer. Data from our previous shake-flask experiments demonstrated that increased intracellular levels of the enzyme have a significant impact on improving polysaccharide production since a two fold enhancement of the final titre of CPS was achieved. However, the strain was extremely unstable, and in order to reproduce the results on a larger scale (2.5-L fermenter), the constant maintenance of selective antibiotic pressure throughout the batch process was crucial (Cimini et al. 2010a).
Currently, more effort is being addressed towards the modification of chromosomal DNA through systems that do not require selective pressure with antibiotic supplementation. Because our previous results were unsuitable for scale up to industrial applications, this work has exploited the potential of increasing the kfoC copy number by using a different engineering strategy and expression system that employed an endogenous copy of the insertion sequence IS2. IS2 is a member of the widespread family of IS3 insertion sequences although it differs from most members of this family in the asymmetry of the transposition mechanism of minicirle formation, in which IRR is the exclusive donor and IRL is always the target (Lewis et al. 2001). The cluster of genes that is responsible for the biosynthesis of CPS in E. coli K4 is characterised by the presence of an IS2 element between the kfoC and kfoD genes. A modified transposable expression cassette was obtained by constructing a new element in which Orfs A and B, of the native IS2 insertion sequence, were replaced by the kfoC gene. In this construct, kfoC was flanked by the two inverted repeats and preceded by the pIRL promoter. A carrier plasmid (pCP04) was then transformed into wt E. coli K4. We posited that during extended growth of this transformed strain, the IRL::kfoC::IRR cassette would be integrated into the host chromosome. After 72 h of growth, the delivery vector was expelled and the engineered cassette was integrated into the genome of E. coli K4. According to real-time PCR experiments, only one copy of the modified IS2 element was transposing in the genome of E. coli K4, and the additional kfoC copy in the recombinant strain induced a 1.8-fold increase of the CPS compared to the wild type (Table 2); moreover, the strain proved its stability by producing the same final amount of polysaccharide after several generations in shake-flasks.
Liu et al. (2009) increased the production of hyaluronic acid (HA) by 34 % through the cultivation of Streptococcus zooepidemicus in the presence of arginine together with cysteine and lysine. They observed that the depletion of such essential amino acids was followed immediately by the end of the exponential phase. Since HA is produced by S. zooepidemicus as a growth-associated product, they extended the exponential phase via addition of three key amino acids and therefore improved the HA yield. The effect of the presence in the standard medium (glycerol-soy) of glutamic acid, arginine and sucrose on the production of K4 polysaccharide was analysed in the present study in 2.5-L batch fermentations. Surprisingly, the final concentration of CPS in the broth was lower compared to that obtained in the medium without supplementation. Growth was promoted for BK4071 and for the control strain by a higher concentration of soy peptone that however did not reflect in higher CPS yields. We previously showed that fructose supplementation increased the amount of CPS produced from E. coli K4 (Restaino et al. 2013). The addition of 0.1 g/L of sucrose, a cheaper source of fructose, however, did not produce the same results. Sucrose catabolism is in fact an extremely variable and poorly described feature among E. coli strains (Sabri et al. 2013). The highest titre of CPS was obtained on the SM medium without any supplementation, and compared to the wt strain, a great improvement of the process productivity was observed since K4 CPS production peaked after only 8 h of growth.
Strain performance was evaluated in fed-batch experiments on 2.5 and 22-L scales. An exponential feeding profile well suited BK4071. The Y K4/X of the processes on the two scales was conserved, demonstrating strain stability and the consistency of the results. The fed-batch performed in the 22-L fermenter, however, resulted in a higher final titre of CPS probably due to the slightly different feeding profile and to the changed reactor configuration (H:D 3:1 vs 2:1) in which the air bubble residence time was longer, thus improving oxygen mass transfer.
Imre and co-workers (2011) modified the IS30 transposon for the expression of a fusion transposase and observed that the modified construct retained the target recognition ability of the wt transposase. However, a higher frequency of integration into new target sites, not preferred by the latter, was also described. The integration and expression of the modified IS2-kfoC transposable cassette in the genome of E. coli K4 was not necessarily predictable; the accidental activation or repression of portions of the recipient cell’s genome that are adjacent to the integration site could in fact have occurred. Our results here, however, have showed that the engineering of IS2 produced an appropriate tool for the overexpression of the kfoC gene. Expression of kfoC in this construct generated a 2.5-fold increase in the production of capsular polysaccharide in fed-batch experiments in a 22-L scale compared to experiments performed with the wt strain, showing the suitability of the system for further investigations in pilot-scale production levels. This represents a first step towards the biotechnological production of chondroitin and chondroitin sulphate for nutraceutical and pharmaceutical applications.
References
Achur RN, Kakizaki I, Goel S, Kojima K, Madhunapantula SV, Goyal A, Ohta M, Kumar S, Takagaki K, Gowda DC (2008) Structural interactions in chondroitin 4-sulfate mediated adherence of Plasmodium falciparum infected erythrocytes in human placenta during pregnancy-associated Malaria. Biochemistry 47(47):12635–12643. doi:10.1021/bi801643m
Bedini E, De Castro C, De Rosa M, Di Nola A, Iadonisi A, Restaino OF, Schiraldi C, Parrilli M (2011) A microbiological-chemical strategy to produce chondroitin sulfate A, C. Angew Chem Int Ed Engl 50:6160–6163. doi:10.1002/anie.201101142
Buchan BW, McLendon MK, Jones BD (2008) Identification of differentially regulated Francisella tularensis genes by use of a newly developed Tn5-based transposon delivery system. Appl Environ Microbiol 74(9):2637–2645. doi:10.1128/AEM.02882-07
Callinan PA, Batzer MA (2006) Retrotransposable elements and human disease. Genome Dyn 1:104–115
Cimini D, De Rosa M, Viggiani A, Restaino OF, Carlino E, Schiraldi C (2010a) Improved fructosylated chondroitin production by kfoC overexpression in E. coli K4. J Biotechnol 150:324–331. doi:10.1016/j.jbiotec.2010.09.954
Cimini D, Restaino OF, Catapano A, De Rosa M, Schiraldi C (2010b) Production of capsular polysaccharide from Escherichia coli K4. Appl Microbiol Biotechnol 85:1779–1787. doi:10.1007/s00253-009-2261-8
Cimini D, De Rosa M, Schiraldi C (2012) Production of glucuronic acid-based polysaccharides by microbial fermentation for biomedical applications. Biotechnol J 7(2):237–250. doi:10.1002/biot.201100242
Cimini D, De Rosa M, Carlino E, Ruggiero A, Schiraldi C (2013) Homologous overexpression of rfaH in E. coli K4 improves the production of chondroitin-like capsular polysaccharide. Microb Cell Factories 12:46–57. doi:10.1186/1475-2859-12-46
Hahn K, Eubanks LM, Testa CA, Blagg BJ, Baker JA, Poulter CD (2001) 1-Deoxy-d-xylulose 5-phosphate synthase, the gene product of open reading frame (ORF) 2816 and ORF 2895 in Rhodobacter capsulatus. J Bacteriol 183:1–11
Hayes F (2003) Transposon-based strategies for microbial functional genomics and proteomics. Annu Rev Genet 3(7):3–29
Hedges DJ, Deininger PL (2007) Inviting instability: transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat Res 616:46–59
Hu ST, Hwang JH, Lee LC, Lee CH, Li PL, Hsieh YC (1994) Functional analysis of the 14 kDa protein of insertion sequence 2. J Mol Biol 236:503–513
Hu ST, Lee LC, Lei GS (1996) Detection of an IS2-encoded 46-kilodalton protein capable of binding terminal repeats of IS2. J Bacteriol 178:5652–5659
Imada K, Oka H, Kawasaki D, Miura N, Sato T, Ito A (2010) Anti-arthritic action mechanisms of natural chondroitin sulfate in humanarticular chondrocytes and synovial fibroblasts. Biol Pharm Bull 33(3):410–414
Imre A, Olasz F, Nagy B (2011) Site directed (IS30-FljA) transposon mutagenesis system to produce nonflagelatted mutants of Salmonella Enteritidis. FEMS Microbiol Lett 317(1):52–59. doi:10.1111/j.1574-6968.2011.02210.x
Izsvak Z, Chuah MKL, VandenDriessche T, Ivics Z (2009) Efficient stable gene transfer into human cells by the Sleeping Beauty transposon vectors. Methods 49:287–297
Lee C, Kim J, Gu Shin S, Hwang S (2006) Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J Biotechnol 123:273–280
Lewenza S, Falsafi RK, Winsor G, Gooderham WJ, McPhee JB, Brinkman FS, Hancock RE (2005) Construction of a mini-Tn5-luxCDABE mutant library in Pseudomonas aeruginosa PAO1: a tool for identifying differentially regulated genes. Genome Res 15:583–589
Lewis LA, Grindley NDF (1997) Two abundant intramolecular transposition products, resulting from reactions initiated at a single end, suggest that IS2 transposes by an unconventional pathway. Mol Microbiol 25:517–529
Lewis LA, Gadura N, Greene M, Saby R, Grindley ND (2001) The basis of asymmetry in IS2 transposition. Mol Microbiol 42(4):887–901
Liu L, Sun J, Xu W, Du G, Chen J (2009) Modeling and optimization of microbial hyaluronic acid production by Streptococcus zooepidemicus using radial basis function neural network coupling quantum-behaved particle swarm optimization algorithm. Biotechnol Prog 25(6):1819–1825. doi:10.1002/btpr.278
Manzoni M, Bergomi S, Molinari F, Cavazzoni V (1996) Production and purification of an extracellularly produced K4 polysaccharide from Escherichia coli. Biotechnol Lett 18(4):383–386
Muenthaisong S, Ujhelly O, Polgar Z, Varga E, Ivics Z, Pirity MK, Dinnyes A (2012) Generation of mouse induced pluripotent stem cells from different genetic backgrounds using Sleeping beauty transposon mediated gene transfer. Exp Cell Res 318:2482–2489
Ninomiya T, Sugiura N, Tawada A, Sugimoto K, Watanabe H, Kimata K (2002) Molecular cloning and characterization of chondroitin polymerase from Escherichia coli strain K4. J Biol Chem 277:21567–21575. doi:10.1074/jbc.M201719200
Pothacharoen P, Siriaunkgul S, Ong-Chai S, Supabandhu J, Kumja P, Wanaphirak C, Sugahara K, Hardingham T, Kongtawelert P (2006) Raised serum chondroitin sulfate epitope level in ovarian epithelial cancer. J Biochem 140(4):517–524
Restaino OF, Cimini D, De Rosa M, De Castro C, Parrilli M, Schiraldi C (2009) High performance capillary electrophoresis analysis of Escherichia coli K4 cell surface polysaccharides: separation and determination of lipopolysaccharide and capsular polysaccharide species and of their respectively de-lipid A defructosylated forms. Electrophoresis 30(22):3877–3883. doi:10.1002/elps.200900279
Restaino OF, di Lauro I, Cimini D, Carlino E, De Rosa M, Schiraldi C (2013) Monosaccharide precursors for boosting chondroitin-like capsular polysaccharide production. Appl Microbiol Biotechnol 97(4):1699–1709
Sabri S, Nielsen LK, Vickers CE (2013) Molecular control of sucrose utilization in Escherichia coli W, an efficient sucrose-utilizing strain. Appl Environ Microbiol 79:478–487
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbour Laboratory Press, New York
Schiraldi C, Alfano A, Cimini D, De Rosa M, Panariello A, Restaino OF, De Rosa M (2012) Application of a 22 L scale membrane bioreactor and cross-flow ultrafiltration to obtain purified chondroitin. Biotechnol Prog 28(4):1012–1018. doi:10.1002/btpr.1566
Shin DW, Lee SM, Shin YR, Ryu SR (2006) Identification of a novel genetic locus affecting ptsG expression in Escherichia coli. J Microbiol Biotechnol 16:795–798
Smith AN, Boulnois GJ, Roberts IS (1990) Molecular analysis of the Escherichia coli K5 kps locus: identification and characterization of an inner-membrane capsular polysaccharide transport system. Mol Microbiol 4:1863–1869
Suzuki K (2008) Chondroitin producing bacterium and method of producing chondroitin. PCT WO 2008/13350 A1
Trilli A, Busiello I, Daly S, Bagatin F (2011) Biotechnological production of chondroitin. USP Application 20120010399
Wang TW, Sun JS, Wu HC, Tsuang YH, Wang WH, Lin FH (2006) The effect of gelatin-chondroitin sulfate-hyaluronic acid skin substitute on wound healing in SCID mice. Biomaterials 27(33):5689–5697
Whelan JA, Russel NB, Whelan MA (2003) A method for the absolute quantification of cDNA using realtime PCR. J Immunol Methods 278:261–269
Wright AC, Powell JL, Kaper JB, Morris JG Jr (2001) Identification of a group 1-like capsular polysaccharideoperon for Vibrio vulnificus. Infect Immun 69:6893–6901
Yu BJ, Kim C (2008) Minimization of the Escherichia coli genome using the Tn5-targeted Cre/loxP excision system. Methods Mol Biol 416:261–277
Acknowledgments
The research was supported by the Ministero dell’Istruzione dell’Università e della Ricerca (MIUR), L.297 project “Produzione biotecnologica di condroitina”.
Author information
Authors and Affiliations
Corresponding authors
Additional information
D. Cimini and S. Fantaccione equally contributed to the work.
Rights and permissions
About this article
Cite this article
Cimini, D., Fantaccione, S., Volpe, F. et al. IS2-mediated overexpression of kfoC in E. coli K4 increases chondroitin-like capsular polysaccharide production. Appl Microbiol Biotechnol 98, 3955–3964 (2014). https://doi.org/10.1007/s00253-014-5506-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5506-0