
Abstract
Rhamnogalacturonan I lyases (RGI lyases) (EC 4.2.2.-) catalyze cleavage of α-1,4 bonds between rhamnose and galacturonic acid in the backbone of pectins by β-elimination. In the present study, targeted improvement of the thermostability of a PL family 11 RGI lyase from Bacillus licheniformis (DSM 13/ATCC14580) was examined by using a combinatorial protein engineering approach exploring additive effects of single amino acid substitutions. These were selected by using a consensus approach together with assessing protein stability changes (PoPMuSiC) and B-factor iterative test (B-FIT). The second-generation mutants involved combinations of two to seven individually favorable single mutations. Thermal stability was examined as half-life at 60 °C and by recording of thermal transitions by circular dichroism. Surprisingly, the biggest increment in thermal stability was achieved by producing the wild-type RGI lyase in Bacillus subtilis as opposed to in Pichia pastoris; this effect is suggested to be a negative result of glycosylation of the P. pastoris expressed enzyme. A ~ twofold improvement in thermal stability at 60 °C, accompanied by less significant increases in T m of the enzyme mutants, were obtained due to additive stabilizing effects of single amino acid mutations (E434L, G55V, and G326E) compared to the wild type. The crystal structure of the B. licheniformis wild-type RGI lyase was also determined; the structural analysis corroborated that especially mutation of charged amino acids to hydrophobic ones in surface-exposed loops produced favorable thermal stability effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pectin is a heteropolysaccharide mainly found in the primary cell wall and in the middle lamella of dicot plants (Willats et al. 1999). It contains the following three subgroups: homogalacturonan (HG), rhamnogalacturonan I (RGI), and rhamnogalacturonan II (Heredia et al. 1995). The structures of pectins are highly heterogeneous and their composition is strongly influenced by their origin (Willats et al. 2006; Wong 2008). The major component HG is a linear homopolymer composed of a backbone of α-(1,4)-d-galacturonic acid residues, they might be methyl esterified at the carboxyl group and/or o-acetylated (O-2 and/or O-3). The highly branched region of pectin RGI has a backbone consisting of repeats of the disaccharide α-(1,2)-l-rhamnose-α-(1,4)-d-galacturonic acid. The galacturonic acid residues can be acetylated and the rhamnose residues substituted at the C4 position. The sidechains vary in size from 1 to more than 50 residues and includes arabinan, galactan, and arabinogalactan (Wong 2008).
The variable composition and, consequently, the different properties of pectin, essentially the degree of methoxylation, make it a desirable material in industry, where it is used as a food additive, as gelling agent, stabilizer, emulsifier, etc. (Laurent and Boulenguer 2003).
Recent work focused on enzymatic conversion of pectin to produce fibers and oligosaccharides with a prebiotic effect. Selective enzymes were used to release rhamnogalacturonan oligosaccharides with different degrees of polymerization (DP) and different functionalities (Thomassen et al. 2011; Holck et al. 2011; Michalak et al. 2012). In fact, the approach holds promise for more than production of prebiotic substances. RGI has been associated with immune modulatory effects (Sakurai et al. 1999), anti-cancer activity (Cheng et al. 2013) and have been suggested to improve biocompatibility of implant materials (Kokkonen et al. 2007).
RGI lyases (EC 4.2.4.-) cleave the α-(1,4) bond between rhamnose and galacturonic acid in the RGI backbone via a β-elimination reaction. They can be used (along with endo-acting RGI hydrolases) to produce oligosaccharides of different DPs (Mutter et al. 1998). The presently known RGI lyases are classified in carbohydrate-active enzyme (CAZY) families 4 and 11. Members of both families have been characterized with respect to the optimum reaction conditions, thermostability and the three-dimensional structure is also known (McDonough et al. 2004; Ochiai et al. 2007a). Bacillus subtilis harbors two family 11 RGI lyases YesW and YesX being endo- and exo-acting, respectively (Ochiai et al. 2007b). The structure of YesW revealed an eight-bladed β-propeller with a deep cleft in the center, which is proposed to contain the active site. The enzyme contains ten calcium ions, of which nine play a structural role in the blades of the propeller, while one is located in the active site and has been suggested to have a role in substrate binding (Ochiai et al. 2007a, 2009). Based on kinetic studies and structural knowledge of a substrate complex with an inactive mutant, the enzymatic function including a detailed reaction model is well understood for the Aspergillus aculeatus rhamnogalacturonan lyase belonging to PL4 (Jensen et al. 2010). Soaking of the crystals of the enzymes from PL11 with rhamnose and galactose has indicated possible carbohydrate binding sites. Interestingly, stripping the enzyme of metal ions by ethylenediaminetetraacetic acid followed by readdition of ions indicated manganese to be a more potent activator of activity, while zinc strongly activated YesX but inhibited YesW (Ochiai et al. 2007b).
We previously characterized a family 11 RGI lyase originating from Bacillus licheniformis that shows 77.7 % sequence identity with YesW and is active and stable at elevated temperatures (half-life of 15 min at 61 °C), after expression in Pichia pastoris (Silva et al. 2011). Enzyme catalyzed processes generally work at lower temperatures compared with noncatalyzed processes. However, it can be advantageous to conduct enzymatic reactions at higher temperatures to, e.g., increase substrate solubility, increase reaction rate, or decrease the risk of microbial contamination (Eijsink et al. 2005). Increased stability extends the lifetime of the enzyme, which in turn decreases cost of substrate processing and expands the range of uses in food industry (Tribst et al. 2013).
Using a semirational mutagenesis strategy based on in silico approaches, sequence alignment, and a homology model and employing site-saturation mutagenesis, we targeted solvent-exposed residues and identified single mutations in seven positions increasing the half-lives at 60 °C by 23–55 % (Silva et al. 2013).
Amino acid residues at the protein surface often make primarily local interactions. It has been suggested that stabilizing mutations in surface-exposed positions are thus more likely to have an additive stabilization effect without affecting the enzymatic function (Perl and Schmid 2002). With this notion, we have in the present study made combinations of up to seven single thermostabilizing mutations in the same genetic backbones and selected the most promising variants. Thermostability of the novel enzymes were analyzed by half-life measurement and circular dichroism (CD). To assess whether catalytic properties were affected by mutation, the optimal reaction conditions were determined. As a benchmark, we used the wild-type B. licheniformis RGI lyase produced in both P. pastoris and B. subtilis. To evaluate our initial strategy for predicting the single mutant sites (Silva et al. 2013) and to obtain the structural background for the increased stability, we have determined the crystal structure of the B. licheniformis RGI lyase WT produced in P. pastoris.
Materials and methods
DNA manipulation and strain construction
Mutagenesis was done by introducing additional mutations into the expression vector pDP66K- RGL, harboring a gene encoding B. licheniformis RGI lyase WT (Uniprot Q65KY4, GenBank accession no. KF373119; Silva et al. 2013). Mutations were introduced using overlapping primer mutagenesis with primers listed in Table 1 and pDP66K-Bl_RGL (WT or mutant as appropriate) as template. Polymerase chain reaction (PCR) products were inserted in pDP66K between NcoI and HindIII. All constructs were sequenced to confirm the mutations and to assure that no unwanted mutations were introduced by PCR. B. subtilis 1A976 (Zhang and Zhang 2011) was transformed with the resulting plasmids as described previously (Silva et al. 2013).
Protein synthesis and purification
For protein synthesis, LB medium supplemented with 50 μg/ml kanamycin was inoculated to OD600 of 0.05 with B. subtilis 1A976 harboring pDP66K-Bl_RGL (WT or mutants) and grown for 23 h, shaking at 37 °C. The supernatant containing the secreted protein was recovered after centrifugation (15 min at 5,000 × g and 4 °C), sterile filtrated using 0.22 μm filters and concentrated by Vivaspin 20 columns (GE Healthcare, Uppsala, Sweden). His-tagged protein was purified using Ni-sepharose columns: His Spin Trap (100 μl) or HiTrap (1 ml) (GE Healthcare). The columns were equilibrated with binding buffer: 40 mM EPPS buffer [4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid; pH 7.4] containing 24 mM imidazole and 500 mM of NaCl. His-tagged protein was eluted with 500 mM imidazole, 40 mM EPPS buffer pH 7.4, and 500 mM NaCl. Finally, imidazole was removed using Vivaspin® 6 columns (10 kDa cut-off, GE Healthcare) by repeated wash with 10 mM EPPS buffer (pH 8.0) containing 10 % glycerol. Protein synthesis of RGI lyase WT (Uniprot Q65KY4; with mutation R3M) in P. pastoris and subsequent purification was done as described previously (Silva et al. 2011). Protein concentration was quantified by measuring the absorbance at 280 nm and using the following extinction coefficient 124,680 M-1 cm-1 from ProtParam (Gasteiger et al. 2005).
Thermostability measurement
The WT and mutant variants were incubated at 60 °C in an Eppendorf Thermomixer® comfort for the time intervals: 0, 5, 10, 15, 20, and 30 min. The concentration of the enzymes was 0.012 mg/ml in 16 mM EPPS buffer and 500 mM NaCl. After incubation 60 μl aliquots were transferred to three microtiterplates (three replicates) and the enzyme activities were measured as described below. The biological parameter half-life (t 1/2) was used to evaluate the degree of thermostability of the mutant variants. The first-order rate constant of irreversible thermal denaturation, k d, was obtained from the slope of the plots of “ln residual activity” vs. “time” and the half-lives were calculated as ln2/k d. The experiment was repeated four times.
Enzyme activity assays
RGI from potato was purchased from Megazyme (Wicklow, Ireland). The general procedure for measuring enzyme activity was to preincubate enzyme and substrate solution at 40 °C for 3 min separately. The reaction mixture (200 μl) comprised 0.750 g/l of RGI from potato, 10 mM EPPS buffer (pH 8.0), 2 mM MnCl2, and purified RGI lyase (0.012–0.024 g/l WT or mutant). The reactions were followed for 10 min at 40 °C in microtiter plates. The enzyme activity was based on measurement of the initial rates by assessing the initial linear increase in absorbance at 235 nm, due to the double bond formed in the reaction product (Infinite M200 Tecan). One unit of enzyme activity was defined as the formation of 1 μmol of product per min at pH 8.0 and 40 °C using 0.750 g of potato RGI per liter. The extinction coefficient used was 5,500 M-1 cm-1.
To determine optimum reaction conditions for RGI lyase activity (temperature and pH), a statistical design approach was used consisting of 11 experiments. MODDE Version 7.0.0.1 (Umetrics, Umeå, Sweden) was used to design the experimental frame and to fit and analyze the data by multiple linear regression analysis. The impact on enzyme activity was tested in the temperature range 50–70 °C and the pH range 7.3–8.3. To test the effect of metal ions, enzyme activity was measured without addition of divalent ions and in the presence of 2 mM MnCl2, 2 mM CaCl2, or 2 mM ZnCl2.
Circular dichroism
All CD measurements were carried out on an AVIV 410 CD spectropolarimeter (Lakewood, NJ, USA) equipped with a temperature control unit. The enzyme was assayed in 10 mM Tris (pH 7.0) and 150 mM NaCl. Far-ultraviolet (UV) CD spectra were recorded with enzyme concentrations of 3–5 μM and thermal transitions with enzyme concentrations of 0.3–0.5 μM. To increase solubility at high temperatures, 1 M Urea was added when performing thermal transitions. Measurements for the wild type and mutants were performed at identical instrument settings to allow direct comparison. Secondary structure contents were estimated from the spectra using the K2D2 server (Perez-Iratxeta and Andrade-Navarro 2008).
Thermal transitions were recorded at 228 nm where the change in ellipticity as a function of temperature [θ] T was most pronounced. Unfolding curves were fitted by Eq. 1:
where α is defined as the fraction of folded molecules at any temperature. [θ]F and [θ]D describe the ellipticity of the fully folded and denatured form, respectively. The temperature at the midpoint of denaturation T m was determined where α = 0.5.
Crystallization
Crystallization was done using the B. licheniformis RGI lyase WT produced in P. pastoris. The protein solution was dialyzed against the crystallization buffer [20 mM Tris (pH 7.5), and 1 mM CaCl2] and concentrated to 43 mg/ml. The protein was N-terminally sequenced commercially (Alphalyse, Odense, Denmark) and its mass was determined using an AutoFLEX MALDI-MS-TOF spectrometer (Bruker Daltonik). An Oryx8 liquid-handling robot (Douglas Instruments) was used to set up INDEX HT, PACT, and JSCG + screens in MRC two-drop plates (Douglas Instruments) with a total drop volume of 0.4 μl and 3:1 and 1:1 ratios of protein solution and reservoir solution. Based upon a hit in INDEX HT conditions, manual setups in VDX plates with 1:1 ratio of protein solution and 15–20 % (w/v) PEG3350 and 150 mM dl-malic acid at pH 7.0 led to formation of clusters of needles, each approx. 8 × 106 μm3 in volume. One of the needles was selected and transferred to cryoprotectant comprised of 16 % glycerol, 16 % ethylen glycol, 18 % sucrose, and 4 % glucose and, subsequently, flash cooled in liquid nitrogen.
Data collection and structure solution
Diffraction images were collected at the European Synchrotron Radiation Facility (ESRF) ID23-2 beamline on the crystal cooled to 100 K (Flot et al. 2010). The strategy devised by EDNA (Incardona et al. 2009) was used for the data collection performed with radiation with a wavelength of 0.873 Å. The diffraction data extended to 2.5 Å resolution and analysis of the data revealed P41212 or P43212 as the possible space groups with one molecule per asymmetric unit. The structure was solved by the molecular replacement method using PHENIX (McCoy et al. 2007) and the YesW rhamnogalacturonan lyase from PL11 as a search model (B. subtilis YesW; PDB entry 2Z8R; Ochiai et al. 2007a). The solution was adapted to the correct sequence after manual building in the tetragonal space group P41212 and automatic introduction of water molecules. During the refinement, the difference electron density revealed the position of ten calcium ions and two glycerol molecules bound in the structure. No residual density was found adjacent to the predicted N-glycosylation sites. The model was finalized by introduction of water molecules, the resulting statistics are shown in Table 2. The structure (4cag) has been deposited in PDB.
Bioinformatics analyses
The RGI lyase produced in P. pastoris is N-glycosylated (Silva et al. 2011). The NGlycPred server with default settings (Chuang et al. 2012) was used to predict possible glycosylation sites.
The previous semi-rational selection of sites to mutate, in RGI lyase, was based on a homology model (Silva et al. 2013). This was re-evaluated based on the solved structure. This entailed prediction of stabilizing mutations using PoPMuSiC version 2.1 (Dehouck et al. 2011) and B-factor iterative test (B-FIT; Reetz et al. 2006).
Results
Combination of single mutations and identification of thermostable variants
We previously identified a number of single mutations that lead to an increase in half-lives at 60 °C ranging from 23 % to 55 % (Silva et al. 2013). The mutants that showed the highest increase in half-lives were all substituted in the same site (E434L/W/F/Y) and of these the E434L mutant displayed the highest thermostability. The mutants G55V and A67P showed only medium improvement. Finally, mutations V541Y/E, G326E, K243G/A, and D158E had only a small positive effect on stability (Silva et al. 2013). Since it has been previously suggested that single mutations in some cases can be additive (Perl and Schmid 2002), we attempted to further improve the thermostability by construction of second-generation mutants containing two to seven mutations. Three of the seven mutated positions, G55, A67, and V541, were relatively close (between 11.2 and 17.3 Å) and therefore would be likely to interact and display a positive or negative effect on thermostability. In contrast, G326E, K243G/A, E434L/W/FY, and D158E were positioned distant from the former three and each other. Based on this, our strategy was to make double mutants based on the three best mutations (E434L, G55V, and A67P) and further combine them with D158E, K243G, G326E, E434W/F/Y, and/or V541Y/E to form new backbones containing 2–7 substitutions. In total, 31 different mutants were produced, purified, and characterized as outlined in Fig. 1.

Results of the thermal stability study: half-life (minutes) for the 31 combined mutants engineered as well as WTBs, WTPp, and E434L
The thermostability of the second-generation mutants was first tested at 60 °C. Wild-type B. licheniformis RGI lyase produced in P. pastoris (WTPp) and in B. subtilis (WTBs) as well as E434L were used as benchmark. Unexpectedly, the half-life of WTBs (16.8 ± 1.6 min) was twofold higher than that of WTPp (8.0 ± 1.2; Fig. 1). The lowest increase in thermostability was seen in double mutant 5 (G55V/A67P) with a half-life of 17.9 ± 2.8 min, which was similar to that of WTBs. The combination of G55V/A67P cancelled out the effect of the single mutations. Comparison of mutant combinations with and without the G55V/A67P mutations revealed a similar trend (compare mutants 22 vs. E434L; 23 vs. 7; 24 vs. 8; 25 vs. 9; 26 vs. 11; 27 vs. 10 in Fig. 1).
The most thermostable variants were mutants 1, 10 and 27 with half-lives of 35.2 ± 2.7, 35.5 ± 2.5 and 35.7 ± 3.4 min, respectively (Fig. 1 and Table 3). They all contained the E434L mutation. In mutant 1 it was combined with the second best single mutation G55V, while in 10 (double mutant) and in 27 (quadruple mutant), it was combined with G326E with and without G55V and A67P, respectively. This indicated that a triple mutant containing the mutations G55V, G326E, E434L might display a further increase in thermostability. In order to test this, we constructed and tested this triple mutant (31) and indeed it demonstrated the highest half-life at 37.6 ± 2.8 min (although not significantly higher than 1, 10, and 27).
Determination of thermodynamic stability by circular dichroism
To further evaluate the mutants, thermodynamic stability was analyzed for the mutants E434L, 1, 10, and 27 as well as WTBs and WTPp by CD. Far-UV CD spectra were recorded from 200 to 250 nm for the six enzymes, and they all showed a characteristic β-sheet spectrum with a minimum around 215 nm (Fig. 2a). The relatively broad minimum suggests minor α-helical contribution. Using the K2D2 server (Perez-Iratxeta and Andrade-Navarro 2008), an α-helical content of 3 % and a β-sheet content of 49 % for all analyzed variants were estimated.

a Far-UV CD spectra of the wild-type and variants E434L, 1, 10, and 27 at 25 °C. b Thermal transition monitored at 228 nm. The data were recorded in 10 mM Tris (pH 7.0) and 150 mM NaCl. To increase solubility at high temperatures, 1 M urea was added when performing thermal transitions
Thermal transitions were recorded for the same variants to compare the effect of amino acid substitutions on thermodynamic stability. In the absence of urea, RGI lyase variants from B. subtilis precipitated at temperatures around 70 °C, while RGI lyase variants from P. pastoris stayed in solution over the whole temperature range. To prevent precipitation and allow for direct comparison, measurements were performed in the presence of 1 M urea under identical conditions for all analyzed RGI lyase variants. Figure 2b shows normalized unfolding monitored at 228 nm. For all variants, we determined T m, i.e., the temperature where the fraction of native enzyme is 50 % (Table 3). A T m of 54.3 ± 0.2 °C was determined for the WTBs. Interestingly, WTPp was destabilized by 10 °C. When comparing the four mutants with WTBs (all expressed in B. subtilis), T m was significantly increased in all mutated variants. The most pronounced increase in T m by 3.3 ± 0.3 °C was attributed to the E434L mutation. In mutant 1, T m was not significantly higher than in the E434L mutant. A significant contribution to stabilization was observed for G326E in that mutant 10 displayed an increase in T m of 0.7 ± 0.2 °C and 4.0 ± 0.3 °C compared to E434L and WTBs respectively. Addition of mutations G55V and A67P (mutant 27) did not significantly affect T m when compared to mutant 10. For short half-life times, slight increases result in relatively large changes in T m, whereas at large t 1/2, T m increases only weakly and approaches a plateau (Fig. 3).

Correlation between the two thermostability measurements (T m and t 1/2) based on our analysis of WT and mutant enzymes. A nonlinear correlation is observed
Optimum reaction conditions
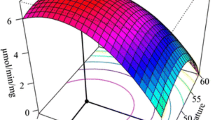
A series of experiments based on a statistical design approach were carried out with the aim to establishing the optimum reaction condition (for highest enzyme activity) with respect to pH and temperature. The surface response plots based on these experiments were obtained for WTBs, WTPp, E434L, and combined mutants (1, 10, and 27) and were shown in Fig. S1. The multivariate regression analysis showed that both pH and temperature had a significant effect on enzyme activity (specific details for each model are shown in Table S1). All models had good correlation coefficients (R 2 between 0.90 and 0.99), together with high values of predictivity (Q 2 ranging from 0.64–0.98) and reproducibility (around 0.99). Therefore, it was possible to conclude that the models were reliable and useful. The statistical analysis of the models also showed that factor resulting from the temperature × temperature interactions influenced enzyme activity whereas the factor pH × pH was less significant for WTPp and WTBs and had no influence in E434L and combined mutants’ model.
For WTPp, it was possible to determine the optimum temperature (57 °C) and pH (7.8; Table 4). This is seen in the surface response plot that consists of a bell-shaped structure (Fig. S1b). The WTBs had higher activity at higher temperatures (61 °C) and at a lower pH (7.3, lowest tested). The pH optimum estimated for the mutants was also 7.3 (Table 4). The observed steepness in the surface response plots of WTBs and mutants suggested that the optimum pH was 7.3 or lower (Fig. S1). Nevertheless, it was possible to estimate the optimum temperature for enzyme activity (Table 4; Fig. S1) that was higher for the mutants than for WTBs. The single mutation had highest activity at 63 °C, while mutant 1 and 27 had the reaction optimum temperature at 64 °C. Finally mutant 10 had the highest optimal temperature (65 °C).
The impact of metal ions MnCl2, CaCl2, and ZnCl2 on enzyme activity was also examined (Table 4). For both WTs and mutants, the activity significantly increased when manganese or calcium was added, while zinc had an inhibitory effect on activity. The WTPp activity was most enhanced in the presence of manganese (163 %), whereas the WTBs activity was highest in the presence of calcium (188 %), when compared to activities in absence of divalent ions (100 %). The metal ion dependence of the mutants was similar to that of WTBs.
Determination of the crystal structure of the RGI lyase expressed in P. pastoris
In order to improve the basis for assessing the impact of mutations affecting thermostability, we solved the structure of the enzyme. This was done using enzyme produced in P. pastoris. We first analyzed the protein by N-terminal sequencing and found indication for insufficient Ste13 processing of the N-terminus in P. pastoris leading to species with two and four additional amino acids in the N-terminus (Fig. S2). The crystal structure was solved by molecular replacement using YesW (PDB entry 2Z8R; Ochiai et al. 2007a) and refined to 2.5 Å resolution (Fig. 4). The final model contained 584 amino acids and did not include the first 5 (+2–4) amino acids and the last C-terminal residue as well as the His6-tag. The fold of the RGI lyase was an eight-bladed β-propeller and was very similar to the other lyases of the PL11 family including B. subtilis YesW. β-sheet was the dominating secondary structural element (34 %), whereas the α-helical content was very low (3 %). The CD analysis reproduced these values well, estimating the content of β-sheet to be 49 % and α-helix to be 3 %. Like YesW, the enzyme contained 10 calcium ions located in positions similar to those observed in YesW. Anomalous difference peaks at six of the ten metal binding sites indicated that they might not be calcium. The mass-spectrometry data support N-glycosylation on the surface of the protein as described previously in Silva et al. (2011; Fig. S3). The NGlycPred server predicted 3 N-glycosylation sites: Asn53, Asn440, and Asn502.

Overall structural fold of RGL11 showing the secondary structure, the ten metal ions (spheres) and the sites mutated successfully (green spheres). α-Helix is shown in magenta, β-sheet is shown in blue, and the loop regions in green. The calcium ions are shown in orange
Re-evaluation of mutagenesis strategy
Previously, with the aim of improving RGI lyase thermostability, we used a homology model, for rational selection of amino acids to target for mutagenesis. The software PoPMuSiC was used to predict the effect of single mutations on stability (Dehouck et al. 2011). Further, we used B-FIT to calculate average B-values based on the structure of B. subtilis YesW. This would serve as an indicator of flexible regions more likely to destabilize the enzyme. With the new crystal structure for the B. licheniformis RGI lyase, we decided to re-evaluate the initial basis for selection criteria based on the YesW crystal structure. The use of PoPMuSiC led to conclude that the beneficial mutations found experimentally were generally not successfully predicted by the software except for G55V, A67P and G326E. Using the new crystal structure for prediction did not improve the consistency with experimental results; in fact, of the three mutations, only A67P was predicted to stabilize the enzyme (Table S2). Instead, the mutations K23F/W (ddG -0.91 and -1.01) and G362P (ddG -1.02) (solvent accessibilities ~ 27 %) were predicted to be the most stabilizing. A multiple alignment showed that the substitution G362P was not frequent, while the other substitutions were not observed. Therefore, it was chosen to not pursue these leads in this study. B-FIT using the structure identified the same regions as when using the structure of the B. subtilis homolog YesW as well as new region namely amino acids 111–116 (KTPDGV). This largely supported the feasibility of using the structure of a close homolog in the absence of a solved structure.
Discussion
The characterization of RGI lyase from B. licheniformis in P. pastoris showed similar results to the ones reported previously (Silva et al. 2011). This enzyme showed highest activity at 57 °C and pH 7.8 which is similar to the optimum reaction conditions described for other members of the PL11 family (Ochiai et al. 2007b; McKie et al. 2001).
When characterizing the WT enzyme produced in B. subtilis and P. pastoris, a difference in thermostability and optimum reaction conditions was observed. The enzyme produced in B. subtilis had the highest activity at higher temperature (61 vs. 57 °C) and lower pH optimum (7.3 vs. 7.8). However, the largest difference was in the thermostability, where WTBs exhibited a twofold higher half-life compared to WTPp. The effect on stability was also confirmed by CD analysis, where the two WTs showed different temperature transitions (Fig. 2) with a 9.9 ± 0.2 °C difference in T m. We previously demonstrated that the enzyme produced in P. pastoris was N-glycosylated (Silva et al. 2011), and this was confirmed in this study, which presents a possible reason for this observation. Protein glycosylation is a common post-translational modification and can influence structural and functional properties in proteins, e.g. kinetic parameters and stability (Helenius and Aebi 2004; Jensen et al. 2013). Usually glycosylated proteins are less prone to thermal inactivation since glycosylation confers structural rigidity to the proteins (Meldgaard and Svendsen 1994). Nevertheless, when recombinant proteins are produced in P. pastoris, glycosylation can affect thermostability positively, negatively or have no effect (Cregg et al. 2000). The α-amylase from an alkalophilic Bacillus provides an example of N-glycosylation-dependent stability reduction. When produced in P. pastoris it had a T m of 76 °C, which was 13 °C lower than the enzyme purified from Bacillus (Tull et al. 2001). RGI lyase is another example where glycosylation leads to a destabilization of the enzyme, resulting in lower T m and smaller t 1/2. The fact that nonglycosylated RGI lyase wild-type precipitates at temperatures around 70 °C, while the glycosylated wild type stays in solution even above 95 °C suggest the glycans to be hydrophilic. These glycans can be thought to mediate protein–solvent interactions, thereby weakening protein intramolecular interactions resulting in destabilizing the enzyme (Bolen and Rose 2008). Additionally, the hydrophilic nature of the glycans might result in a desolvation free energy penalty when folding. Since the RGI lyase originates from B. licheniformis, it could be speculated that the enzyme produced in B. subtilis best reflects the wild-type enzyme.
Not all the single mutations could be combined with an additive effect. Three of the seven mutated positions, Gly55, Ala67, and Val541, were in relatively close vicinity (between 11.2 and 17.3 Å). Combination of Gly55 and Ala67 mutations seemed to cancel out their individual positive effects on thermostability and rendered mutant 5 with a degree of stability similar to that of WTBs.
Our strategy yielded mutants (1, 10, and 27) which displayed an improvement in thermostability over the best single mutant (E434L). These variants, although not significantly different from each other, had half-lives between 35.2 and 35.7 min at 60 °C (Table 3). This was a 2.1- to 2.2-fold improvement in thermostability relative to WTBs. These results also revealed the importance of Gly55Val, Gly326Glu, and Glu434Leu for the thermostability of RGI lyase and confirmed that they were additive. In fact, combining these three mutations (mutant 31) led to the highest half-life recorded although not significantly higher than mutants 1, 10, and 27.
Glutamate 434 is located at the protein surface, but its carboxyl group is not completely solvent-accessible in the native state (see Fig. 4 ).Therefore, E434 might contribute unfavorably to the native state stability by desolvation free energy when folding. Replacing this amino acid with the hydrophobic amino acid leucine would remove this unfavorable free energy contribution, thereby stabilizing the enzyme (Schmid 2011). Glycine 326 is located at the end of a short helix close to the protein surface (see Fig. 4 ). Glycine is known as a helix breaker since its conformational space is much less restricted than for all other amino acids, thereby disrupting secondary structure elements (Pace and Scholtz 1998). By replacing Gly326 with a more restricted amino acid, it was possible to stabilize the enzyme and the beneficial effects for stabilization are additive when in combination with, e.g., the E434L mutation.
Determination of the optimum reaction temperature (Table 3 and Fig. S1), as expected, showed that E434L and mutants 1, 10, and 27 had the highest activities at superior temperatures compared to the WTBs. The highest increase in optimum reaction temperature was observable in mutant 10 with an increment of 4 °C relatively to the WT. CD analysis of the four variants indicated that the main stabilizing effect could be attributed to the replacement of Glu434 with Leu, while the additional mutations introduced in mutants 1, 10, and 27 displayed a minor but also significant stabilizing effect. Figure 3 shows the correlation of T m as a function of t 1/2. For short half-lives, an increase translated into a large improvement in T m, whereas at higher t 1/2, an increase only marginally affected T m. The twofold higher t 1/2 in WTBs compared to WTPp led to an increase in T m of 10 °C, while the twofold higher half-life in E434L compared to WTBs translated into a T m difference of only 3.3 °C.
In this study, the positive effect of calcium and manganese ions on RGI lyase activity was also verified (activity increment 41–88 %, Table 4). Our results hinted on a production host-dependent difference in that WTPp and WTBs (and mutants) displayed a preference for Mn2+ and Ca2+, respectively. Whether this difference is related to the glycosylation of WTPp or, for example, a difference in metal ion incorporation is presently unclear. It is known that enzymes from PL11 exhibit increased activity in the presence of calcium. The positive influence of manganese on RGI lyase activity was only observed in YesW (McKie et al. 2001; Ochiai et al. 2007b).
Apart from our own recent work assessing the thermal stability improvement of different single point mutations in the RGI lyase (Silva et al. 2013), there is only one previous report describing thermal stability improvement of a pectin-modifying lyase by a protein engineering approach (Xiao et al. 2008). Actually, using a T m-guided sequence alignment approach, the half-life at 45 °C of a pectate lyase (EC 4.2.2.2) from Xanthomonas campestris was increased 23-fold by a single mutation (R236F). The mutation also produced an increase in T m of 6 °C (Xiao et al. 2008). In the present work, the thermal stability assessment was done at a much higher temperature (60 °C), which could appear to produce less significant half-life increases than tests at lower temperatures. On the other hand, increasing the thermal stability of an already relatively thermostable enzyme may, in fact, be more demanding since higher temperatures challenge the kinetics of protein folding/unfolding relatively more than lower temperatures. Nevertheless, as Xiao et al. (2008), we conclude that increased hydrophobic desolvation, rather than, e.g., internal increases in disulphide bonds or improved internal hydrogen bonding in the enzyme protein molecule, seem to be the driving force for the observed enzyme protein stability increase. Xiao et al. (2008) did not obtain improvements in T m by combining the favorable, stabilizing R236F mutation with another putatively beneficial mutation (A31G), but did, in fact, observe improvements in the specific activity of the enzyme by combined mutation. They expressed the mutants in E. coli Rosetta 2 cells, so no effects of differences in enzyme protein glycosylation were reported (Xiao et al. 2008).
In conclusion, our approach employed the tenet that mutations of surface-exposed residues are more likely to be additive, but also underscored the relevance of carefully examining additivity by generation of double mutants of most relevant mutations. This allowed the successful design of second-generation mutants further improving the thermostability of B. licheniformis RGI lyase by combination of stabilizing single mutations.
References
Bolen DW, Rose GD (2008) Structure and energetics of the hydrogen-bonded backbone in protein folding. Annu Rev Biochem 77:339–362
Cheng H, Zhang Z, Leng J, Liu D, Hao M, Gao X, Tai G, Zhou Y (2013) The inhibitory effects and mechanisms of rhamnogalacturonan I pectin from potato on HT-29 colon cancer cell proliferation and cell cycle progression. Int J Food Sci Nutr 64:36–43
Chuang GY, Boyington JC, Joyce MG, Zhu J, Nabel GJ, Kwong PD, Georgiev I (2012) Computational prediction of N-linked glycosylation incorporating structural properties and patterns. Bioinformatics 28:2249–2255
Cregg JM, Cereghino JL, Shi J, Higgins DR (2000) Recombinant protein expression in Pichia pastoris. Mol Biotechnol 16:23–52
Dehouck Y, Kwasigroch JM, Gilis D, Rooman M (2011) PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinforma 12:151
Eijsink VG, Gåseidnes S, Borchert TV, van den Burg B (2005) Directed evolution of enzyme stability. Biomol Eng 22:21–30
Flot D, Mairs T, Giraud T, Guijarro M, Lesourd M, Rey V, van Brussel D, Morawe C, Borel C, Hignette O, Chavanne J, Nurizzo D, McSweeney S, Mitchell E (2010) The ID23-2 structural biology microfocus beamline at the ESRF. J Synchrotron Radiat 17:107–118
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, Totowa, NJ, pp 571–607
Helenius A, Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73:1019–1049
Heredia A, Jiménez A, Guillén R (1995) Composition of plant cell walls. Z Lebensm Unters Forsch 200:24–31
Holck J, Lorentzen A, Vigsnæs LK, Licht TR, Mikkelsen JD, Meyer AS (2011) Feruloylated and nonferuloylated arabino-oligosaccharides from sugar beet pectin selectively stimulate the growth of Bifidobacterium spp. in human fecal in vitro fermentations. J Agric Food Chem 59:6511–6519
Incardona MF, Bourenkov GP, Levik K, Pieritz RA, Popov AN, Svensson O (2009) EDNA: a framework for plugin-based applications applied to X-ray experiment online data analysis. J Synchrotron Radiat 16:872–879
Jensen MH, Otten H, Christensen U, Borchert TV, Christensen LLH, Larsen S, Lo Leggio L (2010) Structural and biochemical studies elucidate the mechanism of rhamnogalacturonan lyase from Aspergillus aculeatus. J Mol Biol 404:100–111
Jensen JL, Mølgaard A, Navarro Poulsen JC, Harboe MK, Simonsen JB, Lorentzen AM, Hjernø K, van den Brink JM, Qvist KB, Larsen S (2013) Camel and bovine chymosin: the relationship between their structures and cheese-making properties. Acta Crystallogr D Biol Crystallogr 69:901–913
Kokkonen HE, Ilvesaro JM, Morra M, Schols HA, Tuukkanen J (2007) Effect of modified pectin molecules on the growth of bone cells. Biomacromolecules 8:509–515
Laurent MA, Boulenguer P (2003) Stabilization mechanism of acid dairy drinks (ADD) induced by pectin. Food Hydrocoll 17:445–454
McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674
McDonough MA, Kadirvelraj R, Harris P, Poulsen JC, Larsen S (2004) Rhamnogalacturonan lyase reveals a unique three-domain modular structure for polysaccharide lyase family 4. FEBS Lett 565:188–194
McKie VA, Vincken JP, Voragen AGJ, van den Broek LAM, Stimson E, Gilbert HJ (2001) A new family of rhamnogalacturonan lyases contains an enzyme that binds to cellulose. Biochem J 355:167–177
Meldgaard M, Svendsen I (1994) Different effects of N-glycosylation on the thermostability of highly homologous bacterial (1,3–1,4)-β-glucanases secreted from yeast. Microbiology 140:159–166
Michalak M, Thomassen LV, Roytio H, Ouwehand AC, Meyer AS, Mikkelsen JD (2012) Expression and characterization of an endo-1,4-β-galactanase from Emericella nidulans in Pichia pastoris for enzymatic design of potentially prebiotic oligosaccharides from potato galactans. Enzyme Microb Technol 50:121–129
Mutter M, Renard CM, Beldman G, Schols HA, Voragen AG (1998) Mode of action of RG-hydrolase and RG-lyase toward rhamnogalacturonan oligomers. Characterization of degradation products using RG-rhamnohydrolase and RG-galacturonohydrolase. Carbohydr Res 311:155–164
Ochiai A, Itoh T, Maruyama Y, Kawamata A, Mikami B, Hashimoto W, Murata K (2007a) A novel structural fold in polysaccharide lyases: Bacillus subtilis family 11 rhamnogalacturonan lyase YesW with an eight-bladed beta-propeller. J Biol Chem 282:37134–37145
Ochiai A, Itoh T, Kawamata A, Hashimoto W, Murata K (2007b) Plant cell wall degradation by saprophytic Bacillus subtilis strains: gene clusters responsible for rhamnogalacturonan depolymerization. Appl Environ Microbiol 73:3803–3813
Ochiai A, Itoh T, Mikami B, Hashimoto W, Murata K (2009) Structural determinants responsible for substrate recognition and mode of action in family 11 polysaccharide lyases. J Biol Chem 284:10181–10189
Pace CN, Scholtz JM (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys J 75:422–427
Perez-Iratxeta C, Andrade-Navarro MA (2008) K2D2: Estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol 8:25
Perl D, Schmid FX (2002) Some like it hot: the molecular determinants of protein thermostability. Chem Bio Chem 3:39–44
Reetz MT, Carballeira JD, Vogel A (2006) Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew Chem Int Ed Engl 45:7745–7751
Sakurai MH, Matsumoto T, Kiyohara H, Yamada H (1999) B-cell proliferation activity of pectic polysaccharide from a medicinal herb, the roots of Bupleurum falcatum L. and its structural requirement. Immunology 97:540–547
Schmid FX (2011) Lessons about protein stability from in vitro selections. ChemBioChem 12:1501–1507
Silva IR, Larsen DM, Meyer AS, Mikkelsen JD (2011) Identification, expression, and characterization of a novel bacterial RGI lyase enzyme for the production of bio-functional fibers. Enzyme Microb Technol 49:160–166
Silva IR, Larsen DM, Jers C, Derkx P, Meyer AS, Mikkelsen JD (2013) Enhancing RGI lyase thermostability by targeted single point mutations. Appl Microbiol Biotechnol. doi:10.1007/s00253-013-5184-3
Thomassen LV, Vigsnæs LK, Licht TR, Mikkelsen JD, Meyer AS (2011) Maximal release of highly bifidogenic soluble dietary fibers from industrial potato pulp by minimal enzymatic treatment. Appl Microbiol Biotechnol 90:873–884
Tribst AAL, Augusto PED, Cristianini M (2013) Multi-pass high pressure homogenization of commercial enzymes: effect on the activities of glucose oxidase, neutral protease and amyloglucosidase at different temperatures. Innov Food Sci Emerg Technol 18:83–88
Tull D, Gottschalk TE, Svendsen I, Kramhøft B, Phillipson BA, Bisgård-Frantzen H, Olsen O, Svensson B (2001) Extensive N-glycosylation reduces the thermal stability of a recombinant alkalophilic Bacillus α-amylase produced in Pichia pastoris. Protein Expr Purif 21:13–23
Willats WGT, Gilmartin PM, Mikkelsen JD, Knox JP (1999) Cell wall antibodies without immunization: generation and use of de-esterified homogalacturonan block-specific antibodies from a naive phage display library. Plant J 18:57–65
Willats WGT, Knox P, Mikkelsen JD (2006) Pectin: new insights into an old polymer are starting to gel. Trends Food Sci Technol 17:97–104
Wong D (2008) Enzymatic deconstruction of backbone structures of the ramified regions in pectins. Protein J 27:30–42
Xiao Z, Bergeron H, Grosse S, Beauchemin M, Garron M, Shaya D, Sulea T, Cygler M, Lau PCK (2008) Improvement of the thermostability and activity of a pectate lyase by single amino acid substitutions, using a strategy based on melting-temperature-guided sequence alignment. Appl Environ Microbiol 74:1183–1189
Zhang XZ, Zhang Y-HP (2011) Simple, fast and high-efficiency transformation system for directed evolution of cellulase in Bacillus subtilis. Microb Biotechnol 4:98–105
Acknowledgments
This study was supported by the Danish Strategic Research Council's Committee on Food and Health (FøSu) project “Biological Production of Dietary Fibers and Prebiotics” no. 2101-06-0067. We are grateful to Dorthe Boelskifte for help with the crystallization experiments. We thank the ESRF for synchrotron beamtime and for excellent support by the staff, DanScatt for travel support, and the University of Copenhagen Program of Excellence for funding.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 552 kb)
Rights and permissions
About this article
Cite this article
Silva, I.R., Jers, C., Otten, H. et al. Design of thermostable rhamnogalacturonan lyase mutants from Bacillus licheniformis by combination of targeted single point mutations. Appl Microbiol Biotechnol 98, 4521–4531 (2014). https://doi.org/10.1007/s00253-013-5483-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5483-8