
Abstract
Betulinic acid is a plant-based triterpenoid that has been recognized for its antitumor and anti-HIV activities. The level of betulinic acid in its natural hosts is extremely low. In the present study, we constructed betulinic acid biosynthetic pathway in Saccharomyces cerevisiae by metabolic engineering. Given the betulinic acid forming pathways sharing the common substrate acetyl-CoA with fatty acid synthesis, the metabolic fluxes between the two pathways were varied by changing gene expressions, and their effects on betulinic acid production were investigated. We constructed nine S. cerevisiae strains representing nine combinations of the flux distributions between betulinic acid and fatty acid pathways. Our results demonstrated that it was possible to improve the betulinic acid production in S. cerevisiae while keeping a desirable growth phenotype by optimally balancing the carbon fluxes of the two pathways. Through modulating the expressions of the key genes on betulinic acid and fatty acid pathways, the difference in betulinic acid yield varied largely in the range of 0.01–1.92 mg L−1 OD−1. The metabolic engineering approach used in this study could be extended for synthesizing other triterpenoids in S. cerevisiae.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As a pentacyclic lupane type of triterpenoid, betulinic acid shows great pharmacological properties such as anticancer and anti-HIV activities (Yogeeswari and Sriram 2005). Because of its specific cytotoxicity against tumor cells, betulinic acid was considered to be a future promising anticancer compound (Zuco et al. 2002). Bevirimat, a betulinic acid derivative, has recently been successfully used in phase IIb clinical trials for the treatment of acquired immune deficiency syndrome (Smith et al. 2007). Despite its great potential for clinical applications, the insufficient supply of betulinic acid in its natural hosts is a major obstacle in commercializing this compound. Birch bark is the major plant source for extracting betulinic acid, but the minute amount of betulinic acid in their tissues has limited its production in a large scale for the market (Jäger et al. 2009). Betulin, a precursor of betulinic acid, has been successfully converted to betulinic acid, but some issues remain, including low yields, safety, pollutions, and high costs (Kim et al. 1997). Microbial biotransformation is another approach for converting betulin to betulinic acid, but the conversion efficiency is pretty low, and this approach is also limited by the supply of betulin (Liu et al. 2011).
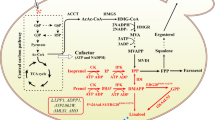
Rapid developments in metabolic engineering and synthetic biology provide alternative approaches for the high production of natural products in microbial hosts (Ajikumar et al. 2010; Paddon et al. 2013; Ro et al. 2006). In many cases, synthetic biology efforts have successfully facilitated the high production of plant monoterpenes, sesquiterpenes, and diterpenes in microorganisms (Kirby and Keasling 2008; Withers and Keasling 2007); however, the metabolic engineering for the microbial synthesis of plant triterpenoids is very limited. To the best of our knowledge, the professional engineering efforts have only been made to produce the beta-amyrin which is the precursor of the oleanolic triterpenoids (Kirby et al. 2008; Madsen et al. 2011). Mainly for the functional characterization of triterpenoid biosynthetic genes, we previously reconstructed the biosynthetic pathway of betulinic acid by introducing AtLUP1 (lupeol synthase) from Arabidopsis and CrAO (amyrin monooxygenase) from Catharanthus roseus into S. cerevisiae, in which S. cerevisiae endogenous ergosterol pathway produces oxidosqualene, the precursor of triterpenoids (Fig. 1) (Huang et al. 2012). However, the overall yield of betulinic acid (around 0.1 mg/L) was pretty low in the previous study. In the current research, we aim to engineer a higher production of betulinic acid in a yeast platform. The engineering of exogenous triterpenoid biosynthesis in S. cerevisiae is mainly based on yeast native mevalonate (MVA) and ergosterol biosynthesis pathways (Fig. 1). Using a beta-amyrin-producing S. cerevisiae strain, several native genes involved in the pathways have been identified as the important targets for exogenous triterpenoid production. For instance, overexpression of the endogenous gene coding for HMG1 (3-hydroxy-3-methylglutaryl coenzyme A reductase), ERG9 (squalene synthase), or ERG1 (squalene monooxygenase) dramatically improved the production of beta-amyrin to variable extents relative to the corresponding control strains (Kirby et al. 2008). On the other hand, the decrease of ERG7 (lanosterol synthase) in sterol biosynthesis attenuated the carbon flux to sterol biosynthesis, while increasing the production of beta-amyrin in S. cerevisiae (Kirby et al. 2008).

Biosynthetic pathways to fatty acid, betulinic acid, and ergosterol in S. cerevisiae. HMG1 3-hydroxyl-3-methylglutaryl-CoA reductase, ERG9 bifunctional farnesyl-diphosphate farnesyltransferase and squalene synthase, ERG1 squalene monooxygenase, LUS1 Arabidopsis thaliana lupeol synthase, CrAO Catharanthus roseus P450 monooxygenase, ERG7 lanosterol synthase, HFA1 acetyl-coenzyme A carboxylase gene
Alongside the MVA and ergosterol pathways, fatty acid biosynthesis seems to be important in engineering exogenous terpenoid biosynthetic pathways in S. cerevisiae (Madsen et al. 2011), as is also true for triterpenoid production. Fatty acids are essential components of the cell membrane, and their biosyntheses share the common precursor acetyl-CoA with ergosterol biosynthesis in S. cerevisiae (Fig. 1). Although there is a competition between fatty acid and ergosterol pathways for acetyl-CoA pool, the increase of HFA1 (the first enzyme in fatty acid synthesis) enhanced the ergosterol content, subsequently leading an obvious improvement in beta-amyrin production in S. cerevisiae expressing a beta-amyrin synthase (Madsen et al. 2011). The mechanism for the positive effect of HFA1 overexpression on ergosterol pathway is not clear but is probably attributable to the alleviation of the intermediate toxic effect from the strengthened upstream MVA pathway (Kizer et al. 2008; Madsen et al. 2011).
Simple overexpression of multibiochemical reactions to alternate host organism often leads to the unbalanced performance with respect to the catalytic efficiency of enzymes and the accumulation of toxic intermediates, as a result of a low yield of desired products and inhibition of cell growth (Ajikumar et al. 2010; Donald et al. 1997; Pitera et al. 2007). Especially, there usually are redox enzymes catalyzing oxidative/reduced reactions in the product pathway. Linear addition of these redox enzymes would perturb the redox balance in cells causing the inhibition of cell growth. Recently, modulation of the transcript levels of the genes in the product pathway, at least in part, has successfully counteracted the unbalance. For example, through modulating the taxol biosynthetic genes by the promoters in different strengths, the fluxes to taxadiene production in Escherichia coli was maximized while no apparent cell growth inhibition was observed (Ajikumar et al. 2010). In this study, a similar approach was applied in engineering betulinic acid production in S. cerevisiae. We investigated whether the metabolic flux balance between fatty acid and betulinic acid biosynthesis could be optimized for betulinic acid synthesis. For fatty acid biosynthetic module, the gene coding for HFA1 was chosen to be modulated which has been identified to be important in producing triterpenoids in S. cerevisiae (Madsen et al. 2011). For betulinic acid module, only three key gene codings for the rate-limiting enzymes of the betulinic acid pathway, HMG1, ERG9, and CrAO, were modulated (Donald et al. 1997; Hampton and Rine 1994; Kennedy and Bard 2001; Madsen et al. 2011). We demonstrated that a reasonable balance between fatty acid and betulinic acid modules was important in engineering betulinic acid production in S. cerevisiae, and the results in this study might be extended in engineering other triterpenoids in microbial hosts.
Materials and methods
The plasmids used in this study are described in Table 1. All the strains used in this study are listed in Table 2. The sequences of the primers used for PCRs (polymerase chain reactions) are shown in the supplemental Table S1.
Plasmid preparations
To replace the native promoter of ERG7 (GenBank accession no. NM_001179202.2) with the MET3 promoter (GenBank accession no. NM_001181668.3), the MET3 promoter and a truncated 5′ segment of ERG7 gene were PCR amplified from the genomic DNA of S. cerevisiae WAT11 using primer pairs 1 and 2, and 3 and 4, respectively. The truncated 5′ segment of ERG7 was placed behind the MET3 promoter by overlapping PCRs with primers 1 and 4, and then cloned into pRS406 at HindШ and XbaI sites to give the construct plJ001 (Table 1). plJ001 was cleaved with BamHI for the integration of P MET3 –ERG7 fusion at the ERG7 locus (Fig. S1). Coupled with the repression of ERG7 gene (Kirby et al. 2008), the ERG1 (GenBank accession no. NM_001181304.1) gene was cloned under a galactose-inducible promoter (GAL10 promoter, GenBank accession no. K02115.1). The open reading frame (ORF) of ERG1 was PCR amplified from the genomic DNA of S. cerevisiae WAT11 using primers 5 and 6, and then was cloned into pESC-LEU at NotI and BglII sites, resulting in plJ002 (Table 1).
To investigate the effect of combinatorial expressions on betulinic acid production in yeast cells, the following plasmids were constructed. Restriction enzyme sites of StuI, SalI, NcoI, and XhoI were introduced into pESC-HIS and pESC-TRP to create pESC-HM and pESC-TM (Fig. S2), respectively. For the expression of LUS1 (GenBank accession no. NM_179572.1), the ORF of LUS1 was amplified with primers 7 and 8, and cloned under the GAL1 (GenBank accession no. K02115.1) promoter at BamHI/NotI sites of pESC-HM leading to the construct plJ003 (Table 1). For the isolation of ADH1 promoter, GPD promoter, TEF promoter, and CYC1 terminator, the genomic DNA of S. cerevisiae WAT11 was used as the template for the PCRs with primers 9 and 10 for ADH1 promoter (GenBank accession no. U33753.1), 11/12 for GPD promoter (GenBank accession no. DQ269148.1), 13/14 for TEF promoter (GenBank accession no. EF210199.1), and 15/16 for CYC1 terminator (GenBank accession no.L11060.1), respectively. To create the expression cassette “pADH1-CrAO-CYC1 terminator” (designated A-CrAO expression box), the ORF of CrAO (GenBank accession no. JN565975.1) was amplified by reverse transcription (RT)-PCRs from the cDNA of C. roseus with the primers 17 and 18 and ligated with ADH1 promoter and CYC1 terminator by overlapping PCRs with the primers 9–16. Similarly, the DNA cassettes, “pGPD-CrAO-CYC1 terminator” (designated G-CrAO expression box) and “pTEF-CrAO-CYC1 terminator” (designated T-CrAO expression box), were amplified with the primers 11–16 and 13–16, respectively. The same approach was applied to prepare the DNA expression cassettes for tHMG1 (GenBank accession no. NM_001182434.1) (A-, G-, or T-tHMG1 expression box) using the primers 19–22, and for ERG9 (GenBank accession no.NM_001179321.1) (A-, G-, or T-ERG9 expression box) using the primers 23–26. The ORF of tHMG1 was amplified from the DNA of S. cerevisiae WAT11 while the ORF of ERG9 was isolated from the genomic DNA of S. cerevisiae CEN.PK 2-1D (Madsen et al. 2011). The CrAO expression box (A-, G-, or T-CrAO expression box) was then cloned into plJ003 at NotI and StuI sites, resulting in plJ004, plJ005, and plJ006, respectively (Table 1). The expression cassette “A-tHMG1 expression box” was digested with StuI/SpeI and subcloned into plJ004 to make the construct plJ007 (Table 1). Similarly, the constructs plJ008 and plJ009 were prepared (Table 1). For the expression of HFA1, the ORF of HFA1 was isolated from the genomic DNA of S. cerevisiae CEN. PK2-1D. Due to the long length of HFA1 ORF (GenBank accession no. NM_001182714.1; the length of the cDNA is 6,372 bp), the three truncated fragments of the HFA1 cDNA were separately amplified using primers 27–34. The 3′ truncated fragment was ligated with CYC1 terminator by overlapping PCRs using primers 33–35. The 5′ truncated fragment was fused with the ADH1, GP,D or TEF promoter, respectively, by overlapping PCRs using the primer 30 paired up with the primers 36–38. Using restriction enzymes followed by ligations (SalI, NcoI, XhoI and NheI), the DNA cassette, “pADH1-HFA1-CYC1 terminator” (designated A-HFA1 expression box), “pGPD-HFA1-CYC1 terminator” (designated G-HFA1 expression box), or “pTEF-HFA1-CYC1 terminator” (designated T-HFA1 expression box), was prepared. The combination of the HFA1 expression cassette (A-, G-, or T-HFA1 expression box) with the ERG9 expression cassette (A-, G-, or T-ERG9 expression box) was constructed in the plasmid pESC-TM, which gave the constructs plJ010, plJ011, plJ012, plJ013, plJ014, plJ015, plJ016, plJ017, and plJ018 (Table 1).
Construction and cultivation of yeast strains
S. cerevisiae WAT11, a derivative of W303–1B, was used as the parent strain to be engineered in this study (Table 2) (Urban et al. 1997). Transformation of Saccharomyces strains was performed by a standard lithium acetate protocol (Gietz and Woods 2002). To investigate the effect of different promoter strengths on betulinic acid biosynthesis, the strains WAT4-A, WAT4-G, and WAT4-T were constructed (Table 2). Of these strains, CrAO gene was cloned under the control of ADH, GPD, and TEF promoters, respectively. For modulating gene expression on fatty acid and betulinic acid biosynthetic pathways, the strains WATM7-AA, WATM7-AG, WATM7-AT, WATM7-GA, WATM7-GG, WATM7-GT, WATM7-TA, WATM7-TG, and WATM7-TT were established (Table 2). The engineered yeast strains were grown at 30 °C either in SD (synthetic defined) medium lacking histidine, tryptophan, leucine, methionine, and uracil, and supplemented with 2 % glucose and 1-mM methionine unless otherwise indicated or in YPDA medium (Burke and Stearns 2000).
A single colony of the engineered strains was inoculated in 2 mL of appropriate medium and grown at 250 rpm and 30 °C in an incubator to OD600 of 2.0. The yeast cultures were then diluted into fresh medium at a ratio of 1:20 and subsequently grown at 30 °C. The yeast growth was evaluated by measuring the cell optical density at 600 nm every 24 h until the seventh day after the subculture.
Extraction and gas chromatography–mass spectrometry (GC–MS) analysis of betulinic acid
The 7th day was the stationary growth phase for all the engineered strains and the logical choice as the sampling time for extracting betulinic acid. Yeast cells were separated from the medium by centrifugation, and the medium was saved for the extraction of betulinic acid. The cell pellets were washed with 1 mL of alkaline buffer (pH 9.0, 50 mM Tris–HCl buffer) resulting in the cell surface fraction. The surface fraction and the medium were all acidified to pH 2 using 2 M HCl, passed through a CNWBOUND LC-C18 column (CNW, Shanghai, China), and extracted with methanol. The methanol extracts were air-dried and derivatized by N, O-Bis (trimethylsilyl) trifluoroacetamide (BSTFA, Sigma-Aldrich) at 80 °C for 30 min prior to GC–MS analysis (Huang et al. 2012). Betulinic acid and ursolic acid standards were purchased from Sigma-Aldrich. Ursolic acid was included into the sample and used as an internal standard.
For the GC–MS analysis, 1 μL of the sample was injected into an HP-5MS column (Agilent) operating at a helium flow rate of 1.2 mL/min. The injection temperature was 250 °C. The oven temperature was held at 80 °C for 2 min after injection and was then ramped to 310 °C at 20 °C/min, and held at 310 °C for 15 min. Full mass spectra were generated for metabolite identification by scanning within the m/z range of 50–600. Compounds were identified by comparing their retention times and mass spectra with those of authentic standards (Huang et al. 2012). Ursolic acid was used as an internal standard to quantify the production of betulinic acid formed by the engineered yeasts using the response factor determined by ursolic and betulinic acid standards on the GC–MS conditions.
Results
Optimization of glucose and galactose ratio for betulinic acid biosynthesis
For constructing the betulinic acid biosynthetic pathway, the LUS1 gene was cloned under the control of a galactose-inducible promoter, and the CrAO gene was placed under the control of a constitutive promoter (TEF, GPD, or ADH promoter). Galactose acts as a gratuitous inducer (Hovland et al. 1989) for all galactose-regulated genes while glucose is a usual carbohydrate for yeast growth. Using S. cerevisiae WAT4-G (Table 2) as the test strain, the overall yield of betulinic acid and the cell growth (estimated by OD600 nm) were compared when 2 % glucose, 2 % galactose, or the mixture of 1 % glucose and 1 % galactose were used as the carbon source. As shown in Fig. 3b, the growth curve of WAT4-G was similar in the presence of the three different sets of the carbohydrates and reached the stationary phase by the third day after the inoculations. To compare the production of betulinic acid, the yeast cultures were collected at the seventh day for this purpose. Betulinic acid yield was extremely low (0.015 mg L−1 OD−1) when the carbon source was 2 % glucose and around 16-fold less than that when the mixture was 1 % glucose and 1 % galactose being the carbon source (0.24 mg L−1 OD−1) (Fig. 2a). The low level of betulinic acid accumulation in the strain cultured by 2 % glucose might be attributed to a strong inhibition of LUS1 expression by 2 % glucose. It is well known that the expression of genes under a galactose-inducible promoter would be strongly repressed by glucose (Hovland et al. 1989). When 2 % galactose was used, the production of betulinic acid was 0.079 mg L−1 OD−1 (Fig. 2a). Thus, the mixture of 1 % glucose and 1 % galactose was considered to be more appropriate for culturing the yeast strains in this study, giving the appearance of a higher accumulation of betulinic acid and no defect of cell growth compared with the other two sets of carbon source (Fig. 2b).

Effects of glucose and galactose ratio on the production of betulinic acid (a) and yeast growth property (b) for the engineered strain WAT4-G (Table 2). GAL of 2 %, the yeast medium containing 2 % galactose; 1 % GAL/1 % GLU, the yeast medium containing 1 % galactose and 1 % glucose; 2 % GLU, the yeast medium containing 2 % glucose. These data are shown as means ± standard deviations for three independent cultures
Effects of different promoter strengths on betulinic acid biosynthesis
To combinatorially modulate gene expressions, the promoters of different strengths are necessary. The ADH1, GPD, and TEF promoters were chosen as they drove the expression of beta-galactosidase in considerably different levels, in which the ADH1 promoter functioned in the weakest manner while GPD or TEF promoter showed a stronger activity (Mumberg et al. 1995). To test the difference of their activities for betulinic acid biosynthesis, the CrAO gene was placed under the control of the ADH1, TEF, and GPD promoters, respectively, and coexpressed with the LUS1 gene that was controlled by a galactose-inducible promoter (GAL1 promoter). The three transgenic yeast strains WAT4-A, WAT4-G, and WAT4-T were created (Table 2). The transgenic yeasts were cultured in appropriate SD medium containing 1 % galactose and 1 % glucose and used for the extraction of betulinic acid at the seventh day of the subculture (see “Method and materials” section). GC–MS analysis showed that the highest concentration of betulinic acid (0.41 mg L−1 OD−1) was detected in the WAT4-A strain in which CrAO was driven by the ADH1 promoter, while the betulinic acid productions from the yeast strains WAT4-G (CrAO was under the control of the GPD promoter) and WAT4-T (CrAO was under the control of the TEF promoter) were 0.24 and 0.009 mg L−1 OD−1, respectively (Fig. 3a). The accumulation pattern of betulinic acid in these strains indicated that the order of the three promoters in strength was ADH1 > GPD > TEF, which was out of our expectations as it was in striking contrast to the previous study (Mumberg et al. 1995). To further compare the promoters’ strengths, the relative transcript level of CrAO was examined by qRT-PCRs, and the results also supported the promoters’ activities described above (Fig. S3). Since the activities of many promoters are carbon source dependent (Pauwels et al. 1999), the difference in the promoters’ activities might be attributed to the difference of carbon source between the current and previous studies (Scholer and Schuller 1994). The growth property of the three transgenic strains was monitored through a time course. As shown in Fig. 3b, they grew in a similar rate before 72 h; however, the growth of WAT4-A was slower compared with the other two strains after the 72-h cultivation. In summary, the overall effect on betulinic acid production was obviously different among the three constitutive promoters by regulating the expression of CrAO.

Effects of different strength promoters on the production of betulinic acid (a) and yeast growth property (b). A WAT4-A (the strain WAT4 harboring pESC-LEU, pESC-TM, plJ004), G WAT4-G (the strain WAT4 harboring pESC-LEU, pESC-TM, plJ005), T WAT4-T (the strain WAT4 harboring pESC-LEU, pESC-TM, plJ006) (Table 2). These data are shown as means ± standard deviations for three independent cultures
Optimization of betulinic acid production by balancing two metabolic fluxes
To divert more carbon flux into betulinic acid biosynthesis from ergosterol biosynthesis (Fig. 1), strains with the downregulation of the ERG7 gene by the MET3 promoter and the concomitant inductions of the ERG1 and LUS1 genes by galactose-inducible promoters were engineered. Based on this genetic background, we further reset the flux balance between betulinic acid and fatty acid biosynthetic pathways by changing the gene expressions using the promoters in different strengths (ADH1, GPD, and TEF promoter). In betulinic acid biosynthesis pathway, the gene coding for HMG1, ERG9, and CrAO were chosen as the betulinic acid module. For the fatty acid module, the gene coding for HFA1 was chosen. Each module was separately cloned under the ADH1, GPD, or TEF promoter, respectively, paired up with the other module, and transformed into the WATM7 strain (Table 2). Eventually, a total of nine strains (WATM7-AA, WATM7-AG, WATM7-AT, WATM7-GA, WATM7-GG, WATM7-GT, WATM7-TA, WATM7-TG, and WATM7-TT) were constructed to vary the fluxes between the two pathways (Table 2). For example, in the engineered strains WATM7-AA, WATM7-AG, and WATM7-AT, the betulinic acid module was placed under the control of the ADH1 promoter while the fatty acid module was directed by the ADH1, GPD, or TEF promoter, respectively (Table 2). The nine kinds of yeast strains were cultured in appropriate SD medium including 1 % galactose/1 % glucose as the carbon source and 1 mM methionine as the inhibitor on the ERG7 expression (Kirby et al. 2008). The betulinic acid production and yeast growth were measured. The yield value (mg L−1 OD−1) for betulinic acid was dramatically different among the nine strains (Fig. 4a). For the strain WATM7-AA, both modules (betulinic acid and fatty acid modules) were cloned under the strongest promoter (ADH1 promoter), but the betulinic acid production of this strain was only 0.049 mg L−1 OD−1 which was 39-fold less than that of the strain WATM7-GT (1.92 mg L−1 OD−1) (Fig. 4a). For the strain WATM7-GT, the betulinic acid module was controlled by the GPD promoter (the GPD promoter showed a moderate strength in this study) while the fatty acid module was placed under the control of the TEF promoter (the TEF promoter was the weakest promoter in this study). Moreover, in comparison to the other strains, the cell growth of the WATM7-AA was severely inhibited whereas the highest betulinic acid producing strain WATM7-GT grew normally or even in a better manner (Fig. 4b). Interestingly, when the betulinic acid module was placed under either the ADH1 or GPD promoter (both promoters showed a relatively stronger activity in this study), the production of betulinic acid increased with the decreased expression of fatty acid module (Fig. 4a). For example, in the strains WATM7-AA, WATM7-AG, and WATM7-AT, the betulinic acid module was cloned under the control of the ADH1 promoter while the fatty acid module was placed under the ADH1, GPD, or TEF promoter, respectively. Logically, from the strain WATM7-AA to WATM7-AT, the expression of fatty acid module decreased due to the difference in promoter strength while the yield of betulinic acid increased from 0.049 to 0.57 mg L−1 OD−1. Similar trend was also found in the strains WATM7-GA, WATM7-GG, and WATM7-GT; the betulinic acid production increased from 0.046 to 1.92 mg L−1 OD−1 (Fig. 4a). Taken together, these results indicated that it was possible to create a higher betulinic acid producing yeast strain by varying the metabolic fluxes between fatty acid and betulinic acid pathways, but maintaining a normal growth phenotype.

Effects of mutually varied expression of two modules (fatty acid and betulinic acid modules) on the production of betulinic acid (a) and yeast growth property (b). AA the strain WATM7-AA, AG the strain WATM7-AG, AT the strain WATM7-AT, GA the strain WATM7-GA, GG the strain WATM7-GG, GT the strain WATM7-GT, TA the strain WATM7-TA, TG the strain WATM7-TG, TT the strain WATM7-TT. For the module expressions in the above strains, see the details in Table 2. These data are shown as means ± standard deviations for three independent cultures
Discussion
Both E. coli and S. cerevisiae are common heterogonous hosts for synthesizing plant-based chemicals through metabolic engineering. The balances on native metabolic fluxes in these hosts are inevitably disturbed by the invasions of exogenous pathways introduced. There are growing evidences to demonstrate that the interaction between existing and heterogonous pathways regulates the formation of desirable molecules. Fatty acid biosynthesis appeared to be one of the native pathways for interacting exogenous terpenoid biosynthesis, which has been demonstrated in both E. coli and S. cerevisiae systems (Kizer et al. 2008; Madsen et al. 2011; Pitera et al. 2007). For this reason, we postulated that the carbon flux balance between fatty acid and MVA biosynthetic pathways could be optimized for diverting more carbon flux into exogenous terpenoid biosynthesis. To test this hypothesis, in the S. cerevisiae strain expressing betulinic acid biosynthetic pathway, we set out to modulate the transcript levels of the key genes on fatty acid and betulinic acid forming pathways to investigate their effects on the production of betulinic acid. The results in this study indeed supported the hypothesis. The mutual variations of the genes on the two pathways considerably affected the production of the end product, betulinic acid, and the property of cell growth. Interestingly, the highest concentration of betulinic acid was observed in the strain WATM7-GT, in which the fatty acid module was controlled by the weakest promoter (a TEF promoter), while the betulinic acid module (ERG9, HMG1, and CrAO) was directed by a moderate promoter (a GPD promoter). Favorably, the strain WATM7-GT showed a similar or even better growth property compared with the other strains. The engineering concept will be very useful for the biological synthesis of other triterpenoids in S. cerevisiae.
Triterpenes are the most versatile class of isoprenoids with more than 30,000 identified compounds (Dzubak et al. 2006). The biological activities of such triterpenes are very diverse and have made them industrially relevant and potent compounds (Dzubak et al. 2006). S. cerevisiae has been successfully applied to the high production of sesquiterpenes (Paddon et al. 2013; Ro et al. 2006; Westfall et al. 2012), diterpenes (Ajikumar et al. 2010; Boghigian et al. 2012), and tetraterpenes (Farmer and Liao 2000; Kim and Keasling 2001; Matthews and Wurtzel 2000). However, there are more challenges in engineering the biosynthesis of triterpenoids in yeast. Triterpenoids are biosynthesized from oxidosqualene which is also the precursor of ergosterol biosynthesis in yeast. Sterol biosynthesis is necessary for yeast growth, and the pathway is tightly regulated (Hampton and Rine 1994; Kennedy and Bard 2001; Leber et al. 2001; Veen et al. 2003). Perhaps, the largest challenge to boost triterpenoid productions is to divert more carbon fluxes from sterol biosynthesis to exogenous triterpenoid pathways. However, the regulatory mechanism on sterol biosynthesis in yeast still remains largely unknown to date. Such a tight sterol regulation might not exist in E. coli or bacteria system. For example, when the ERG1 equivalent gene Se was knocked out, the growth of Stigmatella aurantiaca was not affected (Bode et al. 2003). For the E. coli system, the absence of the tight sterol regulation seems also to be predicted from KEGG pathway database (http://www.genome.jp/kegg/). Therefore, for this reason, it will be expected to be worth pursuing the biological synthesis of triterpenoids in E. coli or bacteria system. To produce triterpenoids in E .coli, the major obstacle is to functionally express P450 enzymes in this microbial host; however, the problem could be resolved by the codon optimization of P450 cDNAs and the alterations on P450’s membrane anchors (Kaspera and Croteau 2006; Schuler et al. 2003; Nelson 1999). One of the successful example is to functionally express a modified amorphadiene oxidase (a P450 in the steps to artemisinin from Artemisia annua) in E. coli, leading to a high titer of artemisinic acid production (Martin et al. 2003). Thus, through similar manipulations on CrAO enzyme (a P450 catalyzing the formation of betulinic acid), it will be promising to engineer betulinic acid biosynthetic pathway in E. coli.
In summary, using S. cerevisiae as the host, we described the efforts to improve betulinic acid production by optimizing the flux balance between fatty acid and betulinic acid forming pathways. This strategy would be promising in synthesizing other triterpenoids in S. cerevisiae.
References
Ajikumar PK, Xiao WH, Tyo KEJ, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G (2010) Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science 330:70–74
Bode HB, Zeggel B, Silakowski B, Wenzel SC, Reichenbach H, Müller R (2003) Steroid biosynthesis in prokaryotes: identification of myxobacterial steroids and cloning of the first bacterial 2,3(S)-oxidosqualene cyclase from the myxobacterium Stigmatella aurantiaca. Mol Microbiol 47:471–481
Boghigian BA, Armando J, Salas D, Pfeifer BA (2012) Computational identification of gene over-expression targets for metabolic engineering of taxadiene production. Appl Microbiol Biotechnol 93:2063–2073
Burke D DDS, Stearns T. (2000) Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. 2000 ed. Burke, D, New York
Donald KAG, Hampton RY, Fritz IB (1997) Effects of overproduction of the catalytic domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase on squalene synthesis in Saccharomyces cerevisiae. Appl Environ Microbiol 63:3341–3344
Dzubak P, Hajduch M, Vydra D, Hustova A, Kvasnica M, Biedermann D, Markova L, Urban M, Sarek J (2006) Pharmacological activities of natural triterpenoids and their therapeutic implications. Nat Prod Rep 23:394–411
Farmer WR, Liao JC (2000) Improving lycopene production in Escherichia coli by engineering metabolic control. Nat Biotechnol 18:533–537
Gietz RD, Woods RA (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350:87–96
Hampton RY, Rine J (1994) Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J Cell Biol 125:299–312
Hovland P, Flick J, Johnston M, Sclafani RA (1989) Galactose as a gratuitous inducer of GAL gene expression in yeasts growing on glucose. Gene 83:57–64
Huang LL, Li J, Ye HC, Li CF, Wang H, Liu BY, Zhang YS (2012) Molecular characterization of the pentacyclic triterpenoid biosynthetic pathway in Catharanthus roseus. Planta 236:1571–1581
Jäger S, Trojan H, Kopp T, Laszczyk MN, Scheffler A (2009) Pentacyclic triterpene distribution in various plants—rich sources for a new group of multi-potent plant extracts. Molecules 14:2016–2031
Kaspera R, Croteau R (2006) Cytochrome P450 oxygenases of Taxol biosynthesis. Phytochem Rev 5:433–444
Kennedy MA, Bard M (2001) Positive and negative regulation of squalene synthase (ERG9), an ergosterol biosynthetic gene, in Saccharomyces cerevisiae. Biochim Biophys Acta-Gene Struct Expression 1517:177–189
Kim SW, Keasling JD (2001) Metabolic engineering of the nonmevalonate isopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol Bioeng 72:408–415
Kim D, Chen ZD, Nguyen VT, Pezzuto JM, Qiu SX, Lu ZZ (1997) A concise semi-synthetic approach to betulinic acid from betulin. Synth Commun 27:1607–1612
Kirby J, Keasling JD (2008) Metabolic engineering of microorganisms for isoprenoid production. Nat Prod Rep 25:656–661
Kirby J, Romanini DW, Paradise EM, Keasling JD (2008) Engineering triterpene production in Saccharomyces cerevisiae-beta-amyrin synthase from Artemisia annua. Febs J 275:1852–1859
Kizer L, Pitera DJ, Pfleger BF, Keasling JD (2008) Application of functional genomics to pathway optimization for increased isoprenoid production. Appl Environ Microbiol 74:3229–3241
Leber R, Zenz R, Schrottner K, Fuchsbichler S, Puhringer B, Turnowsky F (2001) A novel sequence element is involved in the transcriptional regulation of expression of the ERG1 (squalene epoxidase) gene in Saccharomyces cerevisiae. Eur J Biochem 268:914–924
Liu J, Fu ML, Chen QH (2011) Biotransformation optimization of betulin into betulinic acid production catalysed by cultured Armillaria luteo-virens Sacc ZJUQH100-6 cells. J Appl Microbiol 110:90–97
Madsen KM, Udatha G, Semba S, Otero JM, Koetter P, Nielsen J, Ebizuka Y, Kushiro T, Panagiotou G (2011) Linking genotype and phenotype of Saccharomyces cerevisiae strains reveals metabolic engineering targets and leads to triterpene hyper-producers. PLoS One 6:e14763
Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD (2003) Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol 21:796–802
Matthews PD, Wurtzel ET (2000) Metabolic engineering of carotenoid accumulation in Escherichia coli by modulation of the isoprenoid precursor pool with expression of deoxyxylulose phosphate synthase. Appl Microbiol Biotechnol 53:396–400
Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122
Nelson DR (1999) Cytochrome P450 and the individuality of species. Arch Biochem Biophys 369:1–10
Nijkamp JF, van den Broek M, Datema E, de Kok S, Bosman L, Luttik MA, Daran-Lapujade P, Vongsangnak W, Nielsen J, Heijne WHM, Klaassen P, Paddon CJ, Platt D, Kotter P, van Ham RC, Reinders MJT, Pronk JT, de Ridder D, Daran JM (2012) De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact 11:36
Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D, Leavell MD, Tai A, Main A, Eng D, Polichuk DR, Teoh KH, Reed DW, Treynor T, Lenihan J, Fleck M, Bajad S, Dang G, Dengrove D, Diola D, Dorin G, Ellens KW, Fickes S, Galazzo J, Gaucher SP, Geistlinger T, Henry R, Hepp M, Horning T, Iqbal T, Jiang H, Kizer L, Lieu B, Melis D, Moss N, Regentin R, Secrest S, Tsuruta H, Vazquez R, Westblade LF, Xu L, Yu M, Zhang Y, Zhao L, Lievense J, Covello PS, Keasling JD, Reiling KK, Renninger NS, Newman JD (2013) High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496: 528–+
Pauwels K, Abadjieva A, Hilven P, Crabeel M (1999) A strong carbon source effect is mediated by pUC plasmid sequences in a series of classical yeast vectors designed for promoter characterization. Yeast 15:1269–1274
Pitera DJ, Paddon CJ, Newman JD, Keasling JD (2007) Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab Eng 9:193–207
Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MCY, Withers ST, Shiba Y, Sarpong R, Keasling JD (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440:940–943
Scholer A, Schuller HJ (1994) A carbon source-responsive promoter element necessary for activation of the isocitrate lyase gene ICL1 is common to genes of the gluconeogenic pathway in the yeast Saccharomyces cerevisiae. Mol Cell Biol 14:3613–3622
Schuler MA, Werck-Reichhart D (2003) Functional genomics of P450s. Annu Rev Plant Biol 54(1):629–667
Smith PF, Ogundele A, Forrest A, Wilton J, Salzwedel K, Doto J, Allaway GP, Martin DE (2007) Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-O-(3′,3′-dimethylsuccinyl) betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob Agents Chemother 51:3574–3581
Urban P, Mignotte C, Kazmaier M, Delorme F, Pompon D (1997) Cloning, yeast expression, and characterization of the coupling of two distantly related Arabidopsis thaliana NADPH-Cytochrome P450 reductases with P450 CYP73A5. J Biol Chem 272:19176–19186
Veen M, Stahl U, Lang C (2003) Combined overexpression of genes of the ergosterol biosynthetic pathway leads to accumulation of sterols in Saccharomyces cerevisiae. FEMS Yeast Res 4:87–95
Westfall PJ, Pitera DJ, Lenihan JR, Eng D, Woolard FX, Regentin R, Horning T, Tsuruta H, Melis DJ, Owens A, Fickes S, Diola D, Benjamin KR, Keasling JD, Leavell MD, McPhee DJ, Renninger NS, Newman JD, Paddon CJ (2012) Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc Natl Acad Sci U S A 109:111–118
Withers ST, Keasling JD (2007) Biosynthesis and engineering of isoprenoid small molecules. Appl Microbiol Biotechnol 73:980–990
Yogeeswari P, Sriram D (2005) Betulinic acid and its derivatives: A review on their biological properties. Curr Med Chem 12:657–666
Zuco V, Supino R, Righetti SC, Cleris L, Marchesi E, Gambacorti-Passerini C, Formelli F (2002) Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett 175:17–25
Acknowledgments
This work was supported by the National Science and Technology Program of China during the 25th-year plan period (project no. 2012AA02A704) and the grant for 100 Talents Program of the Chinese Academy of Sciences, China (project no. Y129441R01).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 276 kb)
Rights and permissions
About this article
Cite this article
Li, J., Zhang, Y. Increase of betulinic acid production in Saccharomyces cerevisiae by balancing fatty acids and betulinic acid forming pathways. Appl Microbiol Biotechnol 98, 3081–3089 (2014). https://doi.org/10.1007/s00253-013-5461-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5461-1