
Abstract
l-Amino acid oxidase (LAAO) is a flavoenzyme containing non-covalently bound flavin adenine dinucleotide, which catalyzes the stereospecific oxidative deamination of l-amino acids to α-keto acids and also produces ammonia and hydrogen peroxide via an imino acid intermediate. LAAOs purified from snake venoms are the best-studied members of this family of enzymes, although a number of LAAOs from bacterial and fungal sources have been also reported. From a biochemical point of view, LAAOs from different sources are distinguished by molecular mass, substrate specificity, post-translational modifications and regulation. In analogy to the well-known biotechnological applications of d-amino acid oxidase, important results are expected from the availability of suitable LAAOs; however, these expectations have not been fulfilled yet because none of the “true” LAAOs has successfully been expressed as a recombinant protein in prokaryotic hosts, such as Escherichia coli. In enzyme biotechnology, recombinant production of a protein is mandatory both for the production of large amounts of the catalyst and to improve its biochemical properties by protein engineering. As an alternative, flavoenzymes active on specific l-amino acids have been identified, e.g., l-aspartate oxidase, l-lysine oxidase, l-phenylalanine oxidase, etc. According to presently available information, amino acid oxidases with “narrow” or “strict” substrate specificity represent as good candidates to obtain an enzyme more suitable for biotechnological applications by enlarging their substrate specificity by means of protein engineering.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
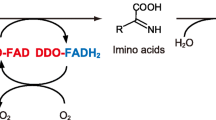
l-Amino acid oxidase (LAAO, EC 1.4.3.2) is a flavin adenine dinucleotide (FAD)-containing flavoenzyme that catalyzes the oxidative deamination of l-amino acids with a strict stereospecificity to give α-keto acids and ammonia; the reduced cofactor (FADH2) then reoxidizes on molecular oxygen, producing hydrogen peroxide (Eq. 1).

This flavooxidase was described 70 years ago by Zeller and Maritz (1944). LAAO activity is widely distributed in nature: it has been identified in a number of snakes, as well as in mammals (from the liver, kidney, brain, mammary gland, and polymorphonuclear leukocytes), insects, mollusks, fishes, algae, fungi, and bacteria species (Lukasheva et al. 2011; Yu and Qiao 2012). A wide range of biological functions is associated with the LAAO reaction in different living organisms; these are generally related to oxidation of the substrate amino acid or to generation of hydrogen peroxide, a reactive oxygen species (Lukasheva et al. 2011). Snake venom LAAOs (SvLAAO) are the best-known members of this enzyme family (Du and Clemetson 2002; Guo et al. 2012). LAAO is a main toxic component of snake venoms since it depletes essential amino acids and binds to the cell surface, generating a high local concentration of hydrogen peroxide (thus, in turn, activating mechanisms of apoptosis) (Ande et al. 2006; Alves et al. 2008). Indeed, LAAO in sea hare inks and giant African snail mucus also protects these organisms against infections. Similarly, the enzyme produced by the fungus Trichoderma inhibits the growth and development of other microorganisms; and in mammalian milk, LAAO showed a potent antibacterial effect, accounting for mastitis prevention. Indeed, we still need to deepen our understanding of the role of bacterial LAAO although it seems to be related to basal catabolism of l-amino acids and α-keto acids production.
LAAO was demonstrated to catalyze the dehydrogenation of the l-amino acid through a hydride transfer mechanism by steering the trajectory of the interacting orbitals—the orbital of the substrate αC–H and the LUMO of the FAD cofactor N(5)—without direct involvement of amino acid functional groups: the active site residues play a fundamental role in substrate recognition, binding, and orientation (Pawelek et al. 2000). The catalyzed reaction and the mechanism of substrate dehydrogenation by LAAO resemble those of d-amino acid oxidase (DAAO, EC 1.4.3.3), which specifically acts on the D-isomers of the substrates (Umhau et al. 2000; Pollegioni et al. 2007). The availability of recombinant DAAO from different sources and a number of protein engineering studies spurred their use for enzyme-based technological applications (see Fantinato et al. 2001; Sacchi et al. 2004; Pollegioni et al. 2008; Pollegioni and Molla 2011). The redox reaction catalyzed by LAAO might be potentially employed in a variety of biotechnological processes too (see "Current biotechnological uses" section) but unfortunately, LAAOs from snake venom have been never overexpressed as recombinant proteins at suitable yields, which prevented engineering and, in turn, biotechnological application of these enzymes.
Most LAAOs exhibit broad substrate specificity, but there are LAAOs with relatively narrower substrate specificity and several members show a very strict preference for a specific l-amino acid. In this review, we summarize the structural and biochemical properties of LAAO activities from microorganisms as well as the advances in biotechnological application based on their substrate preference. For the sake of clarity, l-amino acid deaminases, i.e., the enzymes that oxidize l-amino acids without producing hydrogen peroxide such as the one from Proteus myxofaciens (Pantaleone et al. 2001), will not be discussed as they do not belong to the LAAO protein family, although they might replace “canonical” LAAOs in specific bioconversion processes.
l-Amino acid oxidase
Table 1 summarizes the properties of the principally known microbial LAAOs. The first LAAO activity from a microorganism to be purified and characterized was that from the Gram-positive bacterium Bacillus carotarum (Brearley et al. 1994). This FAD-containing homodimer (2 × 54 kDa) showed the highest k cat,app on neutral and positively charged amino acids l-Leu, l-Lys, l-Arg, l-Met and l-Asn, although the highest specificity constant (k cat,app/K m,app ratio) was apparent for the hydrophobic amino acids l-Phe, l-Trp, and l-Tyr. Specific activity of the pure enzyme on l-Phe was 4.5 U/mg protein. Intriguingly, the enzyme was also reported to oxidize the D-isomers of the substrates (i.e., d-Leu, d-Lys, and d-Ser).
Rhodococcus sp. is a well-known producer of LAAO. In 2002, Rhodococcus opacus DSM 43250 (RoLAAO) was reported to produce 0.1 mg LAAO/g cell with a specific activity on l-Ala of 4.6 U/mg protein (Geueke and Hummel 2002). The purified enzyme was a ~100-kDa homodimer with very broad substrate specificity: among the natural amino acids, only Gly, l-Thr, and l-Pro were not oxidized. The preferred substrate was l-Tyr (V max,app = 9.7 U/mg protein, K m,app = 0.019 mM) and a substrate inhibition effect at increasing substrate concentration was frequently observed (K i in the 1–190-mM concentration range). The overproduction of RoLAAO in E. coli resulted in the accumulation of the recombinant protein as inclusion bodies, while it was produced as soluble protein in Streptomyces lividans (0.05 mg/g cell, 18 U/L of fermentation broth) (Geueke and Hummel 2003). A homodimeric (2 × 51 kDa) LAAO flavoprotein was also isolated from Rhodococcus sp. AIU Z-35-1 (previously classified as lysyl oxidase, see below): it showed significant kinetic parameters with l-Lys (Vmax,app = 32 U/mg protein, specific activity = 19.1 U/mg protein, Km,app = 16 μM), a broad substrate specificity (activity on l-Ala > Nα-Z-l-Lys > l-His > l-Tyr > l-Orn > l-Gln), and a temperature optimum of 50 °C (Isobe and Nagasawa 2007). The main drawback was the very low expression level of fermentation (0.018 mg/L). Recently, the Rhodococcus sp. AIU strain (LAB-3) reached a LAAO productivity of 2.3 mg/L of fermentation (Isobe et al. 2012a, b); the apparent K m was in the 2- to 840-μM range, depending on the amino acid used (the highest affinity was observed for Nα-Z-l-Lys). Rhodococcus sp. AIU LAB-3 LAAO differed from RoLAAO because of the absence of any substrate inhibition effect.
LAAO activity was also identified in marine bacteria Pseudoalteromonas strains and observed to have antibacterial activity against several methicillin-resistant Staphylococcus aureus strains: LAAO from Pseudoalteromonas luteoviolacea was a dimeric ~110-kDa enzyme showing a broad substrate specificity (it was most active on l-Met > l-Gln > l-Leu > l-Phe > l-Glu > l-Trp) (Gómez et al. 2008), while the one isolated from Pseudoalteromonas flavipulchra (60 kDa, pI of 9.4) was mostly active on l-Lys > l-Met > l-Glu > l-Leu > l-Gln > l-Tyr > l-Phe (Chen et al. 2010).
LAAO activity was also isolated from fungi. LAAO was produced by Aspergillus fumigatus up to 56 U/g cell under optimized conditions: the highest activity was obtained with l-Tyr, followed by l-Phe > l-Pro > l-Ser > l-Leu, l-Ala > and l-Asp (13 % vs. l-Tyr) (Singh et al. 2009). The filamentous fungus Trichoderma harzianum ETS 323, used as a biocontrol agent against phytopathogens such as Rhizoctonia solani, produces an extracellular FAD-containing LAAO (2.6 mg/L of fermentation broth) that is active only on l-Phe, l-Lys, l-Glu, and l-Ala (K m in the 6–20-mM range), with a specific activity of 0.4 U/mg protein (Yang et al. 2011a). The enzyme, made of 480 residues, is glycosylated and probably exists in solution in equilibrium between a monomeric 67-kDa and a dimeric 130-kDa form. The enzyme shows antibacterial activity as the result of two mechanisms: by causing membrane permeabilization (through the N-terminal sequence) and by producing reactive oxidative species (triggering cell damage) (Yang et al. 2011b).
Very recently, LAAO activity was identified, too, in the ectomycorrhizal basidiomycete fungi Hebeloma cylindrosporum (HcLAAO), which lives in symbiosis with tree roots of angiosperms and gymnosperms. The recombinant protein was expressed in E. coli BL21(DE3) cells using the pET46 plasmid and Luria Bertani (LB) medium, adding 0.05 mM FAD and 1 mM IPTG at an OD600 nm of 0.5–1.0, and further growing overnight at 16 °C (Nuutinen et al. 2012). The enzyme largely accumulated as inclusion bodies and when isolated from the soluble fraction was inactive: SDS treatment restored the enzymatic activity. The substrate specificity of HcLAAO was very broad: it displayed more than 2 % activity relative to the best substrate with 37 l-amino acids or derivatives (natural and unnatural amino acids). The highest activity was determined for l-Glu, followed by amino acids possessing positively charged or polar side chains (i.e., l-Gln, l-Asn); amino acids with hydrophobic side chains were also accepted as substrates. The K m,app values were in the 0.5- to 6.7-mM range and the highest specificity constants were measured for l-Leu, l-Gln, and l-Glu. The purified enzyme (70 kDa in SDS-PAGE) contained FAD and showed a pH optimum of 8.0 that was not dependent on the amino acids analyzed. The broad substrate specificity of HcLAAO (one of the major soluble proteins of Hebeloma cylindrosporum in the growth conditions) argued for its main role in cellular amino acid catabolism to mineralize the amino acids from nutrient-rich patches of soils, such as pollen, seeds, and animal remnants.
In summary, expression of a recombinant true LAAO activity in E. coli was only achieved in HcLAAO which was produced as inactive enzyme in vitro and converted into the active enzyme form.
l-Aspartate oxidase
In prokaryotes, l-aspartate oxidases (LASPO, EC 1.4.3.16) catalyze the first step of de novo nicotinamide adenine dinucleotide (NAD+) biosynthesis: the product iminosuccinate in vivo is condensed with dihydroxyacetone phosphate (a reaction catalyzed by quinolinate synthase), resulting in the production of quinolinate and, eventually, NAD+ (Tedeschi et al. 1996). In vitro LASPO can use both O2 and fumarate in cofactor re-oxidation, which enables it to perform catalysis under both aerobic and anaerobic conditions. In fact, under aerobic conditions, free LASPO oxidizes l-aspartate to iminosuccinate, which is then nonenzymatically hydrolyzed to oxaloacetate (Eq. 2):

Importantly, LASPO is a member of the succinate dehydrogenase/fumarate reductase family of enzymes and thus different from the LAAO family (see below).
The first study concerning LASPO activity in E. coli (EcLASPO) was reported in 1990 (Seifert et al. 1990). Cloned cDNA was overexpressed in E. coli cells up to about 20 % of the soluble cell protein content (and up to 10 mg/L of fermentation broth) and was purified as a stable monomeric FAD-containing enzyme of 60,284 Da (Table 2). EcLASPO apoprotein bound FAD by a simple second-order process: the interaction was tighter in the presence of glycerol, succinate, or fumarate (K d decreased from 3.8 to 0.67 μM) (Mortarino et al. 1996). EcLASPO was inhibited by the product iminoaspartate (K d of 1.4 μM) as well as by succinate and fumarate (K d of ~0.2 mM) (Mortarino et al. 1996; Tedeschi et al. 1996). Steady-state kinetics was investigated for both the oxidase and the fumarate reductase activity of EcLASPO: with fumarate as electron acceptor, a higher turnover number was apparent than with dioxygen (k cat was 5.6 vs. 2.6 s−1, respectively), but the affinity for oxygen was tenfold higher (K m,O2 = 0.25 mM vs. K m,fumarate = 2.5 mM). K m for l-Asp was 2.7–5.2 mM, and d-Asp and l-Asn were not substrates. Site-directed mutagenesis experiments were performed at positions E43, G44, S45, F47, and Y48 of the putative cofactor binding site, demonstrating that the stretch 43–48 was involved in FAD binding (Mortarino et al. 1996).
The physiological role of LASPO in Bacillus subtilis is also related to the first two steps in the de novo synthesis of NAD. This flavoprotein was overexpressed (up to 10 mg/L of fermentation broth) in E. coli BL21(DE3) cells using the pET-duet plasmid and by growing the cells in LB medium at 28 °C for 5 h after adding 1 mM IPTG (Marinoni et al. 2008). The enzyme was isolated as holoenzyme (K d for FAD = 5 μM) and its quaternary structure in solution depended on the salt concentration. B. subtilis LASPO showed three kinds of enzymatic activity: l-aspartate oxygen oxidoreductase activity, fumarate reductase activity, and l-aspartate-fumarate oxidoreductase activity. Regarding the LASPO activity (assayed using dioxygen as electron acceptor), the k cat,app was 0.18 s−1 and the K m,app for l-Asp was 1.0 mM (values resembling the ones of EcLASPO, see Table 2); a strong product inhibition effect was also apparent (K d was 0.5 and 0.32 mM for oxaloacetate and iminoaspartate, respectively).
Very recently, LASPO from Pseudomonas putida (PpLASPO) was also overexpressed in E. coli BL21(DE3) cells, reaching 10 mg/L of fermentation broth (Leese et al. 2013). The enzyme was purified as active dimeric (~116 kDa) holoenzyme. PpLASPO showed a kinetic efficiency on l-Asp that was tenfold higher than the E. coli counterpart, mainly because of the significantly higher k cat,app (10.6 s−1); a lower activity was detected on l-Asn, for which pronounced substrate inhibition was observed at concentrations >15 mM. Site-directed mutagenesis was carried out on seven positions identified by in silico analysis to be possibly involved in substrate binding: the H244A variant showed a twofold higher k cat,app for l-Asp, but its Km,app was increased approximately 40-fold over the wild-type PpLASPO. Indeed, the R121C substitution increased the solubility of the recombinant protein (with no changes in activity on l-Asp), and the S389A variant PpLASPO showed an increased affinity for l-Glu.
Of potential interest are the enzymes isolated from thermophilic microorganisms, such as Sulfolobus tokodaii (StLASPO) that was overexpressed in E. coli cells by up to 9 % of the total proteins in the crude extract and 13.5 mg/L of fermentation broth (Sakuraba et al. 2008). This flavoenzyme was monomeric (52 kDa) in solution, showed the classical properties of FAD-containing oxidases (and a tight interaction with the cofactor), and possessed a high thermal stability: the enzyme was fully stable at 80 °C (Sakuraba et al. 2008; Bifulco et al. 2013). Moreover, the enzyme showed a higher substrate preference for l-Asp than for l-Asn (the apparent V max/K m ratio is 63-fold higher with the former), the only two amino acids oxidized by StLASPO (Bifulco et al. 2013). Steady-state measurements highlighted a low K m,O2 (0.3 mM, Table 2) in comparison to known amino acid oxidases (Pollegioni et al. 2008). StLASPO was also reported to bind a number of dicarboxylic acids and was weakly inhibited by the product oxaloacetate and by d-Asp. An additional LASPO from a hyperthermophilic archaeon (i.e., Pyrococcus horikoshii) was also efficiently overexpressed in E. coli (7 mg of pure enzyme/L of fermentation broth) (Sakuraba et al. 2002). It differs from StLASPO, with regards to the trimeric oligomeric state (~151 kDa in gel permeation chromatography), higher specific activity (3.2 U/mg protein), and K m,app (8.2 mM) on l-Asp, and in that it oxidizes l-Asp exclusively. In addition, the activity (i.e., the initial rate of the reaction) of this LASPO is not linear when using more than 0.6 μg of enzyme in the assay mixture, a behavior also observed for other LASPOs.
l-Glutamate oxidase
Bacteria from the genus Streptomyces secrete a LAAO specific for l-Glu. The first l-glutamate oxidase (LGO) activity was identified in the culture broth of Streptomyces violascens (4.4 mg of pure protein/L, Table 3) (Kamei et al. 1983). This 60-kDa FAD-containing enzyme showed activity only on l-Glu (60 U/mg protein) and l-Gln (~30 % vs. l-Glu). Most recently, the extracellular LGO was also isolated from Streptomyces sp. Z-11-6 broth (1,600 U/L): 12 mg of pure protein per liter of fermentation broth, with a specific activity of 50 U/mg protein and a K m for l-Glu of 0.34 mM, was obtained (Sukhacheva and Zhuravleva 2004). The molecular mass of the native enzyme in gel permeation chromatography was 95 kDa: it is a tetramer constituted by two α-subunits (25 kDa) and two β-subunits (22.5 kDa).
LGO was also purified from the culture medium of Streptomyces endus (0.2 mg/L) (Böhmer et al. 1989). This dimeric 90-kDa flavoenzyme was strictly specific for l-Glu: specific activity was 6 U/mg and the affinity for the substrate was lower than the one determined for S. violascens LGO (K m = 18 mM). LGO from Streptomyces platensis NTU3304 was expressed in S. lividans 66 cells (grown for 5 days at 28 °C) and was produced both as intracellular and extracellular enzymes reaching up to 8,000 and 4,250 U/L of fermentation broth, respectively (Chen et al. 2001). Owing to this great overproduction, 60 mg of pure enzyme/L with a specific activity of 62.5 U/mg protein could be isolated.
LGO from Streptomyces sp. X-119-6 is a 150-kDa protein constituted by a hexameric α2β2γ2 structure (Arima et al. 2003): in solution it is in equilibrium between 150- and 300-kDa form. Its cDNA was expressed in E. coli JM109 using a pKK223-3 plasmid, growing the cells in 2xYT medium at 22 °C for 24 h after adding 0.5 mM IPTG: 100 mg/L of fermentation broth of the unprocessed, unstable, and marginally active precursor was produced. Proteolysis of the recombinant precursor with a metalloendopeptidase from Streptomyces griseus yielded a mature LGO (optimized conditions reached 100 U/L of fermentation broth and 60 U/mg protein) (Upadhyay et al. 2006) whose properties resembled those of the native enzyme purified from the original source: k cat,app was 75 s−1 (specific activity was 55 U/mg protein) and K m,app was 0.23 mM. Deep analysis of the structure–function relationships in this LGO was obtained by X-ray crystallography (Arima et al. 2009) and by site-directed mutagenesis studies of the active site residues R305, H312, and W564 (Utsumi et al. 2012). While the mutations of H312 and W564 to Ala exerted little influence, the substrate specificity of Streptomyces sp. X-119-6 LGO was altered in the R305 variants: R305L, R305D, and R305K showed an appreciable activity on l-His (V max,app = 12.1 s−1, 5.3 s−1 and 4–8 s−1, respectively) and E305A on both l-His and l-Phe (V max,app = 9.4 and 6.5 s−1, respectively) (Utsumi et al. 2012).
l-Lysine oxidase
Intriguingly, LAAOs isolated from fishes and other sea organisms as well as from various fungal strains preferentially oxidize l-Lys. l-Lysine oxidase (LLYSO) was purified from the aqueous extract of a wheat bran culture of the fungus Trichoderma viride Y244-2 (3 mg enzyme/L) (Kusakabe et al. 1980). This 116-kDa homodimeric flavoenzyme showed a high specific activity on l-Lys (66 U/mg protein) and significantly lower activity on l-ornithine, l-Phe, and l-Arg (18, 8, and 6 % vs. l-Lys, respectively); the K m,app for the substrates was quite high (14 mM for l-Phe and 1.6 mM for dioxygen, Table 4). The properties of LLYSO from different Trichoderma strains have been reviewed (Lukasheva and Berezov 2002).
Recently, LLYSO activity was identified in the Pseudomonas sp. AIU 813 strain: the enzyme expression level was induced by the presence of l-Lys as nitrogen source, reaching ~0.05 U/L of fermentation broth (Isobe et al. 2012a, b). The recombinant enzyme was purified from the original source as a 110-kDa dimeric FAD-containing holoenzyme and was active on l-Lys, l-ornithine, and l-Arg only (100, 31, and 6 % relative activity, respectively; Table 4). Noticeably, its N-terminal sequence resembled that of putative flavin-containing amine oxidases and putative tryptophan 2-monooxygenase, but not that of LAAO.
l-Phenylalanine oxidase
A 140-kDa dimeric phenylalanine oxidase (PAO) showing both oxidative deamination and oxygenative decarboxylation activity of l-Phe was isolated from Pseudomonas sp. P-501 (Ps501-PAO) (Koyama 1984). This enzyme showed the highest specific activity (122 U/mg protein) and amino acid affinity (K m,app = 0.01 mM on l-Phe) among the known amino acid oxidases (compare Tables 1, 2, 3, and 4) and was also active on l-Tyr, l-Met, and l-norleucine. The enzyme fully serves as an oxidase on l-Met, while on l-Phe 80 % of the reaction followed the oxygenase pathway. The nucleotide sequence encoding Ps501-PAO was cloned and overexpressed in E. coli BL21(DE3) cells using a pBluescript-derived vector, growing the cells in tryptose phosphate medium for 4 h at 30 °C after adding IPTG (Suzuki et al. 2004): 40 mg protein/L of fermentation was produced as fully inactive holoenzyme (Table 4). Pronase and trypsin treatment converted the noncatalytic pro-Ps501-PAO to the catalytic competent form (made of a α-subunit of 91 residues and of a β-subunit of 603 residues) whose spectral and kinetic properties resembled those of the native enzyme. The resolution of the Ps501-PAO 3D structure and site-directed mutagenesis studies paved the way for elucidating the catalytic mechanisms of oxidative deamination vs. oxygenative decarboxylation of l-Phe (Ida et al. 2008, 2011): the former was favored in the R143K and Y536F Ps501-PAO variants.
l-Tryptophan oxidase
Lechevaliera aerocolonigenes ATCC 39243 is able to produce the antitumoral antibiotic rebeccamycin, a member of the tryptophan-derived indolocarbazole family (Nishizawa et al. 2005): the biosynthetic pathway comprises the RebO protein, which is a FAD-containing LAAO. The enzyme was overexpressed in E. coli BL21(DE3) cells using the pET24b plasmid and growing the cells in LB medium for 3–4 h at 30 °C after adding 5 mM IPTG and 1 μg/mL FAD. The homodimeric 101-kDa holoenzyme was active on l-Trp and its derivatives only; k cat,app was 0.2 s−1 and K m,app was 1.5 mM (Table 4).
Structural properties
General structure of LAAOs
All LAAOs whose structures were solved are present in solution as homodimers; each monomer which spans from ~55 to 74.7 kDa (for SvLAAOs and Ps501-PAO, respectively) non-covalently binds the FAD cofactor in an extended conformation (Table 5). LAAO monomers fold in two well-defined domains: a FAD-binding domain and a substrate-binding domain which also hosts the residues that form the active site entrance funnel. In Calloselasma rhodostoma l-amino acid oxidase (CrLAAO) and RoLAAO, a third additional small domain, mainly composed of α-helices, is observed (the helical domain). According to the sequence and structure of the FAD-binding domain, LAAOs are classified as members of the large glutathione reductase 2 (GR2) structural subfamily of the FAD-containing proteins (Dym and Eisenberg 2001).
Despite the low sequence identity between different LAAOs (e.g., CrLAAO and RoLAAO share less than 23 % sequence identity), superimposition of the 3D structure of these enzymes reveals that the overall fold of CrLAAO, RoLAAO, and LGO is very similar (RMSD ~ 1.0–1.2 Å), although the primary structure of LGO is 185 residues longer than CrLAAO. As a matter of fact, the additional sequences are either removed during maturation or form surface loops (Fig. 1a, b). Only Ps501-PAO shows some structural differences that are more evident in the substrate-binding domain. The observed structural similarity of both the overall fold and of the active site organization (see below) points to a strong evolutionary relationship among these enzymes (Fig. 1c).

Evolutionary-conserved amino acidic sequences among different LAAOs. a Alignment of five conserved primary sequences in different LAAOs. Identical residues are shaded in dark blue, similar amino acids in light blue. LASPO sequence is shown only in the alignment of region 1 (the region containing the dinucleotide binding motif) because in the other regions the degree of similarity was too low. Additional sequences present in LGO and Ps501-PAO (in comparison to CrLAAO and RoLAAO) are shown in bold. The presence of gaps in these sequences is due to the proteolytic maturation of the proteins. The ends of the chains forming Ps501-PAO and LGO are shown by brackets. The position of these conserved sequences on the primary (top) and tertiary (left, PDB code: 2iid) structure of CrLAAO is shown using different shades of blue. The substrate l-Phe is shown as green spheres and FAD cofactor as yellow sticks. Amino acids conserved in all LAAO sequences are shown as cyan spheres. b 3D structure of LGO (PDB code: 1e2m) showing that the three main additional sequences form surface loops (blue). c Unrooted phylogenetic tree of selected LAAOs cited in this work prepared using the server ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/ and http://www.phylogeny.fr/) (Dereeper et al. 2008). The three LAAOs from snake venom are represented by SvLAAOs label since they are too close in the tree to be individually depicted. Protein whose 3D structure has been solved is shaded in light blue. Some branches have been shortened for graphical representation (dotted lines)
All LAAOs whose 3D structure is known are subject to post-translational modifications (Table 5). Prokaryotic LAAOs are produced as inactive pro-enzymes, which require proteolytic maturation to become activated. During this process, the pro-enzyme of RoLAAO is cleaved at a single site (between residues 45 and 46) (Geueke and Hummel 2002), pro-Ps501-PAO is cleaved at four sites (the mature active Ps501-PAO is composed by two chains formed by residues 16–106 and 109–711) (Ida et al. 2008), and pro-LGO is cleaved at multiple sites, resulting in a mature protein formed by three chains of 42, 17, and 10 kDa (see Tables 3 and 5) that remain closely entangled in the mature protein (Arima et al. 2009). The proteolytic activation, which is triggered by an endopeptidase, occurs after the protein is secreted and represents a strategy to prevent cellular apoptosis due to depletion of l-amino acids and H2O2 production in the cytoplasm. On the other hand, eukaryotic LAAOs show a high level of glycosylation (~3.7 kDa of glycosylation per monomer at two sites—N172 and N361 in CrLAAO): the glycosylation is strongly related to the maturation, secretion, and function of eukaryotic LAAOs in snake venom.
From a structural point of view, LASPO is substantially different from the “classical” LAAOs. The crystal structure was resolved first for the apoprotein form of EcLASPO (Mattevi et al. 1999) and then for the holoenzyme at 2.2- to 2.5-Å resolution (see below) (Bossi et al. 2002). This flavoprotein is present in solution which is in equilibrium between a monomeric and a dimeric form (Table 2). Each monomer is formed by a single polypeptide chain that folds into three distinct domains: a FAD-binding domain, a capping domain, and a helical domain. The helical domain is adjacent to the FAD-binding domain and strongly interacts with it. LASPO shares a considerable homology with the flavoprotein subunits of succinate dehydrogenase and fumarate reductase (Mattevi et al. 1999): the sequence identity between EcLASPO and E. coli fumarate reductase is 36.1 % for the FAD-binding domain, 36.3 % for the capping domain, and 26.9 % for the helical domain (a similar sequence identity is also observed between EcLASPO and the human succinate dehydrogenase). The binding of the FAD cofactor to LASPO apoprotein promotes a conformational change in two short protein segments (residues 44–53 and 104–141) and a 27° rotation of the capping domain with respect to the FAD-binding domain. As a consequence, the capping domain forms new tight interactions with the FAD-binding domain, allowing the complete active site cavity located at the interface to form between the capping domain and the FAD-binding domain. StLASPO from a hyperthermophilic bacterium possesses a 3D structure very similar to the homologue from E. coli (in the apoprotein form); the superposition of the structure of apo-EcLASPO onto the StLASPO one yields a root-mean-square deviation (RMSD) of 1.4 Å (Sakuraba et al. 2008). The main differences concern the conformation of the polypeptide linker that connects the FAD-binding domain to the C-terminal helical domain. The different conformation of this linker in StLASPO results in the formation of an additional β-sheet and five-residue ion-pair network between the FAD-binding and C-terminal domain. The consequent decrease in the flexibility of the C-terminal region of the protein and the increase in the strength of the inter-domain interactions presumably increase protein thermostability.
Active site access
A recurrent feature in LAAOs is the presence of at least two different channels for the two substrates (amino acid and dioxygen) to access the active site of the enzyme: a large channel connecting the re-face of FAD to the solvent that is used for the substrate (amino acid) binding and the product (imino acid) release and a narrow channel that connects the si-face of FAD to the solvent, which is used by O2 to reach the active site (Fig. 2) (Pawelek et al. 2000; Moustafa et al. 2006). As reported for yeast DAAO, this second channel is necessary for oxidation of the reduced FAD by molecular oxygen since access to the cofactor from the main channel is blocked by the product imino acid. In addition, from a mechanistic point of view, the O2 molecule must reach the C(4a)-N(5) positions of the cofactor, which is the ideal location for efficient O2 reactivity (Saam et al. 2010).

Comparisons of the active site funnels between different l-amino acid oxidases and yeast d-amino acid oxidase (RgDAAO). Cross sections of 3D structures of different LAAOs show that the channel for the substrate amino acid (green) and the channel for O2 (pink) connect the cofactor FAD to different (frequently opposite) regions of the protein surface. The substrate channel of Ps501-PAO is occluded by the side chains of several hydrophobic residues (see main text). The lysine residue, close to the si-face of FAD (yellow) and supposed to be central for the modulation of oxygen reactivity is represented in cyan. Ligands, when present, are represented in orange. PDB codes for the structures represented in this figure are reported next to protein abbreviations
In the structure of CrLAAO in complex with 2-aminobenzoate, two inhibitor molecules (in addition to the one bound at the active site) are located in the active site funnel and suggest that substrate trajectory diffusion is probably driven by electrostatic forces which orient the substrate so that the hydrophobic aromatic ring docks to the substrate specificity pocket (Pawelek et al. 2000). A similar situation is also observed in Ps501-PAO. Interestingly, in this latter enzyme, the active site funnel for the substrate is closed when the substrate is bound at the active site. It has been proposed that since the entrance of the funnel is mainly composed of hydrophobic residues, the hydrophobic interactions between these residues and the side chain of the substrate could switch the conformation of the funnel from a closed to an open position. This could represent an evolutionary strategy to further increase the specificity of Ps501-PAO toward the oxidation of large aromatic amino acids (Ida et al. 2008). Similarly to CrLAAO, the O2 channel in Ps501-PAO also reaches the si-face of FAD where two residues, M142 and K478, form a gate. The role of these residues in controlling O2 reactivity was confirmed by site-directed mutagenesis: both M142A and K478A variants show reduced catalytic activity in comparison to the wild-type Ps501-PAO (~10- and ~200-fold, respectively) (Ida et al. 2008). In LGO the substrate channel, which is longer than in CrLAAO, shows a U-shaped form with two entrances, which are approximately 20 Å from the active site. This situation has been observed in additional flavoproteins belonging to the GR2 subfamily (e.g., in polyamine oxidase) (Binda et al. 1999). On the other hand, RoLAAO lacks the main active site funnel since its active site is located in a cleft on the protein surface; consequently, a large part of the side chain of the bound substrate remains exposed to the solvent. In this enzyme, the rate of substrate delivery at the active site is considered “substrate-driven” (Faust et al. 2007).
General active site set-up
The ability of LAAO to discriminate between the two different enantiomers of the substrate lies in the architecture of their active site that conforms to the four-location model proposed by Mesecar and Koshland (2000). According to this model, the enantioselectivity of the enzyme is based on the existence of three binding interactions between the substrate and the active site and one functional “direction”. In the LAAO active site, the substrate is always bound on the re-face of the isoalloxazine moiety of the FAD. The major anchor point is represented by a salt bridge interaction between the α-carboxylate of the amino acid (negatively charged) and the guanidinium group of an arginine of the active site (positively charged) that is located close to the pyrimidine-derivative ring of the isoalloxazine (Fig. 3). This interaction is augmented by an additional hydrogen bond between one of the two oxygen atoms of the carboxylate and the hydroxyl of a tyrosine. The second anchor point is represented by a hydrogen bond between the α-amino group of the substrate and the main chain C=O of the protein belonging to a small residue (glycine or alanine). The third anchor point is formed by the region of the active site placed above the hydrophobic moiety of the FAD isoalloxazine ring: this region, which accommodates the substrate side chain, represents the main structural determinant of the different substrate specificity observed in the various LAAOs (see below). For this reason, most of the active site differences among LAAOs involve residues belonging to this region. The direction is represented by the orientation of the αC–H function that must point to the flavin N(5) in order for efficient hydride transfer to take place. Intriguingly, in both RoLAAO and CrLAAO one oxygen of the substrate α-carboxylate group binds a water molecule that, in turn, is bonded to a highly conserved lysine residue (K326 and K323 in CrLAAO and RoLAAO, respectively) and to the N(5) atom of the flavin. Recently, it has been proposed that this water molecule could be an H+ donor for the activation of O2 during the FAD re-oxidation step (Saam et al. 2010). Overall, the binding mode observed in LAAOs is similar to that observed in DAAO (Mattevi et al. 1996; Umhau et al. 2000; Pollegioni et al. 2002) but with the arginine and the specificity binding pocket mirrored because of the opposed enantioselectivity.

Binding modes and interactions between substrate and the active sites residues of different LAAOs. a Interactions between the α-carboxylate and α-amino group of the substrate and the active site residues of a “generic” LAAO (R = substrate side chain). Numbers in bold refer to CrLAAO sequence. b Comparison of the different interactions between the side chain of the substrate (l-Phe for CrLAAO, Ps501-PAO and RoLAAO, and modeled l-Glu for LGO) and the residues lining the specificity pocket of different LAAOs. c Interactions between l-Asp and the active site residues of EcLASPO. Cofactor FAD is represented in light gray, substrate in dark blue, and active site residues in blue. H bonds are represented by dotted black lines and hydrophobic interactions by small triangles. Numbers in italics refer to residues subjected to site-directed mutagenesis studies in Ps501-PAO (a) (Ida et al. 2008, 2011), in LGO (b) (Utsumi et al. 2012), and in EcLASPO (c) (Tedeschi et al. 2001, 2010)
Active site specificity
As stated above, microbial LAAOs show very different substrate specificity patterns. These properties depend on the different size and polarity of the substrate specificity pocket. CrLAAO preferentially oxidizes aromatic and hydrophobic amino acids; oxidation of polar and basic amino acids proceeds at a much lower rate. In this enzyme the substrate specificity pocket is lined by the hydrophobic side chains of I430, I374, F227, and W465, the imidazole ring of H223, and the aliphatic portion of R322 (Fig. 3b). These hydrophobic residues are conserved in all the eukaryotic LAAOs whose structure has been solved (i.e., the enzymes from Bothrops jararacussu and Agkistrodon halys pallas): accordingly, these enzymes show the same substrate profile as CrLAAO (Ullah et al. 2012). H223 in CrLAAO could also act as a base that deprotonates the α-amino group of the substrate during substrate binding (Moustafa et al. 2006). In fact, the formation of the imino acid product in DAAO was demonstrated to require the α-amino group of the substrate in the neutral form (Umhau et al. 2000). In addition, in CrLAAO the amino acid substrate is bound in a distorted tetrahedral geometry around the substrate αC atom. In this distorted Michaelis complex, the αC atom adopts some sp2 features, achieving a geometry that mimics the imino acid product and thus facilitates the hydride transfer to flavin N(5) (Moustafa et al. 2006).
Ps501-PAO is more specific toward large hydrophobic amino acids in comparison to eukaryotic LAAOs: the best substrates are l-Phe and l-Tyr; l-Met and l-Trp are oxidized at a rate that is ~40 and ~20 % of the rate observed with l-Phe, respectively (Ida et al. 2011). The increased specificity of Ps501-PAO toward aromatic amino acids is due to the replacement of two charged residues (H223 and R322) with two hydrophobic ones (L330 and L619) in the specificity pocket of the latter. Moreover, two additional large hydrophobic active site side chains (F316 and L319) point toward the substrate. Altogether, a total of eight large hydrophobic residues line the substrate specificity pocket, with two of these residues (F617 and W660) forming a hydrophobic sandwich with the benzene ring of the substrate stabilized by H-π interactions (Fig. 3b). Accordingly, the K m of Ps501-PAO for l-Phe is fourfold lower than the one reported for CrLAAO: 13 vs. 50 μM, respectively (Ponnudurai et al. 1994).
RoLAAO oxidizes almost all the 20 proteinogenic l-amino acids. Superimposition of the active sites of Ps501-PAO and RoLAAO reveals that the aromatic cage that surrounds the substrate side chain (formed by residue F617 and W660) in Ps501-PAO is present in the RoLAAO active site, too (formed by residues W426 and W467), but the bond lengths are longer. Apart from the hydrophobic cage, in RoLAAO, the l-Phe benzene ring of the substrate forms two additional weak interactions with F222 and Q228; consequently, a significant portion of the substrate side chain remains exposed to the bulk solvent (Fig. 3b). In RoLAAO almost no restriction regarding the size and polarity of the substrate side chain is present, with the exception of bulky or branched amino acids whose binding is somewhat restricted by the size of the active site cleft. Notwithstanding the few interactions observed in RoLAAO between the substrate and the protein, the affinity for its substrates is similar to those determined for other LAAOs: for instance, the K m value for l-Phe is 22 μM in RoLAAO and 13 μM in Ps501-PAO, respectively (Geueke and Hummel 2002). This demonstrates that most of the ligand-binding energy is generated by the interactions that involve the αCOOH and αNH2 moieties of the substrate.
The 3D structure of LGO was solved without any bound ligand. For this reason we modeled l-Glu in the active site using the orientation of l-Phe in Ps501-PAO as reference. Inspection of the modeled complex shows that the cavity of the active site is narrower than in CrLAAO and that the substrate specificity pocket is significantly more polar (Fig. 3b). Three charged residues (R305, H312, and E617) are within H bond distance from the substrate γCOOH. While the role of E617 is to fix the R305 side chain in an ideal position for substrate binding, the interactions of the other two residues (R305 itself and H312) with the substrate side chain are probably essential in tuning LGO substrate specificity, in agreement with site-directed mutagenesis studies (Utsumi et al. 2012). Indeed, the affinity of the enzyme for the substrate (K m = 5.0 mM) is quite low in comparison to other LAAOs (see Table 4) (Arima et al. 2009). The further LAAO active on acidic amino acids, LASPO, possesses an active site that cannot be superimposed on that of microbial LAAOs (Fig. 3c). This enzyme belongs to the succinate dehydrogenase/fumarate reductase family of flavoproteins and is not evolutionarily related to the classical LAAO members. Accordingly, the physiological role of this protein is different: LASPO is involved in the first step of the biosynthetic pathway of NAD+ in prokaryotes. The most striking feature of the LASPO active site is the presence of a large number of charged side chains involved in a tight electrostatic and H bond network. R386 is functionally equivalent to R90 in CrLAAO: it forms an electrostatic interaction with the substrate αCOOH; this interaction is supported by an additional H bond with H351. Differently from what happens in classical amino acid oxidases, where hydride transfer proceeds from the αC of the substrate, during catalysis in LASPO, the hydride is abstracted from the βC, which is close to the flavin N(5). The sole interaction of the substrate side chain is the H bond with H244. Site-directed mutagenesis experiments were performed at these positions (Tedeschi et al. 2001; Tedeschi et al. 2010). Residues H244, H351, and R386 played a main role in fumarate binding but were not essential for catalysis. On the other end, R290, which is also close to the δCOOH moiety of the substrate, does not seem to play a specific role in l-Asp binding but was mainly involved in catalysis: R290 variants retain the ability to bind the substrate but are not able to oxidize it. It has been proposed that R290 in LASPO acts as a base to extract a proton from substrate αC during catalysis (Tedeschi et al. 2001): proton abstraction from αC is favored by E121, which orients the substrate in a productive binding mode. In fact, EcLASPO variants at position 121 retained fumarate reductase activity and the ability to bind l-Asp but lacked l-aspartate oxidase activity. E121 is a key active site residue distinguishing EcLASPO from all other members of the succinate dehydrogenase/fumarate reductase family (Tedeschi et al. 2010).
Current biotechnological uses
Bioconversion
Enantiomerically pure l-amino acids are widely used in human and animal nutrition (two million tons in 2001). l-Lys, d,l-Met, l-Thr, and l-Trp are added to animal feed because they usually do not contain sufficient amounts of these essential amino acids while, for human nutrition, l-Glu, l-Phe, l-Asp, and Gly are used as food additives. Indeed, several enantiomerically pure l-amino acids are used as intermediates for chemical synthesis of important molecules, e.g., l-Phe and l-Asp are used to produce the sweetener aspartame, l-homoPhe for angiotensin-converting enzymes inhibitors, β-lactam antibiotics, acetylcholinesterase inhibitors, etc. Similarly, d-amino acids are also used as intermediates in the production of pharmaceutical (i.e., d-Phe for the production of ampicillin and amoxicillin) and nutritional (d-Ala for the sweetener alitame) compounds.
Owing to its high specific activity and broad substrate specificity, Rhodococcus sp. AIU Z-35-1 LAAO was employed for the oxidation of a number of l-amino acids: 1 mL solutions of 15 or 50 mM l-Glu, l-Phe, l-Lys, l-Leu, l-His, and l-Arg could be fully converted in 24 h at 30 °C and pH 7.0 using 15 or 45 mU of enzyme, respectively. Indeed, the same LAAO was employed for the resolution of d,l mixtures (100 mM) of citrulline, Gln, homoserine, Lys, Arg, etc. to yield optically pure d-amino acids (Isobe et al. 2010). Resolution of d,l-Ala solutions was also carried out using A. fumigatus LAAO (Singh et al. 2009). Similarly, the same enzyme was employed for the resolution of the racemic mixtures of d,l-Ala, d,l-Phe, d,l-Tyr and d,l-Asp: optically pure products were obtained for d-Ala only (Singh et al. 2011). For resolution of the racemic solutions of aspartate, StLASPO was used because it showed a high thermal and pH stability; furthermore, it was weakly inhibited by the product oxaloacetate and d-Asp (K i = 9.6 mM) and thus represented a valuable tool for bioconversion. A 50 mM d,l-Asp solution was quantitatively converted with a >99.5 % e.e. using quite a low amount of enzyme (300 U/L) (Bifulco et al. 2013).
For the biotransformation of l-DOPA to its α-keto acid (in the presence of catalase to avoid decarboxylation of the product), bacterial RoLAAO outperformed the snake venom homologous C. rhodostoma LAAO (Findrik et al. 2006). RoLAAO showed better kinetic parameters than C. rhodostoma LAAO: lower K m,app (0.02 vs. 0.5 mM) and higher activity (0.33 vs. 0.1 U/mg protein). Complete oxidation of 3.8 mM l-DOPA by 0.3 U/mL in a batch transformation, and using an enzyme membrane reactor, was reached after ~1 h of reaction. R. opacus DSM 43250 was also used for racemic resolution of 7 mM d,l-Phe (Geueke and Hummel 2002). Similarly, one of the first reports concerning the biotransformation of Nε-carboxy(CBZ)-l-Lys into the corresponding keto acid used C. rhodostoma LAAO as well as LLYSO from T. viride and from Providencia alcalifaciens cells (Hanson et al. 1992): with the latter cell extracts, the highest conversion was obtained (95 % yield with 98.5 % optical purity).
Analytical assays and biosensors
LAAO activity has been applied as a biological tool for analytically determining l-amino acid levels by using colorimetric or amperometric biosensors. As an example, StLASPO, in combination with aspartate racemase and l-aspartate dehydrogenase, has been used to assay both d- and l-Asp by a simple colorimetric method (Mutaguchi et al. 2011). With this method, 1–100 μM of l- and d-Asp could be determined in food specimens (i.e., Chinese vinegar) and in biological samples (mouse testes and pig liver).
LAAO purified from goat kidney was recently covalently immobilized onto carboxylated multiwalled carbon nanotubes/nickel hexacyanoferrate/polypyrrole hybrid film electrodeposited on the surface of a carbon electrode (Lata and Pundir 2013). This biosensor, used at a potential of 0.15 V vs. Ag/AgCl, responded within 5 s and yielded a linear relationship between biosensor response and l-Phe concentration in the range 0.5 μM to 100 mM. The biosensor showed a sensitivity of 79.3 nA/cm2 μM and was reused 160 times for determining levels of l-amino acid in fruit juices and alcoholic beverages.
Concerning more specific LAAOs, commercial LGO from Streptomyces sp. X119-6 (6.3 U/mg dry powder) was entrapped in a biocompatible gel layer that retained the enzyme on the surface of a Pt microelectrode previously modified with poly(phenylenediamine) (Tian et al. 2009). The biosensor responded within 10 s and exhibited a linear dependence on l-Glu concentration from 0.5 to 100 μM and a good sensitivity (279 μA/cm2 mM). The biosensor was tested in vivo by recording changes in l-Glu concentration in a specific brain region (the dorsal medullary nucleus of the solitary tract). More recently, we compared the immobilization with poly(ethylene glycol) diglycidyl ether to produce biosensors using three different oxidases, i.e., glucose oxidase, DAAO, and LGO from Streptomyces sp. X119-6. The glutamate biosensor that had a higher K m,app than the free enzyme (but within the range of values obtained with other methods) was used repeatedly in vivo experiments lasting 4–5 h without losing activity (a stable and selective poly-o-phenylenediamine layer was essential to obtain accurate electrochemical recordings throughout the in vivo experiments) and was stored in dry conditions at 4 °C for at least 1 week without losing enzymatic activity (Vasylieva et al. 2011). Later on, Burmeister et al. (2013) concluded that poly(ethylene glycol) diglycidyl ether did not show significant advantages over glutaraldehyde for the immobilization of LGO onto ceramic-based microelectrode arrays. Analogously, LLYSO was also adsorbed on a Pt electrode to assay l-Lys: the lowest detectable concentration was 0.2 μM (Kelly et al. 1998; Saurina et al. 1999).
Diagnostics and therapeutics
Streptomyces sp. Z-11-6 LGO has been employed to develop a photometric assay to determine alanine and aspartate aminotransferase activity in biological samples (Sukhacheva and Zhuravleva 2004). The same application was developed using an amperometric sensing system based on a thin-film, three-electrode system which used immobilized Streptomyces X-119-6 LGO (Upadhyay et al. 2006): the main drawback was that the enzyme also responded to l-Asp and l-Ala. The same method could also be potentially employed to detect l-Glu in food specimens and for monitoring of l-Glu as a neurotransmitter.
LLYSO was shown to suppress DNA, RNA, and protein synthesis in various tumor cell lines in vitro and to affect cell cycle progression: five of the 12 tested tumors were sensitive to the activity of this flavoenzyme; for a detailed review see Lukasheva and Berezov (2002). LAAO was also demonstrated to increase the antitumor activity of melphalan: pretreatment with LAAO (100–200 μg/dose) depleted large neutral amino acids in murine plasma, thus reducing the competition for melphalan transport at the blood–brain barrier, and the subsequent treatment with an anti-LAAO antiserum eliminated the flavoenzyme activity, preventing the degradation of the antineoplastic agent (Moynihan et al. 1997).
Indeed, LLYSO also showed antibacterial and antiviral activity (it inhibited reproduction of type I Herpes simplex virus). The antibacterial and antiprotozoal properties of LAAO activities also represent a promising field for biotechnological application of this flavoenzyme.
Conclusions
While LAAO is one of the most promising flavooxidases for biocatalysis, as well as other novel applications, its practical use has remained limited to date due to several bottlenecks encountered in optimizing it for different applications by directed evolution. The first is due to the poor recombinant expression of active LAAO by an efficient overexpression system; the second is represented by the lack of a suitable in vivo screening assay not affecting cell viability. Setting up a specific laboratory evolution platform to evolve LAAO, as well as similar amino acid oxidase activities, will enable us to engineer substrate specificity and thus to improve biotechnological application of this flavoenzyme.
References
Alves RM, Antonucci GA, Paiva HH, Cintra AC, Franco JJ, Mendonça-Franqueiro EP, Dorta DJ, Giglio JR, Rosa JC, Fuly AL, Dias-Baruffi M, Soares AM, Sampaio SV (2008) Evidence of caspase-mediated apoptosis induced by l-amino acid oxidase isolated from Bothrops atrox snake venom. Comp Biochem Physiol A Mol Integr Physiol 151(4):542–550
Ande SR, Kommoju PR, Draxl S, Murkovic M, Macheroux P, Ghisla S, Ferrando-May E (2006) Mechanisms of cell death induction by L-amino acid oxidase, a major component of ophidian venom. Apoptosis 11(8):1439–1451
Arima J, Tamura T, Kusakabe H, Ashiuchi M, Yagi T, Tanaka H, Inagaki K (2003) Recombinant expression, biochemical characterization and stabilization through proteolysis of an l-glutamate oxidase from Streptomyces sp. X-119-6. J Biochem 134(6):805–812
Arima J, Sasaki C, Sakaguchi C, Mizuno H, Tamura T, Kashima A, Kusakabe H, Sugio S, Inagaki K (2009) Structural characterization of l-glutamate oxidase from Streptomyces sp. X-119-6. FEBS J 276(14):3894–3903
Bifulco D, Pollegioni L, Tessaro D, Servi S, Molla G (2013) A thermostable L-aspartate oxidase: a new tool for biotechnological applications. Appl Microbiol Biotechnol 97(16):7285–7295
Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A (1999) A 30-angstrom-long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure. 7(3):265–276
Böhmer A, Müller A, Passarge M, Liebs P, Honeck H, Müller HG (1989) A novel l-glutamate oxidase from Streptomyces endus. Purification and properties. Eur J Biochem 182(2):327–332
Bossi RT, Negri A, Tedeschi G, Mattevi A (2002) Structure of FAD-bound l-aspartate oxidase: insight into substrate specificity and catalysis. Biochemistry 41(9):3018–3024
Brearley GM, Price CP, Atkinson T, Hammond PM (1994) Purification and partial characterization of a broad range l-amino acid oxidase from Bacillus carotarum 2Pfa isolated from soil. Appl Microbiol Biotechnol 41:670–676
Burmeister JJ, Davis VA, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA (2013) Glutaraldehyde cross-linked glutamate oxidase coated microelectrode arrays: selectivity and resting levels of glutamate in the CNS. ACS Chem Neurosci 4(5):721–728
Chen CY, Wu WT, Huang CJ, Lin MH, Chang CK, Huang HJ, Liao JM, Chen LY, Liu YT (2001) A common precursor for the three subunits of L-glutamate oxidase encoded by gox gene from Streptomyces platensis NTU3304. Can J Microbiol 47(3):269–275
Chen WM, Lin CY, Chen CA, Wang JT, Sheu SY (2010) Involvement of an l-amino acid oxidase in the activity of the marine bacterium Pseudoalteromonas flavipulchra against methicillin-resistant Staphylococcus aureus. Enzym Microb Technol 47:52–58
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:465–469
Du XY, Clemetson KJ (2002) Snake venom l-amino acid oxidases. Toxicon 40(6):659–665
Dym O, Eisenberg D (2001) Sequence-structure analysis of FAD-containing proteins. Protein Sci 10(9):1712–1728
Fantinato S, Pollegioni L, Pilone MS (2001) Engineering, expression and purification of a His-tagged chimeric D-amino acid oxidase from Rhodotorula gracilis. Enzyme Microb Technol 29:407–412
Faust A, Niefind K, Hummel W, Schomburg D (2007) The structure of a bacterial L-amino acid oxidase from Rhodococcus opacus gives new evidence for the hydride mechanism for dehydrogenation. J Mol Biol 367(1):234–248
Findrik Z, Geueke B, Hummel W, Vasić-Rački D (2006) Modelling of l-DOPA enzymatic oxidation catalyzed by l-amino acid oxidases from Crotalus adamanteus and Rhodococcus opacus. Biochem Eng J 27:275–286
Geueke B, Hummel W (2002) A new bacterial l-amino acid oxidase with a broad substrate specificity: purification and characterization. Enzym Microb Technol 31:77–87
Geueke B, Hummel W (2003) Heterologous expression of Rhodococcus opacus l-amino acid oxidase in Streptomyces lividans. Protein Expr Purif 28(2):303–309
Gómez D, Espinosa E, Bertazzo M, Lucas-Elío P, Solano F, Sanchez-Amat A (2008) The macromolecule with antimicrobial activity synthesized by Pseudoalteromonas luteoviolacea strains is an l-amino acid oxidase. Appl Microbiol Biotechnol 79(6):925–930
Guo C, Liu S, Yao Y, Zhang Q, Sun MZ (2012) Past decade study of snake venom l-amino acid oxidase. Toxicon 60(3):302–311
Hanson RL, Bembenek KS, Patel RN, Szarka LJ (1992) Transformation of Nε-CBZ-l-lysine to CBZ-l-oxylysine using l-amino acid oxidase from Providencia alcalifaciens and l-2-hydroxy-isocaproate dehydrogenase from Lactobacillus confusus. Appl Microbiol Biotechnol 37(5):599–603
Ida K, Kurabayashi M, Suguro M, Hiruma Y, Hikima T, Yamomoto M, Suzuki H (2008) Structural basis of proteolytic activation of l-phenylalanine oxidase from Pseudomonas sp. P-501. J Biol Chem 283(24):16584–16590
Ida K, Suguro M, Suzuki H (2011) High resolution X-ray crystal structures of L-phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas sp. P-501. Structures of the enzyme-ligand complex and catalytic mechanism. J Biochem 150(6):659–669
Isobe K, Nagasawa S (2007) Characterization of N α-benzyloxycarbonyl-l-lysine oxidizing enzyme from Rhodococcus sp. AIU Z-35-1. J Biosci Bioeng 104(3):218–223
Isobe K, Fukuda N, Nagasawa S, Saitou K (2010) Enzymes responsible for the conversion of N α-[(benzyloxy)carbonyl]-d-lysine to N α-[(benzyloxy)carbonyl]-d-aminoadipic acid by Rhodococcus sp. AIU Z-35-1. Chem Biodivers 7(6):1549–1554
Isobe K, Satou S, Matsumoto E, Yoshida S, Yamada M, Hibi M, Ogawa J (2012a) Characterization and application of a l-specific amino acid oxidase from Rhodococcus sp. AIU LAB-3. J Biosci Bioeng 115(6):613–617
Isobe K, Sugawara A, Domon H, Fukuta Y, Asano Y (2012b) Purification and characterization of an l-amino acid oxidase from Pseudomonas sp. AIU 813. J Biosci Bioeng 114(3):257–261
Kamei T, Asano K, Suzuki H, Matsuzaki M, Nakamura S (1983) L-glutamate oxidase from Streptomyces violascens. I. Production, isolation and some properties. Chem Pharm Bull 31(4):1307–1314
Kelly S, Curulli A, O’Sullivan C, Guilbault GG, Palleschi G (1998) A new interference-free lysine biosensor using a non-conducting polymer film. Biosens Bioelectron 13(12):1245–1250
Koyama H (1984) Oxidation and oxygenation of l-amino acids catalyzed by a l-phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas sp. P-501. J Biochem 96(2):421–427
Kusakabe H, Kodama K, Machida H, Midorikawa Y, Kuninaka A, Misono H, Soda K (1979) Occurrence of a novel enzyme, l-lysine oxidase with anti-tumor activity in culture of Trichoderma viridae. Agric Biol Chem 43:337–343
Kusakabe H, Kodama K, Kuninaka A, Yoshino H, Misono H, Soda K (1980) A new antitumor enzyme, l-lysine alpha-oxidase from Trichoderma viride. Purification and enzymological properties. J Biol Chem 255(3):976–981
Lata S, Pundir CS (2013) L-amino acid biosensor based on L-amino acid oxidase immobilized onto NiHCNFe/c-MWCNT/PPy/GC electrode. Int J Biol Macromol 54:250–257
Leese C, Fotheringham I, Escalettes F, Speight R, Grogan G (2013) Cloning, expression, characterisation and mutational analysis of l-aspartate oxidase from Pseudomonas putida. J Mol Catal B: Enzymatic 85–86:7–22
Lukasheva EV, Berezov TT (2002) l-Lysine alpha-oxidase: physicochemical and biological properties. Biochemistry (Mosc) 67(10):1152–1158
Lukasheva EV, Efremova AA, Treshalina EM, Arinbarasova AY, Medentzev AG, Berezov TT (2011) L-amino acid oxidases: properties and molecular mechanisms of action. Biochemistry (Mosc) Suppl Ser B: Biomed Chem 5(4):337–345
Marinoni I, Nonnis S, Monteferrante C, Heathcote P, Härtig E, Böttger LH, Trautwein AX, Negri A, Albertini AM, Tedeschi G (2008) Characterization of l-aspartate oxidase and quinolinate synthase from Bacillus subtilis. FEBS J 275(20):5090–5107
Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B (1996) Crystal structure of d-amino acid oxidase: a case of active site mirror-image convergent evolution with flavocytochrome b2. Proc Natl Acad Sci U S A 93(15):7496–7501
Mattevi A, Tedeschi G, Bacchella L, Coda A, Negri A, Ronchi S (1999) Structure of l-aspartate oxidase: implications for the succinate dehydrogenase/fumarate reductase oxidoreductase family. Structure 7(7):745–756
Mesecar AD, Koshland DE Jr (2000) A new model for protein stereospecificity. Nature 403(6770):614–615
Mortarino M, Negri A, Tedeschi G, Simonic T, Duga S, Gassen HG, Ronchi S (1996) l-Aspartate oxidase from Escherichia coli. I. Characterization of coenzyme binding and product inhibition. Eur J Biochem 239(2):418–426
Moustafa IM, Foster S, Lyubimov AY, Vrielink A (2006) Crystal structure of LAAO from Calloselasma rhodostoma with an l-phenylalanine substrate: insights into structure and mechanism. J Mol Biol 364(5):991–1002
Moynihan K, Elion GB, Pegram C, Reist CJ, Wellner D, Bigner DD, Griffith OW, Friedman HS (1997) L-Amino acid oxidase (LOX) modulation of melphalan activity against intracranial glioma. Cancer Chemother Pharmacol 39(3):179–186
Mutaguchi Y, Ohmori T, Sakuraba H, Yoneda K, Doi K, Ohshima T (2011) Visible wavelength spectrophotometric assays of l-aspartate and d-aspartate using hyperthermophilic enzyme systems. Anal Biochem 409(1):1–6
Nishizawa T, Aldrich CC, Sherman DH (2005) Molecular analysis of the rebeccamycin L-amino acid oxidase from Lechevalieria aerocolonigenes ATCC 39243. J Bacteriol 187(6):2084–2092
Nuutinen JT, Marttinen E, Soliymani R, Hildén K, Timonen S (2012) L-Amino acid oxidase of the fungus Hebeloma cylindrosporum displays substrate preference towards glutamate. Microbiology 158:272–283
Pantaleone DP, Geller AM, Taylor PP (2001) Purification and characterization of an l-amino acid deaminase used to prepare unnatural amino acids. J Mol Catal B 11:795–803
Pawelek PD, Cheah J, Coulombe R, Macheroux P, Ghisla S, Vrielink A (2000) The structure of L-amino acid oxidase reveals the substrate trajectory into an enantiomerically conserved active site. EMBO J 19(16):4204–4215
Pollegioni L, Molla G (2011) New biotech applications from evolved D-amino acid oxidases. Trends Biotechnol 29(6):276–283
Pollegioni L, Diederichs K, Molla G, Umhau S, Welte W, Ghisla S, Pilone MS (2002) Yeast D-amino acid oxidase: structural basis of its catalytic properties. J Mol Biol 324(3):535–546
Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G (2007) Physiological functions of d-amino acid oxidases: from yeast to humans. Cell Mol Life Sci 64(11):1373–1394
Pollegioni L, Molla G, Sacchi S, Rosini E, Verga R, Pilone MS (2008) Properties and applications of microbial d-amino acid oxidases: current state and perspectives. Appl Microbiol Biotechnol 78(1):1–16
Ponnudurai G, Chung MC, Tan NH (1994) Purification and properties of the L-amino acid oxidase from Malayan pit viper (Calloselasma rhodostoma) venom. Arch Biochem Biophys 313(2):373–378
Saam J, Rosini E, Molla G, Schulten K, Pollegioni L, Ghisla S (2010) O2 reactivity of flavoproteins: dynamic access of dioxygen to the active site and role of a H+ relay system in d-amino acid oxidase. J Biol Chem 285(32):24439–24446
Sacchi S, Rosini E, Molla G, Pilone MS, Pollegioni L (2004) Modulating d-amino acid oxidase substrate specificity: production of an enzyme for analytical determination of all d-amino acids by directed evolution. Protein Eng Des Sel 17(6):517–525
Sakuraba H, Satomura T, Kawakami R, Yamamoto S, Kawarabayasi Y, Kikuchi H, Ohshima T (2002) l-Aspartate oxidase is present in the anaerobic hyperthermophilic archaeon Pyrococcus horikoshii OT-3: characteristics and role in the de novo biosynthesis of nicotinamide adenine dinucleotide proposed by genome sequencing. Extremophiles 6(4):275–281
Sakuraba H, Yoneda K, Asai I, Tsuge H, Katunuma N, Ohshima T (2008) Structure of l-aspartate oxidase from the hyperthermophilic archaeon Sulfolobus tokodaii. Biochim Biophys Acta 1784(3):563–571
Saurina J, Hernández-Cassou S, Alegret S, Fàbregas E (1999) Amperometric determination of lysine using a lysine oxidase biosensor based on rigid-conducting composites. Biosens Bioelectron 14(2):211–220
Seifert J, Kunz N, Flachmann R, Läufer A, Jany KD, Gassen HG (1990) Expression of the E. coli nadB gene and characterization of the gene product L-aspartate oxidase. Biol Chem Hoppe Seyler 371(3):239–248
Singh S, Gogoi BK, Bezbaruah RL (2009) Optimization of medium and cultivation conditions for L-amino acid oxidase production by Aspergillus fumigatus. Can J Microbiol 55(9):1096–1102
Singh S, Gogoi BK, Bezbaruah RL (2011) Racemic resolution of some dl-amino acids using Aspergillus fumigatus l-amino acid oxidase. Curr Microbiol 63(1):94–99. doi:10.1007/s00284-011-9955-8
Sukhacheva MV, Zhuravleva NI (2004) Properties and prospects of practical use of extracellular l-glutamate oxidase from Streptomyces sp. Z-11-6. Appl Biochem Microbiol 40(2):146–150
Suzuki H, Higashi Y, Asano M, Suguro M, Kigawa M, Maeda M, Katayama S, Mukouyama EB, Uchiyama K (2004) Sequencing and expression of the L-phenylalanine oxidase gene from Pseudomonas sp. P-501. Proteolytic activation of the proenzyme. J Biochem 136(5):617–627
Tedeschi G, Negri A, Mortarino M, Ceciliani F, Simonic T, Faotto L, Ronchi S (1996) L-aspartate oxidase from Escherichia coli. II. Interaction with C4 dicarboxylic acids and identification of a novel L-aspartate:fumarate oxidoreductase activity. Eur J Biochem 239(2):427–433
Tedeschi G, Ronchi S, Simonic T, Treu C, Mattevi A, Negri A (2001) Probing the active site of l-aspartate oxidase by site-directed mutagenesis: role of basic residues in fumarate reduction. Biochemistry 40(15):4738–4744
Tedeschi G, Nonnis S, Strumbo B, Cruciani G, Carosati E, Negri A (2010) On the catalytic role of the active site residue E121 of E. coli l-aspartate oxidase. Biochimie 92(10):1335–1342. doi:10.1016/j.biochi.2010.06.015
Tian F, Gourine AV, Huckstepp RT, Dale N (2009) A microelectrode biosensor for real time monitoring of l-glutamate release. Anal Chim Acta 645(1–2):86–91
Ullah A, Souza TA, Abrego JR, Betzel C, Murakami MT, Arni RK (2012) Structural insights into selectivity and cofactor binding in snake venom l-amino acid oxidases. Biochem Biophys Res Commun 421(1):124–128
Umhau S, Pollegioni L, Molla G, Diederichs K, Welte W, Pilone MS, Ghisla S (2000) The x-ray structure of d-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. Proc Natl Acad Sci U S A 97(23):12463–12468
Upadhyay S, Ohgami N, Kusakabe H, Mizuno H, Arima J, Tamura T, Inagaki K, Suzuki H (2006) Performance characterization of recombinant l-glutamate oxidase in a micro GOT/GPT sensing system. Sens Actuators B 119:570–576
Utsumi T, Arima J, Sakaguchi C, Tamura T, Sasaki C, Kusakabe H, Sugio S, Inagaki K (2012) Arg305 of Streptomyces l-glutamate oxidase plays a crucial role for substrate recognition. Biochem Biophys Res Commun 417(3):951–955
Vasylieva N, Barnych B, Meiller A, Maucler C, Pollegioni L, Lin JS, Barbier D, Marinesco S (2011) Covalent enzyme immobilization by poly(ethylene glycol) diglycidyl ether (PEGDE) for microelectrode biosensor preparation. Biosens Bioelectron 26(10):3993–4000
Yang CA, Cheng CH, Liu SY, Lo CT, Lee JW, Peng KC (2011a) Identification of antibacterial mechanism of l-amino acid oxidase derived from Trichoderma harzianum ETS 323. FEBS J 278(18):3381–3394
Yang CA, Cheng CH, Lo CT, Liu SY, Lee JW, Peng KC (2011b) A novel l-amino acid oxidase from Trichoderma harzianum ETS 323 associated with antagonism of Rhizoctonia solani. J Agric Food Chem 59(9):4519–4526
Yu Z, Qiao H (2012) Advances in non-snake venom L-amino acid oxidase. Appl Biochem Biotechnol 167(1):1–13
Zeller EA, Maritz A (1944) Uber eine neue l-aminosaüre-oxydase. Helv Chim Acta 27:1888–1902
Zhang H, Teng M, Niu L, Wang Y, Wang Y, Liu Q, Huang Q, Hao Q, Dong Y, Liu P (2004) Purification, partial characterization, crystallization and structural determination of AHP-LAAO, a novel l-amino-acid oxidase with cell apoptosis-inducing activity from Agkistrodon halys pallas venom. Acta Crystallogr D Biol Crystallogr 60(5):974–977
Acknowledgments
We thank the support of Fondo di Ateneo per la Ricerca, Centro Grandi Attrezzature (Università dell’Insubria), and Consorzio Interuniversitario per le Biotecnologie CIB.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pollegioni, L., Motta, P. & Molla, G. l-Amino acid oxidase as biocatalyst: a dream too far?. Appl Microbiol Biotechnol 97, 9323–9341 (2013). https://doi.org/10.1007/s00253-013-5230-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5230-1