
Abstract
D-Amino acid oxidase (DAAO) is a biotechnologically relevant enzyme that is used in a variety of applications. DAAO is a flavine adenine dinucleotide-containing flavoenzyme that catalyzes the oxidative deamination of D-isomer of uncharged aliphatic, aromatic, and polar amino acids yielding the corresponding imino acid (which hydrolyzes spontaneously to the α-keto acid and ammonia) and hydrogen peroxide. This enzymatic activity is produced by few bacteria and by most eukaryotic organisms. In the past few years, DAAO from mammals has been the subject of a large number of investigations, becoming a model for the dehydrogenase-oxidase class of flavoproteins. However, DAAO from microorganisms show properties that render them more suitable for the biotechnological applications, such as a high level of protein expression (as native and recombinant protein), a high turnover number, and a tight binding of the coenzyme. Some important DAAO-producing microorganisms include Trigonopsis variabilis, Rhodotorula gracilis, and Fusarium solani. The aim of this paper is to provide an overview of the main biotechnological applications of DAAO (ranging from biocatalysis to convert cephalosporin C into 7-amino cephalosporanic acid to gene therapy for tumor treatment) and to illustrate the advantages of using the microbial DAAOs, employing both the native and the improved DAAO variants obtained by enzyme engineering.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
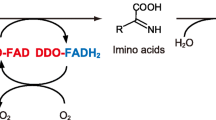
D-Amino acid oxidase (DAAO, EC 1.4.3.3) is a flavine adenine dinucleotide (FAD)-containing flavoenzyme that catalyzes the oxidative deamination of D-amino acids (D-AA) with a strict stereospecificity to give α-keto acids and ammonia; FADH2 then reoxidizes on molecular oxygen, producing hydrogen peroxide (Fig. 1). This flavooxidase was discovered more than 70 years ago by Krebs et al. (1935). Since 1935, DAAO has been the object of a vast mass of investigations, becoming a model for this class of flavoproteins. DAAO belongs to a subgroup of the dehydrogenase-oxidase class of flavoproteins (Massey and Hemmerich 1980): All members of this family react rapidly with oxygen in reduced form, stabilize the red anionic flavin radical via one-electron transfer, produce a flavin N(5)-sulfite adduct, and stabilize the benzoquinoid anionic form of 6- and 8-substituted mercapto- and hydroxyflavins. DAAO activity has been identified in many organisms, ranging from fungi to humans, where it accomplishes distinct physiological functions: from a catabolic role in microorganisms (i.e., DAAO allows yeast cells to grow on D-amino acids) to a regulatory role in the human brain (DAAO is involved in controlling the levels of the neuromodulator D-serine). The multiple physiological roles played by DAAO in the various organisms have been recently reviewed (Pollegioni et al. 2007a).

Catalytic cycle of the reaction catalyzed by D-amino acid oxidase (Porter et al. 1977). The redox state of the FAD cofactor is reported as subscript: ox oxidized; red reduced. E-FADred∼IA is the non-covalent complex between the reduced enzyme form and the imino acid product. IA Imino acid, αKA α-keto acid
The redox reaction catalyzed by DAAO can be employed in a variety of biotechnological processes, such as the production of glutaryl-7-amino cephalosporanic acid (Gl-7-ACA) from cephalosporin C (CephC, by far the most relevant industrial application of DAAO) and of α-keto acids from natural and synthetic D-AAs, the resolution of racemic mixtures of amino acids, the analytical determination of D-AAs, and in the treatment of cancer. Yeast DAAOs are optimal biocatalysts for these enzyme-based technological applications, as they possess a high activity, a tight binding of the coenzyme FAD, and a broad substrate specificity (Pilone and Pollegioni 2002).
Structural and functional studies on DAAO from yeasts and from mammals have suggested that specific physiological functions are implemented through the use of different structural elements controlling access to the active site and substrate/product exchange. Detailed knowledge of the structure–function relationships in DAAO has made it possible to use this enzyme as a protein scaffold to develop enzyme variants with new or improved properties. In these studies, performed both by a rational and a directed evolution approach, DAAO mutants could be produced with altered substrate specificity (e.g., active on both acidic and on unnatural D-AAs) and cofactor binding properties, different oligomeric states, and improved stability: the protein engineering investigations performed on DAAO have been also recently reviewed, see Pollegioni et al. (2007b).
Oxidation of D-amino acids in microorganisms
Bacteria are able to synthesize and degrade D-AAs; D-Ala and D-Glu are fundamental components of the peptidoglycan layer of the bacterial cell wall. Neutral D-AAs, in particular D-Ala, are oxidized to the corresponding α-keto acid by a FAD-containing D-amino acid dehydrogenase (DAAdH) constituted by a 45- and a 55-kDa subunit, in which the D-AA oxidation is not coupled to O2 reduction. The FAD cofactor located on the small subunit receives two electrons from the substrate and transfers them to a sulfur–iron center in the large subunit. In Escherichia coli, DAAdH is a peripheral membrane protein associated with the bacterial inner cell membrane (Olsiewski et al. 1980). Concerning the physiological function of this enzymatic activity in bacteria, two main roles have been identified: (a) DAAdH allows bacteria to grow using D-AAs as the sole carbon, nitrogen, and energy source (DAAdH expression is induced by the presence of D- and L-Ala; Raunio and Jenkins 1973; Deutch 2004), and (b) DAAdH protects cell growth preventing the accumulation of D-AAs and specifically of D-AA analogues that show inhibitory effects on bacterial growth (Yasuda and Tochikubo 1985; David et al. 1989).
About 200 genes coding for putative DAAOs in bacteria have been identified by a bioinformatic analysis. A search in the protein databanks using the BLAST sequence analysis server at NCBI for putative DAAO coding genes identified 11 genes encoding for proteins with a significant sequence identity with yeast DAAOs (ranging from 25 to 31%): All of them belong to Gram-positive Actinobacteria strains, a group of bacteria that comprises some important pathogens, including Mycobacteria and Nocardia. However, sequence similarity (Fig. 2a) is not a parameter that can define their substrate specificity and physiological role(s). To date, only one bacterial DAAO (from A. protophormiae, ApDAAO) has been characterized from a biochemical point of view (Geueke et al. 2007): It is a FAD-dependent flavooxidase (the reoxidation of the reduced flavin by molecular oxygen yields hydrogen peroxide) showing a very high specific activity on D-methionine (180 U/mg protein). In B. subtilis, an oxidase activity, termed glycine oxidase and sharing large primary and tertiary structural similarity with fungal DAAO, has been also identified; however, it is not related to D-AAs catabolism but rather is a component of the thiamine biosynthesis pathway (Settembre et al. 2003; Mörtl et al. 2004). Glycine oxidase catalyses the oxidative deamination of various amines, preferentially those of a small size, and D-amino acids to form the corresponding α-keto acids and hydrogen peroxide. It partially shares substrate specificity with DAAO and sarcosine oxidase (Job et al. 2002).

a Amino acid alignment of microbial DAAOs from the bacteria Rubrobacter xylanophilus (Q1AYM8, putative), Mycobacterium leprae (CAC30966, putative), and Arthrobacter protophormiae (Q7X2D3), from the fungi Fusarium solani (P24552), Trigonopsis variabilis (Q99042), Rhodotorula gracilis (P80324), Candida boidinii (Q9HGY3) and Aspergillus niger (CAK42099, putative) and of glycine oxidase from Bacillus subtilis (O31616). Multiple alignment was performed using ClustalW (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page = npsa_clustalw.html) at the Pole Bioinformatique Lyonnais and optimized based on structural and functional information. Red Identity; green high similarity; blue weak similarity. Active site residues, as shown in c, are highlighted in boxes. The colored stripe shows the position of the amino acids in the 3D structure according to b. b Identification of the position of the conserved regions/residues in the structure of the monomer of DAAO from R. gracilis (1c0p). Bold Active site residue (see c); italics FAD binding. Blue FAD binding domain; green apical domain; purple conserved regions numbered as in a. c Comparison of the active site of DAAO from the yeast Rhodotorula gracilis (1c0p) and the active site of the DAAO from Trigonopsis variabilis (our model). The structural alignment between the amino acid sequences of TvDAAO and of the templates RgDAAO (1c0p), pig kidney DAAO (1kif) and human DAAO (2du8) was optimized using the DeepView - Swiss-PdbViewer application (http://www.expasy.org/spdbv/). The model of the 3D structure of TvDAAO was then built by homology modeling using the Swiss-Model server (http://swissmodel.expasy.org//SWISS-MODEL.html)
Concerning DAAO from eukaryotic microorganisms, direct evidence for the presence of DAAO activity has been reported for a number of fungi, ranging from the basidiomycota Rhodotorula gracilis (Pilone Simonetta et al. 1987) to the following ascomycota: Aspergillus niger (Kishore and Vaidyanathan 1976), Neurospora crassa (Sikora and Marzluf 1982), Candida utilis (Zwart et al. 1983), Trigonopsis variabilis (Kubicek-Pranz and Rohr 1985), Hansenula polymorpha (Sulter et al. 1990), Fusarium solani (Isogai et al. 1990), Candida boidinii (Yurimoto et al. 2000), Fusarium oxysporum (Gabler and Fischer 1999), Verticillium luteoalbum, and Candida parapsilosis (Gabler et al. 2000). The first DAAO complementary DNA (cDNA) from a microorganism, the one encoding for the enzyme from T. variabilis, was reported in 1988 in a patent filed by Asahi Chemical Ind. (Komatsu et al. 1988). Subsequently, the genes for DAAO from F. solani (Isogai et al. 1990) and C. boidinii (Yurimoto et al. 2000) and the cDNA for DAAO from R. gracilis (RgDAAO; Pollegioni et al. 1997) were cloned. These genes encode for proteins of 345–376 residues (markedly, bacterial DAAOs are ∼30 amino acids shorter, Fig. 2a), which have been the object of a number of investigations and applications (see below). By using the primary sequence of RgDAAO as a query, a number of sequences coding for putative DAAOs were identified in the fungi kingdom (the identity degree ranges from 30 to 40%), particularly in filamentous fungi (molds) and yeasts (Pollegioni et al. 2007a). This analysis also identified DAAO paralogue genes coding for proteins structurally and functionally related to DAAO (e.g., D-aspartate oxidase genes) though evolved to play different physiological roles. By aligning their primary structures, six conserved regions were identified (Faotto et al. 1995). Interestingly, the C-terminal tripeptide represents the PTS1 peroxisomal targeting sequence (Ser-(Lys/His/Arg)-Leu); this sequence is absent in bacterial amino acid oxidases, such as glycine oxidase and ApDAAO (Fig. 2a).
Properties of microbial D-amino acid oxidases
The identification of DAAO in microorganisms has been pursued using two different approaches: a traditional approach consisting in the screening for DAAO activity of microorganisms sampled from soil or obtained from collections and a more recent approach that relies on the identification of putative DAAO coding genes based on sequence analysis (see above). Regarding DAAOs from eukaryotic microorganisms, 25 species of yeasts (or filamentous fungi) produce DAAO, and another 14 yeasts possess putative genes coding for DAAOs. Among yeasts, the genera Aspergillus, Candida, and Fusarium are the most represented microorganisms (Fischer 1998). Although a paper reporting on some biochemical properties of a DAAO from A. niger was published in 1976 (Kishore and Vaidyanathan 1976), the first paper reporting on the purification and characterization of DAAO from yeast focused on the enzyme from the ascomycota T. variabilis (Kubicek-Pranz and Rohr 1985; Pollegioni et al. 1993a), followed shortly thereafter by studies of the enzyme from the basidiomycota R. gracilis (Pilone Simonetta et al. 1989a; Pollegioni and Pilone 1992; Pollegioni et al. 1992). Most recently, DAAO from C. boidinii was also cloned, expressed in a homologous system, and characterized (Yurimoto et al. 2001). For a detailed overview of the distribution of DAAO in microorganisms, see Fischer (1998) and Pollegioni et al. (2007a).
General properties
All DAAOs contain a molecule of noncovalently bound FAD per subunit and therefore show the typical spectrum of the flavoprotein oxidases, with two main peaks in the visible region (centered at about 360–380 and 455 nm) due to the FAD cofactor and one large peak in the UV region (centered at 272–274 nm), mainly due to the protein moiety (Casalin et al. 1991; Curti et al. 1992; Schräder and Andreesen 1996; Pollegioni et al. 2007b). During catalysis, two electrons are transferred from the substrate to the cofactor, causing the two absorption peaks in the visible region to disappear due to the formation of the reduced form of the cofactor. Moreover, under specific experimental conditions, a red semiquinonic form of the FAD cofactor can be produced; this form possesses a particular spectrum, with a single peak in the visible region (∼375 nm; Pilone Simonetta et al. 1987; Pilone Simonetta et al. 1989a; Curti et al. 1992). The FAD cofactor bound to DAAO apoprotein is also a fluorophore, with an emission maximum at ∼530 nm when excited at 450 nm (however, its emission is significantly lower than that of the free cofactor; Casalin et al. 1991).
Given the wide range of biotechnological applications of microbial DAAOs, the pH and temperature stability of these enzymes have been extensively investigated. R. gracilis DAAO and T. variabilis DAAO (TvDAAO) show similar pH stability profiles: The enzymes are stable from pH 6.0 to 8.2, above which a slight but continuous decrease in protein stability is observed (at pH 10, the residual activity after 30 min incubation is ∼50%; Pollegioni et al. 2004). C. boidinii DAAO (CbDAAO) shows a higher stability, especially at basic pH values, being fully stable up to pH 10.5 (Yurimoto et al. 2001). ApDAAO possesses a pH profile similar to that of the yeast DAAOs: The bacterial flavoenzyme is stable between pH 6.0 and 9.0 (Geueke et al. 2007). However, major differences in the thermal stability of these proteins are observed. TvDAAO is the most stable enzyme: It is fully stable up to 45°C (∼100% of residual activity after 30-min incubation), whereas thermal inactivation of RgDAAO is observed at temperatures >30°C (after 30-min incubation at 45°C, the activity is completely lost; Pollegioni et al. 2004). The temperature sensitivity of CbDAAO and ApDAAO are very similar to that reported for RgDAAO: after a 30-min incubation period, the residual activity begins to decrease at temperatures >30°C and is completely lost at 50°C (Yurimoto et al. 2001; Geueke et al. 2007). The protein concentration affects stability to a limited extent: At low protein concentrations (0.01–0.2 mg/ml), the time course of protein inactivation is not affected (Pollegioni et al. 2004), while at higher protein concentrations (1.5 to 3.5 mg/ml), a slight increase in the melting temperature is observed (from 55.9 to 56.9°C) (Pollegioni et al. 2003). The presence of exogenous FAD in the incubation buffer significantly protects RgDAAO against inactivation at temperatures >40°C, while no effects are observed on TvDAAO (Pollegioni et al. 2004). Yeast DAAOs can be inactivated by H2O2 (a product of the reaction catalyzed by the enzyme): Partial enzyme inactivation is observed at high H2O2 concentrations (100 mM) and after 2 h of incubation (Pollegioni et al. 2004). As a general rule, the apoprotein forms of DAAO are far less stable than the respective holoenzymes (Casalin et al. 1991; Dib et al. 2006).
Substrate specificity and kinetic mechanism
Microbial DAAOs possess a broad substrate specificity (i.e., they are able to oxidize amino acids possessing side chains of a different size and polarity) and a strict stereoselectivity: l-amino acids are not substrates or competitive inhibitors. ApDAAO, however, represents an exception as it is slightly inhibited by l-Met (Ki = 315 mM; Geueke et al. 2007). Unfortunately, direct comparison of the kinetic parameters among microbial DAAOs is not feasible, as data have been collected using different techniques and under different experimental conditions. We recently re-calculated the published data converting the values cited in the literature for the effects due to the O2 concentration, the temperature (Porter et al. 1977; Curti et al. 1992; Pollegioni et al. 1992; 1993a, b) and the substrate specificity (Pollegioni et al. 2007b). In this work, one DAAO unit is defined as the amount of enzyme that converts 1 μmol of CephC per min at 25°C, as determined by an oxygen electrode and at a pO2 = 0.253 mM (air saturation; Pollegioni et al. 1992).
In general, microbial DAAOs show higher activity (apparent V max at 21% oxygen saturation) on D-AAs with hydrophobic side chains. DAAOs can be divided into two groups regarding their substrate specificity (Tishkov and Khoronenkova 2005): The first group is constituted by enzymes with a preference for amino acids with small apolar side chains (D-Ala is the best substrate for these enzymes), such as DAAOs from F. oxysporum, C. parapsilosis, and C. boidinii. The enzymes from A. niger, V. luteoalbum, R. gracilis, and T. variabilis constitute the second group: The best substrates are D-AAs possessing large hydrophobic side chains such as D-Trp, D-Met, D-Val, and D-Phe (Kishore and Vaidyanathan 1976; Schräder and Andreesen 1996; Tishkov and Khoronenkova 2005; Pollegioni et al. 2007a). Usually, the small amino acid Gly and the charged (acidic or basic) amino acids are poor DAAO substrates, with few exceptions: D-Arg is a good TvDAAO substrate (the specific activity for this substrate is 40% of the specific activity for D-Met) and D-Glu is a good substrate for C. parapsilosis DAAO (the specific activity determined for D-Glu is 54% of the value for D-Ala; Tishkov and Khoronenkova 2005). Interestingly, the substrate specificity of ApDAAO is unique: Its best substrate is D-Met and it also efficiently oxidizes basic amino acids, such as D-Lys and D-Arg (Geueke et al. 2007). Microbial DAAOs are also able to oxidize modified D-AAs, a group of compounds of main interest from a biotechnological point of view; for example, RgDAAO is able to efficiently oxidize CephC (Pollegioni et al. 2004) and D-naphthylalanine (Caligiuri et al. 2006a, b).
From a chemical point of view, experimental evidence from a number of studies indicates that the reaction catalyzed by DAAO consists in the transfer of a hydride ion from the substrate (i.e., the D-AA) to the isoalloxazine moiety of the FAD cofactor (the “reductive half-reaction,” Fig. 1, top). Subsequently, the reoxidation of the cofactor is due to a molecule of O2 that is reduced to H2O2 (the “oxidative half-reaction,” Fig. 1, bottom). Among the DAAOs from microorganisms, only the kinetic mechanisms of RgDAAO and TvDAAO have been clarified (Pollegioni et al. 1993b): The steady state kinetic data, collected using D-Ala and D-Val as substrates, are apparently consistent with a ping-pong (binary complex) mechanism (i.e., they result in a set of parallel lines in the double reciprocal plots). However, a careful investigation of the two half-reactions reveals that both enzymes follow an ordered sequential (ternary complex) mechanism in which the rate of substrate dehydrogenation and reduced flavin re-oxidation are almost irreversible and the rate of flavin reduction during the reductive half-reaction limits turnover. The turnover numbers (with D-Ala as substrate) determined for the microbial DAAOs are high (345 and 52.5 s−1 for RgDAAO and TvDAAO, respectively) as compared to the pig enzyme (12.7 s−1; Porter et al., 1977; Pollegioni et al. 1993b; 1994). This feature makes yeast DAAOs more suitable for biotechnological applications than the mammalian counterparts.
Structure–function relationships
The only 3D structure of a microbial DAAO available is that of RgDAAO, which was solved in 2000 (Umhau et al. 2000), 4 years after the first DAAO structure from pig kidney was deposited in the Protein Data Bank (Mattevi et al. 1996; Mizutani et al. 1996). The overall conformation of the protein monomer from yeast is similar to pig DAAO (and to human DAAO, whose 3D structure was recently solved; Kawazoe et al. 2006), with 281 residues within a 3.5-Å cutoff and a rms deviation of 1.38 Å (Pollegioni et al. 2002). Concerning the active site, all conserved residues are involved in fixating the substrate in the proper orientation for the reaction but not all are directly involved in the chemical step of catalysis (Fig. 2c, top). In RgDAAO, the α-carboxyl moiety of the substrate forms a salt bridge with Arg285 and two hydrogen bonds with Tyr223 and Tyr238. The α-amino group of the substrate forms two hydrogen bonds: the first with Ser335 and the second with an active site water molecule that, in turn, is H-bonded to Asn54 and Gln339 (Umhau et al. 2000). The substrate side chain is accommodated in a relatively wide hydrophobic cavity; the available volume of this cavity is limited by the presence of a bulky methionine (Met213) that plays a key role in determining the substrate specificity of RgDAAO (Sacchi et al. 2002; Caligiuri et al. 2006a). A model of the active site of TvDAAO is shown in Fig. 2c, bottom: Interestingly, Ser228 is present at the position corresponding to Met213 of RgDAAO in TvDAAO.
Notwithstanding the overall structural similarity, systematic comparison of the 3D structure of RgDAAO and mammalian DAAOs reveals the presence of a number of peculiar features that explain some of the major properties distinguishing the two enzymes (Pollegioni et al. 2002). The most evident feature is the presence of a 23-residue-long C-terminal loop (loop βF5-βF6 in RgDAAO) not present in the other DAAOs; this long loop is a key factor for the correct formation of the protein dimer, resulting in a higher stability of the dimeric form in RgDAAO than in the pig kidney enzyme. A further peculiar feature of the RgDAAO structure is the absence of a loop acting as an active site “lid.” In fact, in the mammalian enzyme, the conformational change of this “lid” (the loop βI5-βI6) allows the substrate/product exchange at the active site: Indeed, the dissociation of the product from the enzyme is the rate-limiting step in catalysis (Porter et al. 1977). In RgDAAO, the active site entrance is only partially hindered by the flexible side chain of a tyrosine (Tyr238, Fig. 2c, left), resulting in a faster exchange and a more efficient enzyme (see above).
D-Amino acid oxidase production
In past years, a number of studies have been published on the production of DAAO by fermentation of the original microorganism or as a recombinant protein in different hosts. DAAO is a peroxisomal protein: When induced by D-Ala in R. gracilis cells, peroxisomes size increases by 30% and new peroxisomes are produced (up to 240%; Perotti et al. 1991). This peroxisome proliferation is observed even when DAAO production is induced in C. boidinii cells (reaching an expression level of ∼1.5 U/mg protein growing the cells on glucose and D-Ala; Sakai et al. 1998). Notwithstanding the peroxisomal localization, DAAO was reported to assemble correctly, acquiring the catalytic activity, in the cytosol of H. polymorpha and C. boidinii cells (Sulter et al. 1990; Yurimoto et al. 2000).
Concerning the expression of DAAO in the native microorganism, de novo synthesis of DAAO in peroxisomes can be selectively induced by the presence of D- or D,l-AAs (Pilone Simonetta et al. 1989b; Perotti et al. 1991; Molla et al. 2003). The synthesis is induced by D-Ala, and the presence of the L-isomer in the culture medium prevents induction by inhibiting the D-AA transport by a specific permease: DAAO activity appeared only after the L-isomer was depleted from the medium (Pilone Simonetta et al. 1989b). R. gracilis utilizes D-Ala as a source of both carbon and nitrogen; the maximal DAAO expression (∼1% of total proteins in the crude extract) is obtained when D-Ala is the sole source of nitrogen and glucose of carbon (see Table 1; Molla et al. 2003). As an example, DAAO expression in R. gracilis cells reached up to 770 U/l and 46 U/g cell using 28 or 80 mM D-Ala as inducer (Cambiaghi et al. 1991; Molla et al. 2003). Ammonium sulfate had a negative effect on RgDAAO mRNA translation and on the expression of the enzyme activity (Molla et al. 2003). Analysis of the promoter region of the RgDAAO gene (up to 300 bp upstream of the ATG starting codon) failed to identify putative genetically responsive elements (Alonso et al. 1998). In the methylotrophic yeast C. boidinii, DAAO is involved in growth on D-Ala as the source of carbon, but a different enzyme system metabolizes D-Ala as the source of nitrogen (Sakai et al. 1998; Yurimoto et al. 2000). However, no responsive elements have been identified in the upstream region of the DAAO gene in C. boidinii either (Yurimoto et al. 2000).
In a number of other fungi, DAAO can be induced by D-AAs, although the best inducers differ in each species and do not correspond with the best substrates of the corresponding purified enzyme. In N. crassa cells (where the DAAO activity is related to the sulfur metabolism), for example, the enzyme is induced by the presence of D-Met as the sole sulfur source in the medium (Sikora and Marzluf 1982). The yeast T. variabilis is a good DAAO producer: DAAO activity accounts for up to 225 U/g cell using T. variabilis CBS 4095 and NRRL Y7770 cell strains and D-Ala as inducer (Wei et al. 1989). All D,l-AAs tested (with nonpolar or polar side chains, as well as with negatively or positively charged side groups) induce TvDAAO: D,l-Met, D,l-Ala, D,l-Leu, and D,l-Val gave the highest enzyme production. Moreover, even D-AA analogues that cannot be metabolized induce TvDAAO expression: The best results were obtained with N-carbamoyl-D-alanine, N-acetyl-D-tryptophan, and N-chloroacetyl-D-α-aminobutyric acid (Hörner et al. 1996). When 5 mM N-carbamoyl-D,l-alanine is used as the sole carbon source, an enzyme expression of up to 650 U/g cell was reached (corresponding to 315 U/g cell and 3,450 U/l when cultivated in a fermentor; Fischer 1998). In the presence of zinc ions (up to 140 μM), which are involved in the synthesis of peroxines, TvDAAO synthesis in a bioreactor increased up to 525 U/g cell (Hörner et al. 1996). A high level of biomass can be produced using T. variabilis cells, e.g., the fermentation of the strain CBS 4095 reached a value of about 62 g cell/l using a fed-batch fermentor (and a maximal DAAO production of ∼35 U/g cell at air saturation; Huber et al. 1992). On the other hand, a twofold lower yield in biomass was obtained for the mutant T. variabilis ATCC 20931 strain but with a considerable increase in DAAO production (about 350 U/g cell; Wong and Shen 1990).
The fungus F. oxysporum produced DAAO in a medium containing glucose and ammonium sulfate: D-Ala and D-3-aminobutyric acid were the best inducers and the addition of zinc ions was also required (Table 1). Recently, the bacterial DAAO gene from Arthrobacter protophormiae was expressed in E. coli using a pET21 plasmid. High amounts of the recombinant enzyme were produced (∼1,200 U/g cell and ∼150,000 U/l on D-Met at 30°C), but unfortunately, this bacterial enzyme is practically inactive on both D-Ala and CephC (Geueke et al. 2007).
Concerning the DAAO expression using recombinant microorganisms, the best level of protein expression was achieved for recombinant pig kidney DAAO in E. coli: The enzyme reaches 40% of total soluble protein in the crude extract (Setoyama et al. 1996). However, due to the low activity of the mammalian DAAO, this value only corresponds to 200–300 U/l of fermentation broth. The recombinant RgDAAO was expressed in E. coli by using the pT7-DAAO expression system as a chimeric enzyme with six additional amino acid residues at the N terminus (Molla et al. 1998): With this system, we reached 2,300 U/l and 930 U/g cell, accounting for ∼8% of the total soluble proteins in crude extract. A similar value was obtained for the expression of the F. solani DAAO cDNA in E. coli (Isogai et al. 1990; 1998).
TvDAAO cDNA was expressed in T. variabilis strain CBS4095 (up to 9,000 U/l; Furuya and Matsuda 1995) and in a polyploid mutant of the strain ATCC 10679 (up to 5,000 U/l and 175 U/g cell; Wong and Shen 1997). Furthermore, TvDAAO was expressed in Saccharomyces cerevisiae and Kluyveromyces lactis under the control of the GAL1 promoter (Gonzalez et al. 1997), as well as in Schizosaccharomyces pombe (Table 1; Isoai et al. 2002). The level of TvDAAO expression in S. pombe cells was similar to or slightly lower than that obtained using the natural microorganism (even when using an integrative vector with a multi-copies expression cassette): 550 U/g cell on CephC at 35°C and oxygen saturation (corresponding to ∼80 U/g cell using the conditions reported in Table 1). However, by using a catalase-deficient host strain, the conversion of CephC into Gl-7-ACA was significantly improved (see below). P. pastoris was also used as host strain, achieving intracellular expression of TvDAAO of up to 11,500 U/l (Table 1; Yu et al. 2002). TvDAAO was also expressed in E. coli using a pTrc99A (Lin et al. 2000) and a pET21 plasmid regulated by T5 or T7 promoter (without any tag) and using lactose as an inducer (Hwang et al. 2000). In this latter case, an His-tagged TvDAAO was produced and an expression level of 15% of the total soluble proteins was reached. Recently, chimeric TvDAAO fused with a 12-amino acid peptide at N terminus was expressed up to 35% of the total soluble E. coli proteins when D-Met was added to the growth media to suppress the toxic activity of the enzyme: By adding up to 10 mM D-Met, the volumetric DAAO activity and the specific activity increased 14.5-fold and 3-fold, respectively (Dib et al. 2007). Anyway, when the activity values were calculated on CephC as substrate and at 25°C (see Table 1), the values obtained were similar to those obtained using lactose as inducer (Hwang et al. 2000).
DAAO from C. boidinii was produced at a high level under the control of the alcohol oxidase gene promoter in the original yeast (Yurimoto et al. 2001). The highest level of expression was obtained using an alcohol oxidase depleted strain that had multiple copies (up to eight) of the expression plasmid: Growing the cells on a methanol/glycerol/methylamine medium a DAAO level of ∼32 U/mg protein, which corresponds to ∼30% of the total soluble proteins, was reached.
DAAO was also expressed as an engineered recombinant protein. The first work reporting on a His-tagged TvDAAO (using an expression system derived from pKK223 plasmid) showed that about 90% of the enzyme was produced as an apoprotein (Alonso et al. 1999). This recombinant TvDAAO binds too strongly to metal chelate chromatography supports containing copper or zinc ions, but it was efficiently isolated using a chelate support containing a low amount of cobalt ligands. Subsequently, His-tagged TvDAAO was produced using a pET plasmid as a fully active holoenzyme and up to 15% of total soluble proteins in the crude extract (Hwang et al. 2000). TvDAAO was also expressed as His-tagged protein in Pichia pastoris cells under the control of the alcohol oxidase promoter (Zheng et al. 2006), but the enzyme expression was threefold lower than in the absence of the tag (Yu et al. 2002). An His-tagged RgDAAO protein was also expressed in E. coli up to 3,000 U/l as a fully soluble and active holoenzyme; it was purified in a single step by metal chelate chromatography (82% yield) to a high specific activity, in a stable form, and devoid of side or contamination activities (Fantinato et al. 2001).
TvDAAO was also co-expressed with Gl-7-ACA acylase in E. coli (using a pET plasmid; Luo et al. 2004a). Subsequently, this approach was improved using an E. coli strain that is unable to produce catalase and β-lactamase: a DAAO expression level up to ∼40 U/l was reached (Zheng et al. 2007). These cells could directly convert CephC into 7-ACA with a yield of 74% (see below). A chimeric Gl-7-ACA acylase-linker-DAAO protein that directly converts CephC into 7-ACA has also been produced (although accumulation of a large amount of the Gl-7-ACA intermediate was observed; Luo et al. 2004b).
Applications of microbial D-amino acid oxidases
7-ACA production
The enzymatic conversion of CephC to 7-ACA is of great interest to cephalosporin antibiotics manufacturers: More than 50 semi-synthetic cephalosporins are produced from 7-ACA, with a currently estimated annual worldwide market of ∼200 million US dollars. The traditional method of producing 7-ACA involves the chemical deacylation of CephC in organic solvents, using toxic reagents and requiring laborious procedures: Although the chemical method was optimized and the organic solvents were largely recycled, the increase in production pushed the use of enzymes (Huber et al. 1972; Cabri et al. 1999). In fact, the chemical method has been largely replaced by a two-step enzymatic transformation (Fig. 3): First, DAAO catalyzes the oxidative deamination of CephC to 7-(5-oxoadipoamido) cephalosporanic acid which, in the presence of H2O2, undergoes spontaneous decarboxylation to Gl-7-ACA; subsequently, Gl-7-ACA acylase hydrolyzes Gl-7-ACA to produce 7-ACA. The industrial application of DAAO for oxidation of CephC has been revised recently (Pilone and Pollegioni 2002). For all biotransformation processes employing DAAO, the enzyme was demonstrated to be strongly stabilized by immobilization. Recent results reported that the immobilization of DAAO using highly activated glyoxyl agarose support improved by ∼15,000-fold the stability of the enzyme from R. gracilis while the one from T. variabilis was less affected (Betancor et al. 2003). The catalytic efficiency of an immobilized flavoenzyme such as DAAO, which requires molecular oxygen as electron acceptor, is significantly limited because of the external and internal mass transfer resistances of oxygen to gel matrix. Increasing the partial pressure of oxygen and/or agitating the reaction mixture are normally used to avoid this limitation; however, this decreases the operational stability of the immobilized enzyme. Alternatively, a chimeric enzyme obtained by fusing bacterial hemoglobin from Vitreoscilla (used as an oxygen donor) with RgDAAO significantly enhances the performances of the biocatalyst. The catalytic efficiency of this flavohemoprotein on CephC was 12.5-fold higher than that of immobilized DAAO alone and the operational stability approximately threefold better (Khang et al. 2003).

Alternative pathways for the bioconversion of CephC into 7-ACA: the two-step process catalyzed by D-amino acid oxidase and glutaryl-7-ACA acylase (left; Cambiaghi et al. 1991; Mosbach and Szwajcer 1991; Pilone and Pollegioni 2002) and the one-step process catalyzed by CephC acylase (right; Pollegioni et al. 2005). In the first pathway, the H2O2 produced by the reaction of DAAO is used for the decarboxylation of 7-(5-oxoadipoamido)cephalosporanic acid (a modest addition H2O2 is used industrially to force this step toward glutaryl-7-amino cephalosporanic acid)
DAAO bioconversions can be also performed using whole cells: The diffusion limitations caused by the presence of the cell wall was overcome by permeabilization (Pilone and Pollegioni 2002). In this field, a DAAO catalyst with improved properties was produced by cross-linking T. variabilis cells with polyethyleneimine and glutaraldehyde: Significantly better mechanical properties resulted in an increased number of conversions (the half-life was equal to 142 cycles) (Beĉka et al. 2003). The main obstacle to using whole cells is that other enzyme activities, such as catalase and esterase, decrease both the yield and purity of the resulting Gl-7-ACA product. Among strategies employed to inactivate these enzymes, recently, an E. coli D11 strain unable to produce catalase and β-lactamase was engineered to express both DAAO and Gl-7-ACA acylase simultaneously. The mutation of the catalase gene appears to be an optimal choice (Zheng et al. 2007).
With the aim of lowering the production costs, some investigators focused on a one-step protocol for 7-ACA production. This can be achieved by directly converting CephC to 7-ACA by a single enzymatic activity (Matsuda et al. 1987; Nigam et al. 2005; Pollegioni et al. 2005), by the simultaneous action of DAAO and Gl-7-ACA acylase (Luo et al. 2004b), or by expressing the two enzymes as a single fusion protein (Luo et al. 2004a). The simultaneous use of DAAO and Gl-7-ACA acylase in a single reactor was extensively studied: the main obstacle is the production of H2O2 during the reaction, which inactivates the employed enzymes. The production of 7-ACA by a trienzymatic system (DAAO, Gl-7-ACA acylase, and catalase) was also reported (Lopez-Gallego et al. 2005). This approach has a major drawback in that the rate of hydrolysis by the acylase is comparatively slow with respect to DAAO. However, ∼80% of 7-ACA was produced in 180 min, and only 2.5% of the 7-(5-oxoadipoamido) cephalosporanic acid was not hydrolyzed. An alternative method of producing 7-ACA in a single reactor was developed by using DAAO in permeabilized P. pastoris cells and Gl-7-ACA acylase immobilized on a support. The whole P. pastoris cells contain large amount of catalase and esterase that yielded a significant increase in by-products. This problem was eliminated by inactivating these two enzymatic activities by alkali and heat-treating the cells before bioconversion (Tan et al. 2006). Importantly, the successful application of a new CephC acylase, produced by a combination of rational design and directed evolution strategies, converting CephC into 7-ACA in a one-step process was recently reported, providing a new process for industrially producing 7-ACA (Pollegioni et al. 2005).
Production of α-keto acids and pure l-amino acids
Another area of DAAO application is represented by the conversion of D-AAs to the corresponding α-keto acids, which are valuable and useful compounds. For a review reporting the use of DAAO in this application until 2002, see Pilone and Pollegioni (2002). The use of DAAO was recently optimized to produce pure l-amino acids (l-AA), compounds largely used as feed additives, components of infusion solutions, and starting materials for pharmaceuticals. As an example, immobilized TvDAAO (with soluble catalase from Micrococcus lysodeikticus) was used for the racemic resolution of D,l-Phe (Trost and Fischer 2002). A α-keto acid yield close to 100% and a high degree of purity decreases the costs of down-stream processing enormously; however, the l-Phe recovery corresponds to only 50% of the initial amino acid. Most recently, a process for producing optically pure, unnatural naphthyl-amino acids, crucial tools for modern drug discovery research, by resolution of racemic mixtures was optimized. Using a rational design study, a mutant of RgDAAO was obtained with which all unnatural amino acids tested could be completely resolved (Caligiuri et al. 2006a). The complete recovery of the single L-enantiomer from the racemic mixtures was obtained by combining RgDAAO and two other enzymes, l-aspartate amino transferase and catalase. With this multi-enzymatic system, we could convert the α-keto acid produced by DAAO into the l-AA with a 98% yield in terms of product recovery and a 99.5% enantiomeric excess (Caligiuri et al. 2006b).
Analytical determination of D-AAs (and D-AA analogues)
Up to now, the detection of D-AAs in biological samples has been of relevant interest in particular for the food and beverage industry: D-AAs are components of the peptidoglycan moiety of the bacterial cell wall and their presence in foodstuffs could indicate bacterial contamination (Friedman 1999). Their concentration can be used to measure the nutritional value of food, as D-AAs are not metabolized like the L-isomers are: The racemization of amino acids is influenced by the quality of raw materials and by the food-processing conditions (Marchelli et al. 1992; 1996; Gandolfi et al. 1992). Most recently, determining D- and l-AAs in biological samples has gained significance even for clinical applications since several diseases have been associated with defects in amino acid metabolism. As an example, the D-AAs content in white and gray matter in the brains of Alzheimer disease patients is one to four times higher than that observed in the respective regions of normal brain (D’Aniello et al. 1992). Moreover, D-Ser metabolism is relevant for the functionality of N-methyl-D-aspartate (NMDA) receptors and for disorders associated with altered function, such as schizophrenia, ischemia, epilepsy, and neurodegenerative disorders (Martineau et al. 2006; Pollegioni et al. 2007a). A recent update of the DAAO-based techniques used to detect and quantify D-AAs was reported by (Caldinelli et al. 2006).
Traditional analytical techniques for detecting amino acids are complex laboratory procedures requiring a number of assay steps, additional chemicals, and/or complex instrumentation. The most widespread analytical techniques are gas chromatography or reversed-phase liquid chromatography, based on derivatization of the amino acids (Brückner and Lüpke 1991; Brückner et al. 1991; Brückner and Westhauser 2003). Although chromatographic methods are reliable, reproducible, and very sensitive, they are too slow for the food industry and not suitable for on-line application. Starting in the 1990s, a number of publications reported on the development of biosensors using DAAO coupled to electrochemical transducers for the direct, rapid detection of D-AAs in solution. DAAO sensors have also often been associated with l-amino acid oxidase sensors in estimating the D-AA/l-AA ratio. These sensors are based on a variety of detection principles: hydrogen peroxide detection (Albery et al. 1996; Yao and Wasa 1998; Sacchi et al. 1998; Váradi et al. 1999), tetrathiafulvalinium tetracyanoquinodimethanide electrodes (Albery et al. 1987), screen-printed electrodes with the enzyme immobilized on rhodinized carbon (Sarkar et al. 1999), or on different kinds of carbon paste electrodes (Wu et al. 2004), or on a fiber optic biosensor (Zhujun et al. 1995). These sensors frequently were obtained by combination with a second enzyme, such as horse radish peroxidase (Kulys and Schmid 1991; Kacaniklic et al. 1994; Dominguez et al. 2001) or pyruvate oxidase (Inaba et al. 2003). In this latter case, a sensor based on determining the amount of oxygen consumed by means of an oxygen electrode was developed for monitoring fermentation processes. A further means of detecting D-AAs was obtained by combining the separation efficiency of HPLC with the specificity of enzymes and the sensitivity of an electrochemical detector: In this way, an extremely specific determination of D-AAs was obtained even on complex matrices like the ones encountered in food and tissue analysis (Voss and Galensa 2000; Oguri et al. 2005; Hamase et al. 2005).
In some cases, DAAO amperometric biosensors were also applied to enantioselectively analyze clinically relevant compounds such as D-pipecolic acid (a marker of liver cirrhosis and chronic hepatic encephalopathy; Stefan et al. 2003a) or for enantiopurity tests of chiral drugs, such as D,l-carnitine and D,l-methotrexate (Stefan et al. 2003b, c). An example is represented by R-perindopril (also known by its proprietary name Coversyl) assay; this molecule is a long-acting ACE (angiotensin-converting enzyme) inhibitor, mainly used to treat hypertension and heart failure. An amperometric biosensor based on DAAO was developed (van Staden et al. 2000): The detection limit was ∼10 nmol/l of R-perindopril and the main interfering species were D-AAs such as D-Pro.
All the DAAO-based sensors reported so far use the commercial DAAO from pig kidney. These sensors often showed a low stability (the enzyme instability is mainly due to the weak binding of the FAD cofactor to the apoprotein moiety; Parkin and Huiltin 1979; Wu et al. 2004), a low turnover number, and inactivity on acidic D-AAs. The first two problems have been solved by our group using RgDAAO absorbed on a graphite electrode (Sacchi et al. 1998). The last limitation was addressed by a rational approach for the design and production of the M213R RgDAAO mutant active on D-Asp (Sacchi et al. 2002). Although the catalytic efficiency of the mutant enzyme on neutral D-amino acids decreased dramatically, and it is practically inactive on basic D-AAs, M213R RgDAAO was exploited in the quantification of acidic and neutral D-AAs, a relevant parameter, for example, in rose wine or Parmigiano cheese (Marchelli et al. 1996). Neither the wild-type RgDAAO nor the M213R mutant is, however, useful for determining the total D-AAs concentration in food samples. For this reason, we recently used a directed evolution approach to produce RgDAAO variants with an increased activity on all, and particularly on basic and acidic D-AAs (Sacchi et al. 2004). Among the selected RgDAAO variants, two mutants (L118H and T60/Q144R/K152E RgDAAO) showed a remarkably improved catalytic efficiency on all D-AAs and were applied to analytically determine the total D-AA content in biological samples (Sacchi et al. 2004). Most recently, even TvDAAO was applied to determine D-AA content by developing an oxygen-independent assay based on the reaction with 2,6-dichloroindophenol as an artificial electron acceptor (Trampitsch et al. 2005). These results indicate that microbial DAAOs could efficiently replace the pig kidney enzyme in biosensor devices and also that improved electrodes can be achieved by using protein engineering approaches (i.e., rational and irrational mutagenesis strategies).
Analytical applications of the apoprotein form of DAAO
Interesting analytical applications were also developed by exploiting the apoprotein form of DAAO (apo-DAAO), e.g., using an amplification cascade to detect hydrolase enzymes (such as alkaline phosphatase, AP). This application is based on the property, common to a family of artificial substrates of several hydrolases, that their hydrolysis produces a prosthetic group that can bind a detector apoenzyme, thus providing a sensitive amplification of the original signal (Rabin et al. 1995). In the specific case, an AP activity catalyzes the formation from a prosthetogen, specifically by dephosphorylating the substrate flavin adenine dinucleotide phosphate (FADP), of the prosthetic group FAD. The combination of the FAD prosthetic group with an apoenzyme, precisely with apo-DAAO, yields the corresponding holoenzyme form, which in the presence of suitable substrates (such as D-Ala, D-Met, and D-Pro) produces hydrogen peroxide. Finally, H2O2 can be quantified by the horseradish peroxidase-mediated conversion of 3,5-dichloro-2-hydroxybenzenesulfonic acid and 4-aminoantipyrine to a colored product. Suitable apoenzymes include those obtained from glucose oxidase, l-amino acid oxidase, xanthine oxidase, etc. However, DAAO is the preferred apoenzyme, as it is very shelf-stable and can be used as a “single pot” system (i.e., all components can be added as a single mixing to the analyte-linked AP). It is noteworthy that the prosthetogen FADP does not show any significant activity as a prosthetic group, thus avoiding a background signal, and does not interfere with the formation of the holoenzyme from apoenzyme and FAD. The FADP-based enzyme amplification cascade has been used in several applications, coupling it with a variety of different assays, such as hybridization or immunoassays. A first example is represented by the detection of a wide range of analytes, adapting high-sensitivity immunoassays for quantification via colorimetric detection. An analyte can be represented by thyrotropin, known also as thyroid-stimulating hormone (Obzansky et al. 1991). In this case, an antibody–thyrotropin complex is recognized by a dethiobiotinylated secondary antibody and subsequently is reacted with a streptavidin-AP conjugate. Biotin is then used to release the conjugate, and AP is quantified by applying the FADP-based amplification cascade (the reaction mixture contains FADP, apo-DAAO, D-Pro, horseradish peroxidase, and 3,5-dichloro-2-hydroxybenzenesulfonic acid). A second example is represented by a hybridization assay used for detecting a single-strand target nucleic acid (Harbron 2002). Still, the use of microbial DAAOs (whose interaction with the FAD coenzyme is at least tenfold stronger than with the mammalian protein) could improve the practical use of this technique.
Therapeutic uses
The observation made at the end of the 1990s that the administration of D-AA-containing solutions to cancer patients improves the nutritional status and inhibits tumor cell growth suggested there is a relationship between DAAO activity and tumors (Sasamura et al. 1999). Furthermore, the observation that DAAO activity is extremely low in liver tumors indicated that the enzyme could play an inhibitory role on cell growth (Sasamura et al. 2002). RgDAAO was thus used for a new gene-directed enzyme prodrug therapy, an anti-neoplastic treatment strategy designed to overcome the systemic toxicity of chemotherapy by specifically expressing a foreign enzyme in malignant cells that converts a nontoxic prodrug into a cytotoxic metabolite. Stegman and coworkers expressed an active truncated RgDAAO mutant (lacking the type-1 peroxisomal targeting sequence, see Fig. 2a) in the cytosol of rat 9L glioma cells. The overexpression of cytoplasmatically targeted DAAO conferred toxicity upon D-Ala exposure (Stegman et al. 1998), which was attributed to the induction of oxidative stress by the overproduction of H2O2, a reactive oxygen species (ROS). Interestingly, H2O2 released by this treatment was toxic to both proliferating and quiescent cells: This is an attractive feature because a substantial fraction of cells within solid tumors may be quiescent. The anti-tumoral activity of DAAO was subsequently confirmed by Fang et al. (Fang et al. 2002; Fang et al. 2004). They even succeeded in improving the effectiveness of this therapeutic strategy by conjugating the enzyme (from pig kidney) with polyethylene glycol: PEG-DAAO showed better pharmacokinetic parameters and selectively accumulated in the tumor tissue. Furthermore, human colon carcinoma SW480 cells pretreated with pegylated zinc propoporphyrin (a potent inhibitor of Heme oxygenase I, an enzyme that protects tumor cells against oxidative stress) become more sensitive to insults caused by cytotoxic agents.
Concerning ischemia, the neuronal damage is largely caused by massive stimulation of NMDA receptors because of D-Ser release by damaged cells: exogenously applied DAAO can be used to prevent this damage (Katsuki et al. 2004). In fact, DAAO was previously shown to deplete D-Ser in brain slices without affecting the levels of other amino acids, including glycine (Mothet et al. 2000). In this specific application, a commercial preparation of mammalian DAAO was added to cortical slices from rat: DAAO at 1–30 U/l provided concentration-dependent protection against neuronal damage induced by 20 min of simulated ischemia. For this purpose, and for tumor treatment, microbial DAAOs (and in particular engineered variants most active at low oxygen concentration) would be more efficient in controlling D-Ser levels/inducing oxidative stress, thus representing a novel therapeutic tool for the treatment of various human pathologies.
Conclusions
DAAO is employed in numerous biotechnological applications, which have been described in this work; indeed, its importance is increasing by the day as new applications are continuously being discovered. Oddly, in many of these applications the enzyme from pig kidney is used, which shows properties not favorable for these purposes. For many of these applications, microbial DAAOs are more efficient. In fact, they can be produced in large quantity, which makes them cost competitive, and show properties (such as activity, substrate specificity, enantioselectivity, stability, etc.) more suitable for the applications. Furthermore, the modern methods of genetic engineering combined with an increasing knowledge of the structure–function relationships make it possible to produce enzyme variants with new and evolved properties. This will promote the exploration of novel applications for DAAO, not limited to the industrial sector: This will certainly be the major field of development in next few years.
References
Albery WJ, Barlett PN, Bycroft M, Craston DH, Driscoll BJ (1987) Amperometric enzyme electrodes. Part III. A conducting salt electrode for the oxidation of four different flavoenzymes. J Electroanal Chem 218:119–126
Albery WJ, Hutchins JEC, Uttamlal M (1996) An L-amino acid sensor for beer fermentation. J Appl Electrochem 26:243–248
Alonso J, Barredo JL, Díez B, Mellado E, Salto F, Garcia JL, Cortés E (1998) D-amino-acid oxidase gene from Rhodotorula gracilis (Rhodosporium turoloides) ATCC 26217. Microbiology 144:1095–1101
Alonso J, Barredo JL, Armisèn P, Díez B, Salto F, Guisán JM, Garcia JL, Cortés E (1999) Engineering the D-amino acid oxidase from Trigonopsis variabilis to facilitate its overproduction in Escherichia coli and its downstream processing by tailor-made metal supports. Enzyme Microb Technol 25:88–95
Beĉka S, Ŝkrob F, Plháĉková K, Kujan P, Holler P, Kyslik P (2003) Cross-linked cell aggregates of Trigonopsis variabilis: D-amino acid oxidase catalyst for oxidation of cephalosporin C. Biotechnol Lett 25:227–233
Betancor L, Hidalgo A, Fernández-Lorente G, Mateo C, Rodríguez V, Fuentes M, López-Gallego F, Fernández-Lorente R, Guisan JM (2003) Use of physicochemical tools to determine the choice of optimal enzyme: stabilization of D-amino acid oxidase. Biotechnol Prog 19:784–788
Brückner H, Lüpke M (1991) Determination of amino acid enantiomers in orange juices by chiral phase capillary gas chromatography. Cromatographia 31:123–128
Brückner H, Wittner R, Godel H (1991) Fully automated high-performance liquid chromatographic separation of DL-amino acids derivatized with o-phtaldialdehyde together with N-isobutyryl-cysteine. Cromatographia 32:383–388
Brückner H, Westhauser T (2003) Chromatographic determination of L- and D-amino acids in plants. Amino Acids 24:43–55
Cabri W, Verga R, Cambiaghi S, Bernasconi E (1999) Environmentally friendly production of 7-ACA. Chimica e Industria, 81:461–464
Caldinelli L, Motteran L, Sacchi S, Piubelli L, Boselli A, Mothet JP, Pollegioni L, Pilone MS (2006) Detection of D-amino acids by D-amino acid oxidase. In: Konno R, Brückner H, D’Aniello A, Fisher G, Fujii N, Homma H (eds) D-amino acids: a new frontier in amino acid and protein research—pratical methods and protocols, chapter 2.6. Nova Biomedical Books, New York, pp 135–168
Caligiuri A, D’Arrigo P, Rosini E, Tessaro D, Molla G, Servi S, Pollegioni L (2006a) Enzymatic conversion of unnatural amino acids by yeast D-amino acid oxidase. Adv Synth Catal 348:2183–2190
Caligiuri A, D’Arrigo P, Gefflaut T, Molla G, Pollegioni L, Rosini E, Rossi C, Servi S (2006b) Multistep enzyme catalysed deracemisation of 2-naphthyl alanine. Biocatal Biotrans 24:409–413
Cambiaghi S, Tomaselli S, Verga R (1991) Enzymatic process for preparing 7-aminocephalosporin acid and derivatives. European Patent 0496993
Casalin P, Pollegioni L, Curti B, Pilone MS (1991) A study on apoenzyme from Rhodotorula gracilis D-amino acid oxidase. Eur J Biochem 197:513–517
Curti B, Ronchi S, Pilone MS (1992) D- and L- amino acid oxidases. In: Müller F (ed) Chemistry and biochemistry of flavoenzymes, vol. III. CRC Press, Boca Raton, pp 69–94
D'Aniello A, Vetere A, Fisher GH, Cusano G, Chavez M, Petrucelli L (1992) Presence of D-alanine in proteins of normal and Alzheimer human brain. Brain Res 592:44–48
David HL, Clavel-Seres S, Clement F, Lazlo A, Rastogi N (1989) Methionine as methyl-group donor in the synthesis of Mycobacterium avium envelope lipids, and its inhibition by D,L-methionine, D-norleucine and D,L-norleucine. Acta Leprol 7:77–80
Deutch CE (2004) Oxidation of 3,4-dehydro-D-proline and other D-amino acid analogues by D-alanine dehydrogenase from Escherichia coli. FEMS Microbiol Lett 238:383–389
Dib I, Slavica A, Riethorst W, Nidetzky B (2006) Thermal inactivation of D-amino acid oxidase from Trigonopsis variabilis occurs via three parallel paths of irreversible denaturation. Biotechnol Bioeng 94:645–654
Dib I, Stanzer D, Nidetzky B (2007) Trigonopsis variabilis D-amino acid oxidase: control of protein quality and opportunities for biocatalysis through production in Escherichia coli. Appl Environ Microbiol 73:331–333
Dominguez R, Serra B, Revieo J, Pingarron JM (2001) Chiral analysis of amino acids using electrochemical composite bienzyme biosensors. Anal Biochem 298:275–282
Fang J, Sawa T, Akaike T, Maeda H (2002) Tumor-targeted delivery of polyethylene glycol-conjugated D-amino acid oxidase for antitumor theraphy via enzymatic generation of hydrogen peroxide. Cancer Res 62:3138–3143
Fang J, Sawa T, Akaike T, Greish K, Maeda H (2004) Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int J Cancer 109:1–8
Fantinato S, Pollegioni L, Pilone MS (2001) Engineering, expression and purfication of a His-tagged chimeric D-amino acid oxidase from Rhodotorula gracilis. Enzyme Microb Technol 29:407–412
Faotto L, Pollegioni L, Ceciliani F, Ronchi S, Pilone MS (1995) The primary structure of D-amino acid oxidase from Rhodotorula gracilis. Biotechnol Lett 17:193–198
Fischer L (1998) D-amino acid oxidase in biotechnology. Recent Res Devel Microbiol 2:295–317
Friedman M (1999) Chemistry, nutrition, and microbiology of D-amino acids. J Agric Food Chem 47:3457–3479
Furuya K, Matsuda A (1995) Trigonopsis transformant producing D-amino acid oxidase. United States Patent US5453374
Gabler M, Fischer L (1999) Production of a new D-amino acid oxidase from the fungus Fusarium oxysporum. Appl Environ Microbiol 65:3750–3753
Gabler M, Hensel M, Fischer L (2000) Detection and substrate selectivity of new microbial D-amino acid oxidases. Enzyme Microb Technol 27:605–611
Gandolfi I, Palla G, Delprato L, De Nisco F, Marchelli R, Salvadori C (1992) D-amino acids in milk as related to heat treatments and bacterial activity. J Food Sci 57:377–379
Geueke B, Weckbecker A, Hummel W (2007) Overproduction and characterization of a recombinant D-amino acid oxidase from Arthrobacter protophormiae. Appl Microbiol Biotechnol 74:1240–1247
Gonzalez FJ, Montes J, Martin F, Lopez MC, Ferminan E, Catalan J, Galan MA, Dominguez A (1997) Molecular cloning of TvDAO1, a gene encoding a D-amino acid oxidase from Trigonopsis variabilis and its expression in Saccharomyces cerevisiae and Kluyveromyces lactis. Yeast 13:1399–1408
Hamase K, Konno R, Morikawa A, Zaitsu K (2005) Sensitive determination of D-amino acids in mammals and the effect of D-amino acid oxidase activity on their amount. Biol Pharm Bull 28:1578–1584
Harbron S (2002) Hybridization assay for detecting a single-stranded target nucleic acid in which excess probe is destroyed. United States Patent US6423492
Hörner R, Wagner F, Fischer L (1996) Inducion of the D-amino acid oxidase from Trigonopsis variabilis. Appl Environ Microbiol 62:2106–2110
Huber F, Chauvetee R, Jackson B (1972) Preparative methods for 7-aminocephalosporanic acid and 6-aminopenicillanic acid. In: Flynn E (ed) Cephalosporins and penicillins, chemistry and biology. Academic, New York, pp 27–48
Huber FM, Vicenzi JT, Tietz AJ (1992) High yielding culture conditions for the biosynthesis of D-amino acid oxidase by Trigonopsis variabilis. Biotechnol Lett 14:195–200
Hwang TS, Fu HM, Lin LL, Hsu WH (2000) High-level expression of Trigonopsis variabilis D-amino acid oxidase in Escherichia coli using lactose as inducer. Biotechnol Lett 22:655–658
Inaba Y, Mizukami K, Hamada-Sato N, Kobayashi T, Imada C, Watanabe E (2003) Development of a D-alanine sensor for the monitoring of a fermentation using the improved selectivity by the combination of D-amino acid oxidase and pyruvate oxidase. Biosens Bioelectron 19:423–431
Isoai A, Kimura H, Reichert A, Schorgendorfer K, Nikaido K, Tohda H, Giga-Hama Y, Mutoh N, Kumagai H (2002) Production of D-amino acid oxidase (DAO) of Trigonopsis variabilis in Schizosaccharomyces pombe and the characterization of biocatalysts prepared with recombinant cells. Biotechnol Bioeng 80:22–32
Isogai T, Ono H, Ishitani Y, Kojo H, Ueda Y, Kohsaka M (1990) Structure and expression of cDNA for D-amino acid oxidase active against cephalosporin C from Fusarium solani. J Biochem (Tokyo) 108:1063–1069
Isogai T, Ono H, Kojo H (1998) D-amino acid oxidase of F. solani and methods for its recombinant production. United States Patent US5773272
Job V, Marcone L, Pilone MS, Pollegioni L (2002) Glycine oxidase from Bacillus subtilis: characterization of a new flavoprotein J. Biol. Chem. 277:6985–6993
Kacaniklic V, Johansson K, Marko-Varga G, Gorton L, Jönsson-Pettersson G, Csöregi E (1994) Amperometric biosensors for detection of L- and D-amino acids based on coimmobilized peroxidase and L- and D- amino acid oxidase in carbon paste electrodes. Electroanalysis 6:381–390
Katsuki H, Nonaka M, Shirakawa H, Kume T, Akaike A (2004) Endogenous D-serine is involved in induction of neuronal death by N-methyl-D-aspartate and simulated ischemia in rat cerebrocortical slices. J Pharmacol Exp Ther 311:836–844
Kawazoe T, Tsuge H, Pilone MS, Fukui K (2006) Crystal structure of human DAAO: context-dependent variability of the backbone conformation of the VAAGL hydrophobic stretch located at the si-face of the flavin ring. Protein Sci 15:2708–2717
Khang Y, Kim I, Hah Y, Hwangbo J, Kang K (2003) Fusion protein of Vitreoscilla hemoglobin with D-amino acid oxidase enhances activity and stability of biocatalyst in the bioconversion process of cephalosporin C. Biotechnol Bioeng 82:480–488
Kishore G, Vaidyanathan CS (1976) Purification and properties of D-amino acid oxidase of Aspergillus niger. Indian J Biochem Biophys 13:216–222
Komatsu K, Sugiura K, Matsuda A, Yamamoto K (1988) D-amino acid oxidase gene. Japan Patent 63071180
Krebs HA (1935) Metabolism of amino-acids: deamination of amino-acids. Biochem J 29:1620–1644
Kubicek-Pranz EM, Rohr M (1985) D-amino acid from the yeast Trigonopsis variabilis. J Appl Biochem 7:104–113
Kulys J, Schmid RD (1991) Bienzyme sensors based on chemically modified electrodes. Biosens Bioelectron 6:43–48
Lin L, Chien HR, Wang W, Hwang T, Fu H, Hsu W (2000) Expression of Trigonopsis variabilis D-amino acid oxidase gene in Escherichia coli and characterization of its inactive mutants. Enzyme Microb Technol 27:482–491
Lopez-Gallego F, Batencor L, Hidalgo A, Mateo C, Fernandez-Lafuente R, Guisan JM (2005) One-pot conversion of cephalosporin C to 7-aminocephalosporanic acid in the absence of hydrogen peroxide. Adv Synth Catal 347:1804–1810
Luo H, Yu H, Li Q, Shen Z (2004a) Cloning and co-expression of D-amino acid oxidase and glutaryl-7-aminocephalosporanic acid acylase genes in Escherichia coli. Enzyme Microb Technol 35:514–518
Luo H, Li Q, Yu H, Shen Z (2004b) Construction and application of fusion proteins of D-amino acid oxidase and glutaryl-7-aminocephalosporanic acid acylase for direct bioconversion of cephalosporin C to 7-aminocephalosporanic acid. Biotechnol Lett 26:939–945
Marchelli R, Dossena A, Palla G, Audhuy-Peaudecerf M, Lefeuvre S, Carnevali P, Freddi M (1992) D-amino acids in reconstituted infant formula: a comparison between conventional and microwave heating. J Sci Food Agric 59:217–226
Marchelli R, Dossena A, Palla G (1996) The potential of enantioselective analysis as a quality control tool. Trends Food Sci Technol 7:113–119
Martineau M, Baux G, Mothet JP (2006) D-Serine signalling in the brain: friend and foe. Trends Neurosci 29:481–491
Massey V, Hemmerich P (1980) Active-site probes of flavoproteins. Biochem Soc Trans 8:246–257
Matsuda A, Matsuyama K, Yamamoto K, Ichikawa S, Komatsu KI (1987) Cloning and characterization of the genes for two distinct cephalosporin acylases from Pseudomonas strain. J Bacteriol 169:5815–5820
Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B (1996) Crystal structure of D-amino acid oxidase: a case of active site mirror-image convergent evolution with flavocytochrome b2. Proc Natl Acad Sci USA 93:7496–7501
Mizutani H, Miyahara I, Hirotsu K, Nishina Y, Shiga K, Setoyama C, Miura R (1996) Three-dimensional structure of porcine kidney D-amino acid oxidase at 3.0 Å resolution. J Biochem (Tokyo) 120:14–17
Molla G, Vegezzi C, Pilone MS, Pollegioni L (1998) Overexpression in Escherichia Coli of a recombinant chimeric Rhodotorula Gracilis D-amino acid oxidase. Protein Expr Purif 14:289–294
Molla G, Motteran L, Piubelli L, Pilone MS, Pollegioni L (2003) Regulation of D-amino acid oxidase expression in the yeast Rhodotorula gracilis. Yeast 20:1061–1069
Mörtl M, Diederichs K, Welte W, Molla G, Motteran L, Andriolo G, Pilone MS, Pollegioni L (2004) Structure-function correlation in glycine oxidase from Bacillus subtilis. J Biol Chem 279:29718–29727
Mosbach K, Szwajcer E (1991) A D-amino acid oxidase and method for isolation thereof. European Patent EP0211033B1
Mothet JP, Parent AT, Wolosker H, Brady RO Jr, Linden DJ, Ferris CD, Rogawaski MA, Snyder SH (2000) D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA 97:4926–4931
Nigam VK, Kundu S, Ghosh P (2005) Single-step conversion of cephalosporin C to 7-aminocephalosporanic acid by free and immobilized cells of Pseudomonas diminuta. Appl Biochem Biotechnol 126:13–21
Obzansky DM, Rabin BR, Simons DM, Tseng SY, Severino DM, Eggelte H, Fisher M, Harbron S, Stout RW, Di Paolo MJ (1991) Sensitive, colorimetric enzyme amplification cascade for determination of alkaline phosphatase and application of the method to an immunoassay of thyrotropin. Clin Chem 37:1513–1518
Oguri S, Nomura M, Fujita Y (2005) A new strategy for the selective determination of D-amino acids: enzymatic and chemical modifications for pre-column derivatization. J Chromatogr A 1078:1–58
Olsiewski PJ, Kaczorowski GJ, Walsh C (1980) Purification and properties of D-amino acid dehydrogenase, an inducible membrane-bound iron-sulfur flavoenzyme from Escherichia coli B. J Biol Chem 255:4487–4494
Parkin K, Huiltin HO (1979) Immobilization and characterization of D-amino acid oxidase. Biotechnol Bioeng 21:939–953
Perotti ME, Pollegioni L, Pilone MS (1991) Expression of D-amino acid oxidase in Rhodotorula Gracilis under induction conditions: a biochemical and cytochemical study. Eur J Cell Biol 55:104–113
Pilone Simonetta M, Vanoni MA, Casalin P (1987) Purification and properties of D-amino acid oxidase, an inducible flavoenzyme from Rhodotorula gracilis. Biochim Biophys Acta 914:136–142
Pilone Simonetta M, Pollegioni L, Casalin P, Curti B, Ronchi S (1989a) Properties of D-amino-acid oxidase from Rhodotorula gracilis. Eur J Biochem 180:199–204
Pilone Simonetta M, Verga R, Fretta A, Hanozet GM (1989b) Induction of D-amino-acid oxidase by D-alanine in Rhodotorula gracilis grown in defined medium. J Gen Microbiol 135:593–600
Pilone MS, Pollegioni L (2002) D-amino acid oxidase as an industrial biocatalyst. Biocatal Biotransform 20:145–159
Pollegioni L, Pilone MS (1992) Purification of Rhodotorula gracilis D-amino acid oxidase. Protein Expr Purif 3:165–167
Pollegioni L, Falbo A, Pilone MS (1992) Specificity and kinetics of Rhodotorula gracilis D-amino acid oxidase. Biochim Biophys Acta 1120:11–16
Pollegioni L, Butò S, Tischer W, Ghisla S, Pilone MS (1993a) Characterization of D-amino acid oxidase from Trigonopsis variabilis. Biochem Mol Biol Int 31:709–717
Pollegioni L, Langkau B, Tischer W, Ghisla S, Pilone MS (1993b) Kinetic mechanism of D-amino acid oxidases from Rhodotorula gracilis and Trigonopsis variabilis. J Biol Chem 268:13850–13857
Pollegioni L, Fukui K, Massey V (1994) Studies on the kinetic mechanism of pig kidney D-amino acid oxidase by site-directed mutagenesis of tyrosine 224 and tyrosine 228. J Biol Chem. 269:31666–31673
Pollegioni L, Molla G, Campaner S, Martegani E, Pilone MS (1997) Cloning, sequencing and expression in E. coli of a D-amino acid oxidase cDNA from Rhodotorula gracilis active on cephalosporin C. J Biotechnol 58:115–123
Pollegioni L, Diederichs K, Molla G, Umhau S, Welte W, Ghisla S, Pilone MS (2002) Yeast D-amino acid oxidase: structural basis of its catalytic properties. J Mol Biol 324:535–546
Pollegioni L, Iametti S, Fessas D, Caldinelli L, Piubelli L, Barbiroli A, Pilone MS, Bonomi F (2003) Contribution of the dimeric state to the thermal stability of the flavoprotein D-amino acid oxidase. Protein Sci 12:1018–1029
Pollegioni L, Caldinelli L, Molla G, Sacchi S, Pilone MS (2004) Catalytic properties of D-amino acid oxidase in cephalosporin C bioconversion: a comparison between proteins from different sources. Biotechnol Prog 20:467–473
Pollegioni L, Lorenzi S, Rosini E, Marcone GL, Molla G, Verga R, Cabri W, Pilone MS (2005) Evolution of an acylase active on cephalosporin C. Protein Sci 14:3064–3076
Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G (2007a) Physiological functions of D-amino acid oxidases: from yeast to humans. Cell Mol Life Sci 64:1373–1394
Pollegioni L, Sacchi S, Caldinelli L, Boselli A, Pilone MS, Piubelli L, Molla G (2007b) Engineering the properties of D-amino acid oxidases by a rational and a directed evolution approach. Curr Protein Pept Sci 8:600–618
Porter DJ, Voet JG, Bright HJ (1977) Mechanistic features of the D-amino acid oxidase reaction studied by double stopped flow spectrophotometry. J Biol Chem 252:4464–4473
Rabin BR, Harbron S, Eggelte HJ, Hollaway MH, Holloway A (1995) Amplification assay for hydrolase enzymes. United States Patent US5445942
Raunio RP, Jenkins WT (1973) D-alanine oxidase from Escherichia coli: localization and induction by L-alanine. J Bacteriol 115:560–566
Sacchi S, Pollegioni L, Pilone MS, Rossetti C (1998) Determination of D-amino acids using a D-amino acid oxidase biosensor with spectrophotometric detection. Biotech Techn 12:149–153
Sacchi S, Lorenzi S, Molla G, Pilone MS, Rossetti C, Pollegioni L (2002) Engineering the substrate specificity of D-amino acid oxidase. J Biol Chem 277:27510–27516
Sacchi S, Rosini E, Molla G, Pilone MS, Pollegioni L (2004) Modulating D-amino acid oxidase substrate specificity: production of an enzyme for analytical determination of all D-amino acids by directed evolution. Protein Eng Des Sel 17:517–525
Sakai Y, Yurimoto H, Matsuo H, Kato N (1998) Regulation of peroxisomal proteins and organelle proliferation by multiple carbon sources in the methylotrophic yeast Candida boidinii. Yeast 14:1175–1187
Sarkar P, Tothill IE, Setford SJ, Turner APF (1999) Screen-printed amperometric biosensors for the rapid measurement of L- and D-amino acids. Analyst 124:865–870
Sasamura T, Matsuda A, Kokuba Y (1999) Effects of D-methionine-containing solution on tumor cell growth in vitro. Arzneimittelforschung 49:541–543
Sasamura T, Matsuda A, Kokuba Y (2002) Determination of D-amino acid oxidase activity in tumor cells. Ann Clin Biochem 39:595–598
Schräder T, Andreesen JR (1996) Properties and chemical modification of D-amino acid oxidase from Trigonopsis variabilis. Arch Microbiol 165:41–47
Setoyama C, Miura R, Nishina Y, Shiga K, Mizutani H, Miyahara I, Hirotsu K (1996) Crystallization of expressed porcine kidney D-amino acid oxidase and preliminary X-ray crystallographic characterization. J Biochem (Tokyo) 119:1114–1117
Settembre EC, Dorrestein PC, Park JH, Augustine AM, Begley TP, Ealick SE (2003) Structural and mechanistic studies on ThiO, a glycine oxidase essential for thiamin biosynthesis in Bacillus subtilis. Biochemistry 42:2971–2981
Sikora L, Marzluf GA (1982) Regulation of L-amino acid oxidase and of D-amino acid oxidase in Neurospora crassa. Mol Gen Genet 186:33–39
Stefan RI, Nejem RM, van Staden JF, Aboul-Enein HY (2003a) Biosensors for the enantioselective analysis of pipecolic acid. Sens Actuators B Chem 94:271–275
Stefan RI, Bokretsion RG, van Staden JF, Aboul-Enein HY (2003b) Simultaneous determination of L- and D-carnitine using a sequential injection analysis/amperometric biosensors system. J Pharm Biomed Anal 33:323–328
Stefan RI, Bokretsion RG, van Staden JF, Aboul-Enein HY (2003c) Simultaneous determination of L- and D-methotrexate using a sequential injection analysis/amperometric biosensors system. Biosens Bioelectron 19:261–267
Stegman LD, Zheng H, Neal ER, Ben-Yoseph O, Pollegioni L, Pilone MS, Ross BD (1998) Induction of cytotoxic oxidative stress by D-alanine in brain tumor cells expressing Rhodotorula gracilis D-amino acid oxidase: a cancer gene therapy strategy. Hum Gene Ther 9:185–193
Sulter GJ, Waterham HR, Goodman JM, Veenhuis M (1990) Proliferation and metabolic significance of peroxisomes in Candida boidinii during growth on D-alanine or oleic acid as the sole carbon source. Arch Microbiol 153:485–489
Tan Q, Song Q, Wei D (2006) Single-pot conversion of cephalosporin C to 7-aminocephalosporanic acid using cell-bound and support-bound enzymes. Enzyme Microb Technol 39:1166–1172
Tishkov VI, Khoronenkova SV (2005) D-Amino acid oxidase: structure, catalytic mechanism, and practical application. Biochemistry (Moscow) 70:40–54
Trampitsch C, Slavica A, Riethorst W, Nidetzky B (2005) Reaction of Trigonopsis variabilis D-amino acid oxidase with 2,6-dichloroindophenol: kinetic characterisation and development of an oxygen-independent assay of the enzyme activity. J Mol Catal B Enzym 32:271–278
Trost EM, Fischer L (2002) Minimization of by-product formation during D-amino acid oxidase catalyzed racemate resolution of D/L-amino acids. J Mol Catal B Enzym 19–20:189–195
Umhau S, Pollegioni L, Molla G, Diederichs K, Welte W, Pilone MS, Ghisla S (2000) The x-ray structure of D-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. Proc Natl Acad Sci U S A 97:12463–12468
van Staden JF, Stefan RI, Aboul-Enein HY (2000) Amperometric biosensor based on D-aminoacid oxidase for the R-perindopril assay. Fresenius J Anal Chem 367:178–180
Váradi M, Adányi N, Szabó EE, Trummer N (1999) Determination of the ratio of D- and L-amino acids in brewing by an immobilised amino acid oxidase enzyme reactor coupled to amperometric detection. Biosens Bioelectron 14:335–340
Voss K, Galensa R (2000) Determination of L- and D-amino acids in foodstuffs by coupling of high-performance liquid chromatography with enzyme reactors. Amino Acids 18:339–352
Wei CJ, Huang JC, Tsai YP (1989) Study on the synthesis of D-amino acid oxidase in Trigonopsis variabilis in continuous culture. Biotechnol Bioeng 34:570–574
Wong B, Shen YQ (1990) Enzymatic production of 7-amino cephalosporanic acid. World Intellectual Property Organization WO9012110A1
Wong B, Shen YQ (1997) Production of 7-amino cephalosporanic acid with D-amino acid oxidase and deacylase. United States Patent US5618687
Wu X, van Wie BJ, Kidwell DA (2004) An enzyme electrode for amperometric measurement of D-amino acids. Biosens Bioelectron 20:879–886
Yao T, Wasa T (1998) HPLC detection of L- and D-amino acids by use of immobilized enzyme electrodes as detectors. Anal Chim Acta 209:259–264
Yasuda Y, Tochikubo K (1985) Germination-initiation and inhibitory activities of L- and D-alanine analogues for Bacillus subtilis spores. Modification of methyl group of L- and D-alanine. Microbiol Immunol 29:229–241
Yu J, Li DY, Zhang YJ, Yang S, Li RB, Yuan ZY (2002) High expression of Trigonopsis variabilis D-amino acid oxidase in Pichia pastoris. J Mol Catal, B Enzym 18:291–297
Yurimoto H, Hasegawa T, Sakai Y, Kato N (2000) Physiological role of the D-amino acid oxidase gene, DAO1, in carbon and nitrogen metabolism in the methylotrophic yeast Candida boidinii. Yeast 16:1217–1227
Yurimoto H, Hasegawa T, Sakai Y, Kato N (2001) Characterization and high-level production of D-amino acid oxidase in Candida boidinii. Biosci Biotechnol Biochem 65:627–633
Zheng H, Wang X, Chen J, Zhu K, Zhao Y, Yang Y, Yang S, Jiang W (2006) Expression, purification, and immobilization of His-tagged D-amino acid oxidase of Trigonopsis variabilis in Pichia pastoris. Appl Microbiol Biotechnol 70:683–689
Zheng H, Zhu T, Chen J, Zhao Y, Jiang W, Zhao G, Yang S, Yang Y (2007) Construction of recombinant Escherichia coli D11/pMSTO and its use in enzymatic preparation of 7-aminocephalosporanic acid in one-pot. J Biotechnol 129:400–405
Zhujun Z, Zhilong G, Wangbai M (1995) An enzyme-based fiber optic biosensor for determination of D-amino acids in serum. Microchem J 52:131–138
Zwart KB, Overmars EH, Harder W (1983) Significance of microbodies in the metabolism of L-aspartate in Candida utilis. FEMS Microbiol Lett 19:273–279
Acknowledgements
This work was supported by grants from FAR to G. Molla, M. S. Pilone, and L. Pollegioni and from Fondazione CARIPLO to L. Pollegioni. We apologize to those whose work we were unable to cite owing to space limitations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pollegioni, L., Molla, G., Sacchi, S. et al. Properties and applications of microbial D-amino acid oxidases: current state and perspectives. Appl Microbiol Biotechnol 78, 1–16 (2008). https://doi.org/10.1007/s00253-007-1282-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1282-4