
Abstract
We report here the in vivo expression of the synthetic transposase gene himar1(a) in Streptomyces coelicolor M145 and Streptomyces albus. Using the synthetic himar1(a) gene adapted for Streptomyces codon usage, we showed random insertion of the transposon into the streptomycetes genome. The insertion frequency for the Himar1-derived minitransposons is nearly 100 % of transformed Streptomyces cells, and insertions are stably inherited in the absence of an antibiotic selection. The minitransposons contain different antibiotic resistance selection markers (apramycin, hygromycin, and spectinomycin), site-specific recombinase target sites (rox and/or loxP), I-SceI meganuclease target sites, and an R6Kγ origin of replication for transposon rescue. We identified transposon insertion loci by random sequencing of more than 100 rescue plasmids. The majority of insertions were mapped to putative open-reading frames on the S. coelicolor M145 and S. albus chromosomes. These insertions included several new regulatory genes affecting S. coelicolor M145 growth and actinorhodin biosynthesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Members of the order Actinomycetales are the most productive bacteria with respect to the synthesis of bioactive metabolites. These bacteria have been in the focus of industrial application for many decades, e.g., in the production of tetracycline, erythromycin, and vancomycin. Recent whole genome sequencing programs have revealed that the biosynthetic potential of Actinomycetales has been greatly underexplored with traditional approaches. A common feature of all actinobacterial genomes is a length of about 4–11 Mb and the presence of approximately 20–30 gene clusters encoding the synthesis of secondary metabolites. With the advent of next-generation DNA sequencing techniques, we can access the huge amount of genetic information, which awaits development into new chemical and biological entities. Therefore, efficient methods for the functional genes characterization are of great importance.
Transposition became a powerful tool in genetic analysis, with applications in creating insertional knockout mutations, generating gene–operon fusions to reporter functions, providing physical or genetic landmarks for the cloning of adjacent DNAs, and locating primer binding sites for DNA sequence analysis (Damasceno 2010; Petzke and Luzhetskyy 2009; Weaden and Dyson 1998). The number and uses of transposons continue to expand into new fields such as genomics and transcriptomics. In vivo transposon-based strategy is a valuable tool to identify functions of a number of genes and to construct random mutant libraries for diverse applications.
Several in vivo transposon mutagenesis systems have been applied for actinobacteria, such as the native actinobacteria transposons IS6100 (Weaden and Dyson 1998), Tn4560 (Ikeda 1993), Tn1792 (Herron 1999), or the foreign transposon Tn5. A recently developed transposon based on Tn5 hypertransposase synthetic encoding gene has been shown as an efficient tool for the generation of random mutants in streptomycetes (Petzke and Luzhetskyy 2009). It combines very high transposon mutagenesis frequency with fast detection of insertion loci. Tn5 transposon insertions are however slightly biased to GC-rich sequences (Fernandez-Martinez 2011). Therefore, using different transposon systems in addition to Tn5 could minimize gaps in chromosome coverage attributed to insertion-site biases. The transposon we established for random in vivo mutagenesis in streptomycetes is a derivative of the transposon Himar1, a mariner family element isolated from the horn fly Haematobia irritans. The mariner transposon has a remarkable lack of host specificity as it does not require any host-specific factors for transposition (Lampe 1996). The Himar1 transposon has previously been shown to have little site specificity in vitro and in vivo (Rubin 1999; Maier 2006). Apart from showing a preference for AT-rich DNA, Himar1 only requires the presence of a TA dinucleotide for insertion (Craig 1997). Even in members of the actinobacteria with the highest GC contents, it is most likely that at least one TA dinucleotide is present in each gene. Himar1 has been shown to transpose in distantly related insects, even more distantly related protozoa, vertebrate cells, and many Gram-positive and Gram-negative bacteria, while most transposons are limited to their own host range (Rubin 1999).
As of yet, in vivo application of the Himar1 transposon in Mycobacteria showed randomness without recognizable sequence determinants (Rubin 1999). Highly efficient stable integrations were also reported in Francisella tularensis, suggesting that comprehensive transposon libraries could be generated with the procedure (Maier 2004). In vivo mutagenesis of Burkholderia pseudomallei was reported to be highly efficient with up to 44 % integration success (Rholl 2008). Use of a mutant Himar1 transposase in Escherichia coli was also shown to be efficient by increasing the activity up to 50 times over the native transposase (Lampe 1999). For phenotypic analysis, a Himar1 mutagenesis approach in Leptospira biflexa produced randomly distributed mutations which could be screened for phenotypes affecting several aspects of metabolism and physiology (Louvel 2005). It was also shown that transgenes flanked by inverted terminal repeats could be integrated into the Myxococcus xanthus genome in a MycoMar transposon, suggesting that mariner family elements could be used to deliver large gene clusters (Fu 2008). Nonetheless, there are no reports to date of successful application of mariner transposons in streptomycetes.
In this paper, we introduce a transposon mutagenesis system based on the synthetic Himar1 encoding transposase gene under Streptomyces specific promoter. Several aspects make this system superior for generating mutant libraries among other applications: Himar1 offers an unparalleled efficiency and randomness of the transposon insertions; the minitransposon is equipped with loxP and rox sites allowing excision of transposon resistance markers by Cre and/or Dre recombinase and their further reutilization in the same genetic background; pSG5 replicon allows to eliminate the replicative plasmid after mutagenesis; the presence of the R6Kγ origin of replication allows fast and easy insertion loci identification; possibility to generate transposon mutant libraries for single or multiple insertion mutants with a slight method modification.
Materials and methods
Bacterial strains and media
Streptomyces coelicolor M145 and Streptomyces albus J1074 were used to express the synthetic himar1 gene. E. coli DH5α (Hanahan 1983) was used for cloning, and the non-methylating E. coli ET12567/pUZ8002 was used to drive conjugative transfer of non-methylated DNA to Actinobacteria as described previously (Luzhetskyy 2006). E. coli TransforMax™ EC100D™ pir-116 (Epicentre, Madison) was used for transfer and recovery of rescue plasmids derived from chromosomal DNA. Cultivation of all E. coli strains was performed as described previously (Sambrook 1989). For conjugation, Streptomyces strains were grown on mannitol soy flour agar plates (MS agar); for standard cultivation, S. coelicolor M145 and S. albus J1074 were either grown on HA agar plates or on R2YE agar plates. For liquid cultivation purposes, tryptone soy broth (TSB) was used. For antibiotic phenotyping R2YE, NL5 and minimal medium (MM) were used (Kieser 2000). All seed cultures were started from frozen mycelia stock and cultured in flasks with a 10-cm coil to promote dispersed growth. Apramycin, spectinomycin, and hygromycin were added to the media to final concentrations of 50, 100, and 100 μg/mL, respectively.
Plasmid construction
The plasmids used in this work are listed in Table 1. DNA manipulation and cloning were carried out according to standard protocols (Sambrook 1989). Plasmid constructs were confirmed by DNA sequencing. The synthetic gene himar1, flanked by HindIII and XbaI restriction sites, was synthesized by GenScript (New Jersey, USA) and provided on the plasmid pHimar1. This plasmid was digested with HindIII and XbaI. The fragment containing the synthetic himar1 gene was cloned into pAL1 and pNL1 (Fedoryshyn 2008a) to yield pALHim and pNLHim.
Four plasmids with different minitransposons were constructed, two containing the apramycin resistance gene aac(3)IV, one containing the spectinomycin resistance gene aadA(1), and one containing the hygromycin resistance gene hph (pHTM, pHAM, pHSM, and pHAH, respectively). All plasmids contained the R6Kγ origin, and inserts were flanked on each side by ITR sites.
Construction of pHTM
The gene aac(3)IV was amplified from pIJ773 with forward primer 5′-acgtaccgaattcggttcatgtgcagctccatcagc-3′ and the reverse primer 5′-acgtacgaattcatgagctcagccaatcgactgg-3′ containing EcoRI restriction sites. The PCR product was cloned into the plasmid pITRΔNheI (derivated from pITR, GenScript, New Jersey, USA) by EcoRI restriction sites, leading to pITRΔNheIaac. PvuII was used to excise a fragment from pITRΔNheIaac containing the aac(3)IV gene and the R6Kγ origin, all flanked by ITR sites. This fragment was cloned into pALHim restricted with EcoRV, yielding pHTM (Fig. 1a).

Maps of four Himar1-containing plasmids. The plasmids contain the following shared features: ITR, inverted terminal repeat for Himar1; oriT, origin of plasmid transfer; R6Kori, origin of replication in E. coli for rescue plamids; hph, hygromycin resistance marker. Plasmids pHTM, pHAH, and pHAM contain aac(3)IV, apramycin resistance marker. pHSM contains aadA(1),spectinomycin resistance marker. Plasmids pHTM, pHSM, and pHAH contain pSG5rep, temperature-sensitive replicon in Streptomyces, and tipAp, thiostreptone-inducible promoter. Plasmid pHAM contains Pr-φC31, φC31integrase promoter. Plasmids pHTM and pHSM contain rox, recognition sites for Dre recombinase. Plasmids are not drawn to scale
Construction of pHSM
In a similar procedure, the gene aadA(1) from pHP45Ω (Table 1) was excised by EcoRI digest and cloned into the pITRΔNheI, giving pITRΔNheIaad. pITRΔNheIaad was then digested with PvuII for blunt end cloning into the pALHim EcoRV site, giving pHSM (Fig. 1b).
Construction of pHAH
The gene hph was amplified by PCR using pAL1 as a template, the forward primer 5′-ccccctctagagaataggaacttcggaatagg-3′ with a XbaI site (in italics) and the reverse primer 5′-ccccccaattgg-ggtcgcagggcgtgcccttgggctccccgggcgcgtaccgtatttgcagtaccagcgt-3′ with a MunI site. The amplified fragment was cloned into pTn5Oks (ShineGene Molecular Biotech, Inc.) via MunI and XbaI restriction sites leading to pTn5Okshph. An EcoRV fragment was cut from pTn5Okshph containing the hph gene and the R6Kγ origin, all flanked by ITR sites, and cloned into pNLHim restricted with EcoRV, yielding pHAH (Fig. 1c).
Construction of pHAM
The promoter of φC31integrase was amplified from pSET152 (Bierman 1992) using the forward primer5′-aaaagatctccccgtgccggagcaatcgc-3′ and the reverse primer 5′-aaaaagcttatgtcggcgaccctacgccc-3′. The PCR product was digested with BglII and HindIII enzymes and cloned into the respective sites of pKCLP2, yielding pKCLP2phiCpr. The promoterless transposase of the Himar1 transposon was recovered as HindIII BamHI fragment from pHTM and cloned into the respective sites of pKCLP2phiCpr, yielding p31Him. The Himar transposon was excised from pITRΔNheIaac as an EcoRV fragment and cloned into the EcoRV site of p31Him to make pHAM (Fig. 1d).
Rescue plasmid generation and recovery
To generate rescue plasmids, genomic DNA was isolated from S. coelicolor M145 and S. albus J1074 wild-type strains and corresponding transposon mutants grown for 2 days at 28 °C in 30 mL TSB liquid cultures (Kieser 2000). The DNA was either digested with BamHI, NcoI, NotI, PstI, or SacII and self-ligated with T4-DNA ligase. Where required, the DNA was transformed into E. coli TransforMax™ EC100D™ pir-116 electrocompetent cells by electroporation (E. coli pulser Bio-Rad™). Plasmids were recovered with the Wizard® Plus SV Minipreps DNA Purification System (Promega™). Chromosome–Himar1 junction sequences were determined using the sequencing primer pMODfor (5′-ccaacgactacgcactagccaac-3′) for S. coelicolor M145 mutants and pTn5Oksfor (5′-attcaggctgcgcaactg-3′) for S. albus mutants.
Expression of Dre recombinase
Spores of S. coelicolor M145 transposon mutants were conjugated with the pUWL-Dre plasmid, containing the Dre-recombinase gene. Exconjugants were collected and inoculated to 100 mL TSB with 50 μg/mL thiostrepton and 200 μg/mL phosphomycin. Cultures were grown for 3 days. Aliquots were plated onto 50 μg/mL thiostrepton HA agar and grown for 3 days to sporulation. Spores were collected and inoculated to TSB with thiostrepton 2 days at 28 °C. Aliquots were plated onto MS agar with an appropriate antibiotic and grown for 3 days at 28 °C.
Southern hybridization of genomic DNA
Southern blot analysis was carried out on genomic DNA of S. coelicolor M145 and S. albus J1074 Himar1-transposon mutants. Genomic DNAs were digested with BamHI for S. coelicolor M145 and with NcoI for S. albus J1074 mutants, separated by agarose gel electrophoresis, and transferred to a positively charged nylon membrane. A 0.8-kb aac(3)IV gene from pHTM (for S. coelicolor M145 mutants) and 0.4-kb R6Kγ origin from pTn5Oks (for S. albus mutants) were prepared and used as template for probe labeling with digoxigenin-dUTP by the random priming method. Hybridization, washing, and signal detection were carried out as described by Roche Diagnostics.
Results
Construction of the transposon vectors containing a synthetic Himar1 transposase gene
Actinobacteria usually have a GC content more than 70 %; therefore, their codon usage differs from that of most other bacteria. To circumvent the problem of inefficient translation of codons for which the corresponding amino-acyl-tRNA molecules are in limited supply or absent in, the genetic code of the transposase gene was modified in silico to make a synthetic Himar1 transposase gene (GenScript, New Jersey, USA). Three vectors for its expression were constructed. The synthetic gene was cloned to pAL1 and pNL1, giving pALHim and pNLHim, respectively. pHSM and pHTM were obtained by cloning the apramycin and spectinomycin resistance genes linked to the R6Kγ origin flanked by ITR (transposase recognition sequence) sites to pALHim (Fig. 1a, b). pHAH was obtained by cloning the hygromycin resistance gene linked to the R6Kγ origin flanked by ITR sites to pNLHim (Fig. 1c). All plasmids carry the oriT, the thiostrepton-inducible promoter, tipA and the temperature-sensitive replicon, pSG5rep. It allows to transfer the plasmids from an E. coli host to different Streptomyces species by conjugation, to induce an expression of the transposase gene, and to select against the plasmids when the cultivation temperature is raised to 39 °C.
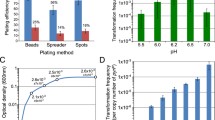
Most replicative transposon delivery vectors contain the temperature-sensitive pSG5 replicon, which is not supported in some Streptomyces strains. In such cases, suicide vectors can substitute replicative vectors. To establish such a system, the transposase gene expression should start immediately after vector introduction into the recipient cell. To accomplish this, we used the promoter of the φC31 integrase from pSET152 since this integrative plasmid does not replicate, and without rapid expression of the integrase gene, it would be lost like a suicidal vector. We constructed pHAM based on the suicide vector pKCLP2. This vector contains a Himar1 transposase encoding gene under the control of the φC31 integrase promoter, the Himar1 transposon and origin from the oriT. After introduction of pHAM in S. albus, transposon mutants were obtained with a frequency of between 10−3and 10−4 (based on input recipient spores). This means that the transposase gene under the φC31 integrase promoter expresses early enough to permit the transposition from the backbone of nonreplicative plasmid.
Himar1 mutagenesis of S. coelicolor M145 and S. albus J1074
We applied our system in S. coelicolor M145 with pHTM and pHSM. The plasmids were introduced into S. coelicolor M145 by intergeneric conjugation. The exconjugants were carried through several cultivation stages (Fig. 2). Aliquots of cultures were plated on HA agar plates, and spores were recovered and diluted to 10−7 and plated out again. Single colonies were obtained and transferred to HA agar plates containing apramycin and spectinomycin. Transposon mutants of S. coelicolor M145 were also plated to HA agar plates containing hygromycin and cultivated at 28 °C for 7 days. Of the 264 tested mutants with the apramycin resistance marker, a single colony was found to be hygromycin resistant. Eighty-eight mutants with the spectinomycin resistance marker were tested. All were hygromycin sensitive. To assess the stability of Himar1 insertions, ten randomly selected apramycin-resistant mutants and two spectinomycin-resistant mutants were grown in liquid TSB cultures for approximately 100 generations in the absence of antibiotic selection. The cultures were plated onto their respective antibiotic R2YE agar and were all found to be antibiotic resistant. These results demonstrate the effectiveness of the synthetic hyperactive Himar1-based system for transposon mutagenesis. The same system of random transposon mutagenesis was applied in the commonly used heterologous host strain S. albus J1074.

Schematic diagram depicting the steps for obtaining a transposon mutant library of S. coelicolor M145 with pHTM/pHSM and S. albus J1074 with pHAH
Identification of the insertion loci and analysis of integration frequency
Rescue plasmids for S. coelicolor M145 and S. albus mutants were generated and sequenced to identify the loci of transposon insertion. Sequencing started from the transposon outwards into the genomic DNA. Thereby, approximately 800 bp of the connecting chromosomal DNA could be identified. Sequence data were used for BLAST analysis against the genomic DNA sequence of S. coelicolor A(3)2 and S. albus J1074 [Electronic supplementary material (ESM) Tables S1 and S2]. The transposon insertions have been randomly distributed throughout the genomes of S. coelicolor M145 or S. albus.
Southern blot analysis was carried out to determine if a given transposon insertion was unique. All but one mutant of S. coelicolor M145 showed multiple transposon integration sites (Fig. 3a). Conversely, only one unique transposon integration site was detected for each of the S. albus J1074 mutants (Fig. 3c). This difference could be attributed to the inability to induce the thiostrepton-inducible promoter during the exponential growth phase of S. albus.

Hybridization membrane after Southern blot hybridization of Himar1-mutants; 1, 2, 12 positive control (pHTM/NdeI); 20, 29 positive control (pTn5Oks/HindIII); 13 negative control (wild-type S. coelicolor M145 DNA); 14, 21 negative control (wild-type S. albus J1074 DNA); 3–11 S. coelicolor M145 transposon mutants (tipAp induced); 15–19 S. coelicolor M145 transposon mutants (tipAp uninduced); 22–28 S. albus J1074 transposon mutants (tipAp induced)
While multiple gene disruptions caused by multiple transposon insertions could give rise to new and interesting phenotypes or reduce the number of colonies, one would need to screen for a specific phenotype, the presence of two or more insertions in the genome of S. coelicolor M145 transposon mutants complicates relating an observed phenotype to the disrupted gene. We therefore developed a method to increase the number of mutants with unique transposon cassette in the genome (Fig. 2). Cultivation conditions optimized to cause loss of the plasmid in the early stage of cultivation increased the frequency of S. coelicolor M145 mutants with single insertion (Fig. 3b).
Excision of the apramycin resistance gene using dre recombinase
To assess Dre-mediated marker excision, selected apramycin resistance mutants were conjugated with pUWL-Dre containing a thiostrepton resistance marker, and Dre excision was performed as previously described (Herrmann 2012). As expected, apramycin-sensitive colonies were readily obtained with marker excision efficiencies reaching 100 %. All of the marker-free mutants were verified by PCR analysis.
Identification of regulatory genes in S. coelicolor M145 involved in secondary metabolite production
After transposon mutagenesis, a mutant library with a variety of mutant phenotypes was obtained. Four transposon mutants of S. coelicolor M145 showing impaired actinorhodin production were selected for further analysis. Rescue plasmids were recovered from the mutants and sequenced. The BLAST analysis of four S. coelicolor M145 mutants revealed sequences identical to genes SCO3812 encoding a putative gntR-family transcriptional regulator, SCO4197 encoding a putative MarR family regulator, SCO4198 encoding a putative DNA binding protein, and to SCO4192 encoding a hypothetical protein (http://streptomyces.org.uk; Bentley 2002).The corresponding mutants showed a different phenotype on R2YE, MM, and NL5 agar plates compared to the wild type. To ensure that the observed phenotype was due to the identified orfs (SCO3812, SCO4197, SCO4198, and SCO4192), we inactivated these genes via homologous recombination in a clean genetic background of S. coelicolor M145 (ESM Table S3). The obtained mutants of S. coelicolor M145 did not produce actinorhodin on the R2YE agar and overproduced this antibiotic on the MM and NL5 agar plates, in contrast to the wild-type strain (ESM Figs. S1 and S2). Actinorhodin production was blocked upon substitution of glucose in MM with sucrose or glycerol. Addition of glycerol to MM induced yellow pigment production by all mutants (ESM Fig. S1). In contrast to the wild type, all four mutants showed actinorhodin production on NL5 medium where glutamine was used as a carbon source. Addition of glycerol, glucose, or sucrose to the NL5 medium blocked actinorhodin production by all mutants (ESM Fig. S2).
Another two transposon mutants which were analyzed contained insertions in the SCO3390 and SC03919 genes, encoding for a putative two-component sensor kinase and putative LysR-family transcriptional regulator. We generated both knockouts via homologous recombination (ESM Table S3) yielding the strain S. coelicolor M145 B04 (with disrupted SC03919) and the strain S. coelicolor M145 A07 (with disrupted SCO3390). Both mutants showed slight actinorhodin overproduction on the RY2E agar, while S. coelicolor M145 B04 in contrast to the wild type and S. coelicolor M145 A07 produces actinorhodin on the MM (ESM Fig. S1). The actinorhodin production capabilities of S. coelicolor M145 B04 are very similar to the four S. coelicolor M145 mutants with inactivated SCO3812, SCO4197, SCO4198, and SCO4192.
The last transposon mutant showing impaired actinorhodin production contained insertion in the SCO5222 gene encoding a putative lyase. We have performed the respective gene inactivation experiments to prove the phenotypes (ESM Table S3). However, the mutant did not show any differences in antibiotic production (ESM Figs. S1 and S2). Obviously, actinorhodin production impairment was caused by some additional insertions.
Discussion
Implementing a transposon mutagenesis system generally requires optimization of several parameters. To obtain an efficient transposon system, we combined an optimized synthetic gene with high GC content encoding the Himar1 transposase and several resistance markers as minitransposons. The availability of hygromycin, spectinomycin, and apramycin resistant genes within minitransposons might be important when working with different actinobacteria being resistant to some of those antibiotics. There is a limited choice of efficient resistance markers which can be used for genetic manipulation of actinobacteria. Therefore, apramycin, hygromycin, and/or spectinomycin resistance tagging of mutants can significantly impair downstream genetic applications such as complementation, double-mutant isolation, or heterologous gene overexpression in the same genetic background. This obstacle was overcome by flanking the minitransposons with a Dre recombinase-excisable resistance marker.
The development of the Himar1 random mutagenesis system with the R6Kγ origin has a distinct advantage over other transposon systems that do not offer the possibility to replicate rescue plasmids in a foreign host. Rescue plasmid sequencing proved to be a quick and convenient way to map several insertion loci. TransforMax™ EC100D™ pir-116 electrocompetent E. coli cells constitutively express the π protein (pir gene product) for replication of plasmids containing the R6Kγ origin of replication. Upon digestion of Streptomyces Himar1 mutant chromosomal DNA, several fragments of various sizes were formed. The enzyme used was confirmed to not cut within the antibiotic resistance marker or any other part of the transposon that would disrupt rescue plasmid replication or sequencing. Given the frequency of SacII sites in the chromosome (on average every 570 bp), ligated rescue plasmids were statistically small and efficient to transform. The fragment containing the transposon and a piece of gene downstream of the sequencing primer binding site was simply ligated end to end to give a rescue plasmid that could be efficiently transformed to E. coli TransforMax™. Naturally, plasmids of several different sizes and religation possibilities are present, but only those with an R6Kγ origin are replicated providing an opportunity to identify a transposon insertion locus. The Himar1 minitransposon system is not only an efficient tool for identifiable random mutagenesis (Fig. 4) but also a means of delivering useful markers or recognition sites into the chromosome.

Distribution of loci of insertion for Himar1 (triangles) and Tn5 (squares; Petzke and Luzhetskyy 2009) transposons in the S. coelicolor chromosome (a) and for Himar1 (rhombs) transposons in the S. albus chromosome (b)
The Himar1 minitransposons were designed with unique integrated loxP and I-SceI sites. Introducing loxP sites into the chromosome generates several possibilities to experiment with multigene recombinations using the Cre recombinase (Fedoryshyn 2008b), while the presence of I-SceI sites provides the possibility to facilitate homologous recombination in a certain locus (Siegl 2010). Transposon delivery vectors are very important during transposon mutagenesis. So far, only replicative vectors were used for introduction of transposons in Streptomyces cells. This has a few serious drawbacks, which reduce efficacy of mutagenesis dramatically. One of such problems is an early transposition effect. This problem arises due to the absence of a tight regulation of transposase gene expression and a low frequency of transposition vector delivery in Strepomyces cells. After the vector was introduced into the cell, the transposons jump into the chromosome even without induction of the transposase gene expression, as even a low level of its expression is sufficient for transposition. In the worst case, the number of individual mutants after mutagenesis will correspond to the number of transposon vector containing clones used in mutagenesis, or all the clones will have several transpositions among which one will be the same in most clones. Another problem arises when two subsequent transposon mutagenesis have to be applied. After the first round of mutagenesis, the vector should be cured in order to introduce the new vector with a new transposon. To avoid this unnecessary step, we have delivered the Himar1 transposon into the cells on the suicide vector pKCLP2. In this case, every clone arising after conjugation is an independent transposon mutant. Using a suicidal vector solves the problem with early transposition events, multiple insertions of transposons, and vector curing.
By Southern blot analysis, the general frequency of insertion into the genome of S. coelicolor M145 for the Himar1 transposon was determined. We observed both multiple and single transposon insertions, depending on the conditions during Himar1 library preparation. Each method has its distinct advantages. Multiple insertions caused by high-frequency transposition present advantages for phenotype screening and discovery. A combination of more than one insertion could give rise to phenotypes like antibiotic overproduction that would otherwise not be seen with a single insertion. On the other hand, the presence of two or more insertions complicates the identification of a targeted DNA locus responsible for the mutant phenotype. Alternatively, where a specific phenotype is desired in order to discover the gene mutation responsible for it, multiple insertions would make it more likely that the applicable gene mutation occurred, reducing the number of colonies that would have to otherwise be screened. Having a single insertion is important for linking a mutant phenotype with the certain targeted gene.
To demonstrate the usefulness of our system, we identified several regulatory proteins involved in actinorhodin and prodigiosin biosynthesis. The inactivation of the first group of genes (SCO3812, SCO4197, SCO498, and SCO4192) leads to the complete abolishment of the actinorhodin production on rich R2YE medium and to activation of its production on minimal medium. Thus, all four corresponding proteins act as activators of antibiotic production if the strain grows on rich media, and as repressors if the strain is grown on minimal medium. Addition of sucrose and glycerol diminished the stimulatory effect of the mutations on actinorhodin production. This shows that the proteins encoded by SCO3812, SCO4197, SCO4198, and SCO4192 are part of the regulatory cascade sensing these two carbon sources.
In summary, we have demonstrated that the modified Himar1 transposon generates random mutants in Streptomycetes with a high efficiency in vivo. The presence of the R6Kγ origin of replication in the transposon enables rapid identification and cloning of transposon insertion sites in rescue plasmids. Due to the rox sites flanking the resistance marker of the transposon, it is possible to remove the marker efficiently from the chromosome and reutilize it in further experiments. To demonstrate the usefulness of our transposon system, we identified four novel regulators responsible for actinorhodin production in S. coelicolor M145. In addition, the mutants were shown to be genetically stable. It is very probable that this transposon will be active in many bacterial strains containing GC-rich DNA (other Actinobacteria, myxobacteria) and will serve as an efficient system for different forward genetic approaches.
References
Bentley SD, Chater KF, Cerdeño-Tárraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. doi:10.1038/417141a
Bierman M, Logan R, O’Brien K, Seno ET, Rao RN, Schoner BE (1992) Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49
Blaesing F, Mühlenweg A, Vierling S, Ziegelin G, Pelzer S, Lanka E (2005) Introduction of DNA into Actinomycetes by bacterial conjugation from E. coli—an evaluation of various transfer systems. J Biotechnol 120:146–161. doi:10.1016/j.jbiotec.2005.06.023
Craig NL (1997) Target site selection in transposition. Annu Rev Biochem 66:437–474. doi:10.1146/annurev.biochem.66.1.437
Damasceno JD, Beverley SM, Tosi LR (2010) A transposon toolkit for gene transfer and mutagenesis in protozoan parasites. Genetica 138:301–311. doi:10.1007/s10709-009-9406-7
Fedoryshyn M, Petzke L, Welle E, Bechthold A, Luzhetskyy A (2008a) Marker removal from actinomycetes genome using Flp recombinase. Gene 419:43–47. doi:10.1016/j.gene.2008.04.011
Fedoryshyn M, Welle E, Bechthold A, Luzhetskyy A (2008b) Functional expression of the Cre recombinase in actinomycetes. Appl Microbiol Biotechnol 78:1065–70. doi:10.1007/s00253-008-1382-9
Fernandez-Martinez LT, Del Sol R, Evans MC, Fielding S, Herron PR, Chandra G, Dyson PJ (2011) A transposon insertion single-gene knockout library and new ordered cosmid library for the model organism Streptomyces coelicolor A3(2). Antonie Van Leeuwenhoek 99:515–522. doi:10.1007/s10482-010-9518-1
Fu J, Wenzel SC, Perlova O, Wang J, Gross F, Tang Z, Yin Y, Stewart F, Müller R, Zhang Y (2008) Efficient transfer of two large secondary metabolite pathway gene clusters into heterologous hosts by transposition. Nucleic Acids Res 36:e113. doi:10.1093/nar/gkn499
Gust B, Challis GL, Fowler K, Kieser T, Chater KF (2003) PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. ProcNatlAcadSci U S A 100:1541–1546. doi:10.1073/pnas.0337542100
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi:10.1016/S0022-2836(83)80284-8
Herrmann S, Siegl T, Luzhetska M, Petzke L, Jilg C, Welle E, Erb A, Leadlay PF, Bechthold A, Luzhetskyy A (2012) Site-specific recombination strategies for engineering actinomycete genomes. Appl Environ Microbiol 78:1804–1812. doi:10.1128/AEM.06054-11
Herron PR, Evans MC, Dyson PJ (1999) Low target site specificity of an IS6100-based mini-transposon, Tn1792, developed for transposon mutagenesis of antibiotic-producing Streptomyces. FEMS Microbiol Lett 171:215–221. doi:10.1111/j.1574-6968.1999.tb13435.x
Ikeda H, Takada Y, Pang CH, Tanaka H, Omura S (1993) Transposon mutagenesis by Tn4560 and applications with avermectin-producing Streptomyces avermitilis. J Bacteriol 175:2077–2082
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. John Innes Foundation, Norwich
Lampe DJ, Churchill ME, Robertson HM (1996) A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J 15:5470–5479
Lampe DJ, Akerley BJ, Rubin EJ, Mekalanos JJ, Robertson HM (1999) Hyperactive transposase mutants of the Himar1 mariner transposon. Proc Natl Acad Sci U S A 96:11428–11433. doi:10.1073/pnas.96.20.11428
Louvel H, Saint Girons I, Picardeau M (2005) Isolation and characterization of FecA- and FeoB-mediated iron acquisition systems of the spirochete Leptospira biflexa by random insertional mutagenesis. J Bacteriol 187:3249–3254. doi:10.1128/JB.187.9.3249-3254.2005
Luzhetskyy A, Fedoryshyn M, Gromyko O, Ostash B, Rebets Y, Bechthold A, Fedorenko V (2006) IncP plasmids are most effective in mediating conjugation between Escherichia coli and streptomycetes. Genetika 42:595–601. doi:10.1134/S1022795406050036
Maier TM, Havig A, Casey M, Nano FE, Frank DW, Zahrt TC (2004) Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl Environ Microbiol 70:7511–7519. doi:10.1128/AEM.70.12.7511-7519.2004
Maier TM, Pechous R, Casey M, Zahrt TC, Frank DW (2006) In vivo Himar1-based transposon mutagenesis of Francisella tularensis. Appl Environ Microbiol 72:1878–1885. doi:10.1128/AEM.72.3.1878-1885.2006
Petzke L, Luzhetskyy A (2009) In vivo Tn5-based transposon mutagenesis of Streptomycetes. ApplMicrobiolBiotechnol 83:979–986. doi:10.1007/s00253-009-2047-z
Prentki P, Krisch HM (1984) In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29:303–313
Rholl DA, Trunck LA, Schweizer HP (2008) In vivo Himar1 transposon mutagenesis of Burkholderia pseudomallei. Appl Environ Microbiol 74:7529–7535. doi:10.1128/AEM.01973-08
Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ (1999) In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc Natl Acad Sci U S A 96:1645–1650. doi:10.1073/pnas.96.4.1645
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Siegl T, Petzke L, Welle E, Luzhetskyy A (2010) I-SceI endonuclease: a new tool for DNA repair studies and genetic manipulations in streptomycetes. Appl Microbiol Biotechnol 87:1525–1532. doi:10.1007/s00253-010-2643-y
Weaden J, Dyson P (1998) Transposon mutagenesis with IS6100 in the avermectin-producer Streptomyces avermitilis. Microbiology 144:1963–1970. doi:: 10.1099/00221287-144-7-1963
Acknowledgments
This work was supported by a DFG grant (Lu1524/2-1) to AL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Bohdan Bilyk and Stephen Weber contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 232 kb)
Rights and permissions
About this article
Cite this article
Bilyk, B., Weber, S., Myronovskyi, M. et al. In vivo random mutagenesis of streptomycetes using mariner-based transposon Himar1 . Appl Microbiol Biotechnol 97, 351–359 (2013). https://doi.org/10.1007/s00253-012-4550-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4550-x