
Abstract
Immunotoxins are rationally designed cancer targeting and killing agents. Disulfide stabilized antibody Fv portion—toxin conjugates (dsFv-toxin) are third generation immunotoxins containing only the antibody fragment variable portions and a toxin fused to the VH or VL. Pseudomonas exotoxin fragment (PE-38) is a commonly used toxin in immunotoxin clinical trials. dsFv-toxin purification was previously published, but the recovery was not satisfactory. This report describes the development of a cGMP production process of the dsFv-toxin that incorporated a novel purification method. The method has been successfully applied to the clinical manufacturing of two dsFv-PE38 immunotoxins, MR1-1 targeting EGFRvIII and HA22 targeting CD22. The two subunits, VL and VH PE-38 were expressed separately in Escherichia coli using recombinant technology. Following cell lysis, inclusion bodies were isolated from the biomass harvested from fermentation in animal source component-free media. The dsFv-toxin was formed after denaturation and refolding, and subsequently purified to homogeneity through ammonium sulfate precipitation, hydrophobic interaction and ion-exchange chromatography steps. It was shown, in a direct comparison experiment using MR1-1 as model protein, that the recovery from the new purification method was improved three times over that from previously published method. The improved recovery was also demonstrated during the clinical production of two dsFv-PE38 immunotoxins—MR1-1 and HA22.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The technology of immunotargeting and immunotherapy of tumors using specific antibodies was developed in the 1980s (Bird and Walker 1991). Immunoglobulin fragments generated either by enzymatic cleavage or by genetic engineering contain only the antigen recognition variable region of a normal antibody. Lack of the constant region (Fragment, crystalizable, or Fc) eliminates Fc-mediated binding, reduces immunogenicity, improves the rate of tumor penetration, and promotes rapid clearance from normal tissue (Reisfeld and Gillies 1996). In these antibody fragments, variable regions of the heavy and light chains (VH and VL) are linked through either disulfide bond (dsFv) or peptide linker (scFv) (Reiter et al. 1994; Huston et al. 1988; Bird et al. 1988; Pastan et al. 2006). Despite their intrinsic lower affinity and rapid clearance, antibody variable fragments were shown to have excellent selectivity with high tumor/normal tissue binding ratios. These unique characteristics provide great potentials for scFv and dsFv to be used as specific tumor cells targeting drug (Yokata et al. 1992; Bird and Walker 1991).
Antibody-toxin conjugates (immunotoxin) were developed to target and kill cancer cells efficiently by taking advantage of the antibody’s specificity and toxin’s cyto-toxicity (FitzGerald et al. 2011). Pseudomonas exotoxin (PE) secreted by Pseudomonas aeruginosa has been coupled to a variety of monoclonal antibodies (Kuan and Pastan 1996; Reiter 2001) and shown to inhibit protein synthesis by catalyzing the ADP-ribosylation of elongation factor 2 (Iglewski and Kabat 1975). PE conjugated immunotoxin is 5- to 5,000-fold more cytotoxic than other toxin conjugates such as ricin A chain (Pirker et al. 1985; Bjorn et al. 1986). The cell binding domain of PE is removed by genetic engineering in order to reduce toxicity to normal cells. Various PE immunotoxins including anti-IL-2 Tac-PE38 (LMB-2, anti-CD25), anti-CD22-PE38 (BL22, CAT-3888; HA22, CAT-8015), anti-LeY PE (LMB-1), transferrin-CRM107, anti-mesothelin variable fragment (Fv)-PE38 (SS1P), and IL-4 (38-37)-PE38KDEL have been evaluated and shown in vivo anti-tumor activity in animal models (FitzGerald et al. 1988; Frankel et al. 2000; Kreitman et al. 2009; Pai et al. 1996; Zhang et al. 2011). Immunotoxins have been involved in many ongoing or completed clinical trials (Frankel et al. 2000; Pastan et al. 2006; Kreitman 2006; Kreitman et al. 2009; Wayne et al. 2010; Kreitman et al. 2012).
Despite the promising results from preclinical and clinical studies with immunotoxins, several problems have impeded the wider application of immunotoxins in cancer therapy. The problems include lack of high affinity, heterogeneity of antigen expression on tumor cell surfaces, and difficulties in immnuotoxin production. Progress has been made in improving immunotoxin affinity and activity (Frankel 2002; Pastan et al. 2006). Hot spot mutagenesis and phage display have been applied to identify high-affinity, adequately tumor-selective ligands. Critical tumor cell proteins such as oncogene products are targeted to generate immunotoxin. Although recombinant immunotoxins may be expressed as secreted protein in the periplasm or harvested from cytoplasm of E. coli, better yields of purified immunotoxins may be achieved by recovery of recombinant protein in soluble inclusion bodies (Kreitman et al. 2009). The isolation and purification methods for immunotoxins were developed previously for several immunotoxins in clinical trials, such as LMB-2 (Kreitman et al. 2000; Batra et al. 1990; Chaudhary et al. 1989) and BL22 (Kreitman et al. 2001; Mansfield et al. 1997a, 1997b). The published production methods are similar for scFv-PE38 (LMB-2) and dsFv-PE38 (BL22). Basically, the immunotoxins are expressed in E. coli as inclusion bodies, an insoluble form of protein aggregation. The proteins are solubilized with a denaturing buffer (urea or guanidine–HCl). The refolding occurs in a redox environment in order to promote correct disulfide bond formation. Following refolding, two ion exchange and one size exclusion chromatography steps are applied to the immunotoxin purification (Batra et al. 1990; Buchner et al. 1992). The overall yield of the entire process was low, for instance 2 % in one case (Lorimer et al. 1996), and production cost was high.
MR1-1 is a recombinant disulfide bond stabilized single-chain antibody-immunotoxin that binds specifically to a mutant of the epidermal growth factor receptor, EGFRvIII, found on tumor cells including glioblastomas, breast carcinomas, and others (Wikstrand et al. 1995; Lorimer et al. 1995). EGFRvIII contains an in-frame deletion of NH2-terminal amino acid residues 6-273 from the extracellular domain of EGFR that results in a 145-KD molecule with a unique primary sequence characterized by an inserted glycine residue at the fusion junction (Humphrey et al. 1988; Yamazaki et al. 1990; Wikstrand et al. 1998). MR1-1 was selected from a phage display library derived from the parent antibody-MR1 by random complementarity determining region mutagenesis (Lorimer et al. 1996; Beers et al. 2000). MR1-1 binds to EGFRvIII with higher affinity than does the parent antibody MR1. The VH in MR1-1 is fused to a truncated mutant form of Pseudomonas exotoxin A (PE38KDEL) that was modified to remove the natural cell-binding domain. VH-PE38 is linked with the VL variable region via disulfide bonding. In vitro and in vivo (animal model) studies showed that anti-EGFRvIII toxin possessed excellent tumor retention and rapid clearance from normal tissues, which makes MR1-1 as a potential immunotherapeutic agent (Kuan et al. 1999; Archer et al. 1999; Wikstrand et al. 1999; Kuan et al. 2000).
RFB4 is a murine antibody specifically targeting CD22, a B lineage antigen strongly expressed on hairy cell leukemia (HCL) cells and less strongly on chronic lymphocytic leukemia (CLL) cells. The variable domains of RFB4 were used in the construction of dsFv-PE38 immunotoxin BL22, currently in clinical trials for treatment of HCL, CLL, and pediatric acute lymphoblastic leukemia. Three amino acid changes were introduced into BL22 VH portion to create a new immunotoxin HA22 that has 10-fold higher affinity to CD22 and 10- to 100-fold higher potency against CLL (Bang et al. 2005). Similarly to MR1-1 and BL22, HA22 is a heterodimer of VL and VH-PE38 linked by a disulfide bond.
GLP and cGMP manufacturing of immunotoxin at large scale to support toxicology studies and clinical trials has been a challenging task due to its low production yield and high cost. In the conventional process for dsFv-toxin production, the VH and VL are expressed separately in the form of inclusion bodies in E. coli. The inclusion bodies are solubilized with strong denaturant and subsequently linked via disulfide bond during a refolding process under optimal redox conditions. L-arginine is added to suppress incorrect polypeptide interactions leading to aggregation (Buchner et al. 1992). Nonetheless, the overall refolding efficiency is low and a large portion of VH and VL are not correctly folded. Often times VH molecules form homodimers that are difficult to be separated from VH–VL heterodimers due to their similar physiochemical properties. The refolded immunotoxin is purified through two consecutive strong anion-exchange chromatographic steps and a size exclusion chromatographic step (Mansfield et al. 1997a, b; Buchner et al. 1992; Kuan et al. 2000). MR1-1 laboratory-scale production and BL22 clinical manufacturing followed this conventional method (Kreitman et al. 1999). While both immunotoxins were successfully made and used in preclinical and clinical applications, overall production yields were low.
In this report, we demonstrated a new recovery and purification method that utilized ammonium sulfate precipitation followed by column chromatography. The new method was successfully applied in clinical manufacturing of MR1-1 and HA22, and the improvement on yield was confirmed in cGMP production.
Materials and methods
Fermentation of MR1-1 and HA22 heavy and light chain (VH and VL) and inclusion bodies preparation
Plasmids (pRB1399 and pRB1499) expressing MR1-1 heavy chain and light chain are described previously (Brinkmann et al. 1991; Kuan et al. 2000). Plasmids (pRB902 and pRB698) expressing HA22 heavy chain and light chain were provided by Dr. Ira Pastan. E. coli BL21(DE3) cells were transformed with the plasmids individually and Master Cell Banks that were qualified for clinical manufacturing were prepared. Fermentation conditions were the same for MR1-1 and HA22 VH and VL, except for induction time. Cells were grown in modified superbroth (12 g soytone, 24 g yeast extract, 5 ml glycerol, 3.8 g KH2PO4, and 12.5 g K2HPO4 per liter) containing 2 % glucose (w/v), 0.05 % MgSO4 (w/v), 20 μg/ml chloramphenicol, and 30 μg/ml Kanamycin. An overnight culture was used to inoculate the fermentor (New Brunswick Scientific) at 5 %. MR1-1 VH and VL expression was induced with 1 mM IPTG (isopropyl β-d-thiogalactoside) for 2 h when the optical density at 600 nm (OD600) reached 6 + 2. HA22 VH and VL expression was induced when OD600 reached 6 + 2 with 1 mM IPTG for 3 h. Cells were harvested by centrifugation and the paste was kept at −80 °C if not immediately subjected to lysis. Cells were resuspended in TES buffer (50 mM Tris–HCl, pH 7.4, 20 mM EDTA, 100 mM NaCl) at 16 ml/per gram (wet weight) cell paste ratio. Cells were lysed with a pressure homogenizer (Gaulin) followed by centrifugation at 17,696×g for 50 min. The pellet (inclusion bodies) was washed extensively by tangential flow filtration using 0.2 μm MiniKros Membrane Module (Spectrum) with TE buffer (50 mM Tris–HCl, pH 7.4, 20 mM EDTA) containing 2.5 % Triton X-100 followed by washing with the same buffer without Triton X-100. Inclusion bodies were kept at −80 °C.
Inclusion bodies solubilization and protein refolding
The refolding procedure follows a previous protocol (Buchner et al. 1992) with modification. Briefly, the VH and VL inclusion bodies were dissolved separately in solubilization buffer (6 M Guanidine–HCl, 0.1 M Tris–HCl, pH 8.0, 2 mM EDTA) at 10 mg/ml protein concentration. Proteins were reduced by the addition of DTE (dithioerythritol, Sigma) to 10 mg/ml final concentration and incubated for 8 h at room temperature with gentle shaking. Reduced VH and VL solution were then mixed at 1:1 molar ratio. The mixture was slowly diluted 100-fold into refolding buffer (0.1 M Tris, 0.5 M L-arginine–HCl, 0.9 mM oxidized form of glutathione, 2 mM EDTA; pH 10.2) with adequate mixing. The refolding reaction was kept at 4 °C for 36–48 h without agitation. After neutralization with 6 M HCl to pH 7.5, the refolded protein was concentrated 10 times and diafiltered to 20 mM Tris–HCl, pH 7.4, 100 mM urea by tangential flow filtration using a 30-KDa molecular weight cut off (MWCO) membrane (Millipore Pellicon 2).
Purification of MR1-1 by conventional consecutive anion-exchange chromatography and size exclusion chromatography
Refolded protein solution was loaded onto a Q-Sepharose Fast Flow (GE HealthCare) column equilibrated with buffer A (20 mM Tris, pH 7.4, 1 mM EDTA). After sufficient washing with 0.1 M NaCl in Buffer A, proteins were eluted with a gradient from 0.1 to 0.3 M NaCl in Buffer A. The main peak was diluted five times with buffer A and loaded onto a Source 15 Q (GE HealthCare) column. The protein was eluted by a 30 column volume (CV) NaCl gradient (0.1–0.3 M) in buffer A. The product-containing fractions were pooled based on SDS-PAGE analysis. After concentration by tangential flow filtration (TFF), the protein was loaded to a Superdex S200 (GE HealthCare) column pre-equilibrated with phosphate-buffered saline (PBS). Loading volume was less than 2 % of the column volume and the flow rate was between 10 and 30 cm/h. MR1-1 purified bulk product was sterile filtered through a 0.2-μm membrane.
Purification of MR1-1 and HA22 by ammonium sulfate precipitation, hydrophobic interaction chromatography, and anion exchange chromatography
Ammonium sulfate precipitation
For both MR1-1 and HA22, concentrated ammonium sulfate solution was added to the refolded protein solution to a final concentration of 1.25 (for MR1-1) or 1.0 M (for HA22) (NH4)2SO4. The solution was kept at 4 °C for >0.5 h. After centrifugation at 15,000×g, MR1-1 protein was recovered in the supernatant. Concentrated ammonium sulfate solution was then added to the supernatant to 2 M to precipitate the MR1-1 and HA22. The product containing pellets were separated from supernatant by centrifugation for long-term storage. Prior to the Phenyl HP column the protein precipitate was dissolved in 1.25 M (for MR1-1) or 1.0 M (for HA22) (NH4)2SO4 solution.
Phenyl HP hydrophobic interaction chromatography
For MR1-1, the protein solution in 1.25 M (NH4)2SO4 was adjusted to pH 8.0 and loaded onto a Phenyl HP (GE HealthCare) column at a ratio of 3 mg/ml column volume. The column was washed by 5–10 CV of 20 mM Tris buffer (pH 8.0) containing 1.25 M (NH4)2SO4, and eluted with a 20-CV gradient from 1.25 M to 0 M (NH4)2SO4 in 20 mM Tris buffer (pH 8.0). The fractions containing MR1-1 were pooled based on SDS-PAGE analysis. For HA22, the protein solution in 1.0 M (NH4)2SO4 was adjusted to pH 8.0 and loaded onto a Phenyl HP column at a ratio of 3 mg/ml column volume. The column was washed by 5–10 CV of 20 mM Tris buffer (pH 8.0) containing 1.0 M (NH4)2SO4, and eluted with a 20-CV gradient from 1.25 M to 0 M (NH4)2SO4 in 20 mM Tris buffer (pH 8.0). The fractions containing HA22 were pooled based on SDS-PAGE analysis.
Q Sepharose Fast Flow chromatography
For both MR1-1 and HA22, Phenyl HP pool was diluted 10 times with 20 mM Tris pH 7.4 buffer, and loaded onto a Q Sepharose Fast Flow column. After washing with 10 CV of washing buffer (20 mM Tris, pH 7.4, 0.1 M NaCl), target protein was eluted with 20 mM Tris buffer (pH 7.4) containing 0.3 M NaCl. The purified protein was concentrated and diafiltered against PBS pH 7.4 buffer, and sterile filtered through a 0.2-μm membrane.
Protein concentration, purity, MR1-1 activity assay, and the endotoxin assays
The protein concentration was determined by Coomassie plus assay (Pierce). The MR1-1 and HA22 purity was determined by high-performance liquid chromatography (HPLC) size exclusion chromatography. The MR1-1 activity was evaluated by in vitro cytotoxicity assay described previously (Lorimer et al. 1996). Endotoxin level was determined using a Kinetic-QCL Limulus Amebocyte Lysate assay kit (Lonza).
Results
Cell growth and expression of MR1-1 VH-toxin and VL in BL21(DE3) cells
E. coli BL21 (DE3) containing VH or VL was cultured in batch-process fermentation with soytone-based modified superbroth plus 2 % glucose, 0.05 % MgSO4, and appropriate antibiotics. IPTG induction conditions were evaluated and results showed that 1 mM IPTG induction for 2–3 h was optimal (data not shown) for both light and heavy chain expressions. The culture media contained sufficient nutrients to promote cell growth to a final OD600 of 15–25. During the fermentation, the pH was monitored but not actively controlled. In a typical run, the pH dropped from 7.0 to 6.8 due to the accumulation of organic acid production during early growth phase. The slight pH shift did not impede the cell growth. Dissolved oxygen was maintained at 30 % by adjusting the agitation and aeration rate. No oxygen supplementation was necessary during the fermentation. Samples were taken every hour for OD600 measurement. A typical fermentation during clinical manufacturing took about 3 + 0.5 h to reach OD600 6 + 2 to begin induction. A representative VL fermentation profile at 80 L scale is shown as Fig. 1. At an 80 L production scale, 57 L of production medium was inoculated with 6.7 % of overnight cultured seed. IPTG addition occurred after 3 h and the fermentation was continued for 2.5 h post induction. The final OD was 25.9.

MR1-1 VL fermentation profile. A representative 80 L MR1-1 VL fermentation profile is shown. The X-axis represents elapsed fermentation time (minutes), The Y-axis represents agitation (rpm), temperature (°C), DO (%), pH, CO2 (%), and O2 (slpm), OD600 values with different colors
Fermentation biomass was harvested by centrifugation and the cell pellets were stored at −80 °C. The expressions were analyzed by SDS-PAGE with Coomassie blue staining (Fig. 2). VL with an apparent molecular weight of 9 KDa was expressed upon induction for 1 and 2 h (Fig. 2, lanes 4 and 5; lane 3 before induction). VH with an apparent molecular weight of 54 KDa was expressed in responding to IPTG induction for 1 and 2 h (Fig. 2, lanes 7 and 8; lane 6 before induction). End of production cells were directly sequenced to confirm that they carried the respective VH or VL plasmid 100 % homologous to the reference sequence. The entire fermentation process was designed to be cGMP compliant.

MR1-1 VL and VH expression in E. coli. BL21(DE3) cells containing VH-PE38 or VL were grown in fermentor until OD600 reached around 6–8; 1 mM IPTG was added to induce proteins for 2 h. Samples were taken at different time points (before induction, 1 and 2 h post induction). Equal amount of cells at each time point were subjected to SDS-PAGE analysis (4–12 % NuPAGE) after reduced by dithiotreitol and heated for 10 min on a 70 °C heating block. The gel was stained with Coomassie blue. Lane 1 is the molecular weight standards (Invitrogen). Lane 2 is MR1-1 protein standard (VH ∼ 54 KDa; VL ∼ 9 KDa). Lanes 3 to 5 VL samples at before induction, 1 h post induction and 2 h post induction. Lanes 6 to 8 VH samples at before induction, 1 h post induction, and 2 h post induction
MR1-1 recovery, inclusion bodies preparation, and protein refolding
VH or VL cells were lysed by pressure homogenization as described in “Materials and methods”. The inclusion bodies were isolated and dissolved in strong denaturant. The MR1-1 refolding process was carried out at different scales. In a representative clinical manufacturing run, 190 g of each of VH and VL cell paste (wet weight) was lysed. Inclusion bodies isolated by centrifugation were resuspended in 1 L of TE buffer containing 2.5 % Triton X-100, and washed with 6 L of the same buffer followed by 10 L of TE buffer using a MiniKros Tangential Flow Membrane Module (0.2 μm, 1.0 ft2). Inclusion bodies were solubilized in 300 ml solubilizing buffer and yielded 5.0 g of VH and 5.1 g of VL in protein solutions. The purity of both VH and VL inclusions were estimated by SDS-PAGE density analysis (Figs. 4 and 5, lanes 4 and 5) to be around 50 and 25 % pure, respectively. Both VH and VL were diluted to 10 mg/ml in solubilization buffer; 266.7 ml of VH and 133.3 ml of VL (1:1 molar ratio) were reduced by addition of DTE and gently mixed. The mixture was then slowly diluted into 40 L of refolding buffer while mixed with an overhead mixer. After being kept at 4 °C for 40 h, the solution was neutralized to pH 7.5 with 6 N HCl, concentrated to 4 L, and subsequently diafiltered against 22.5 L of dialysis buffer by 30 KDa MWCO TFF. The diafiltered solution was centrifuged and 0.45 μm filtered to remove precipitated material; 2.67 g of protein in 3.6 L solution was recovered at this stage. Results from SDS-PAGE showed significant refolding efficiency. Among the impurities there were a substantial amount of VH monomers. Some VH dimers and aggregates were also observed (Figs. 4 and 5, lane 6). The refolded MR1-1 protein was 47.2 % pure based on HPLC–size exclusion chromatography (HPLC-SEC) analysis.
Evaluating ammonium sulfate precipitation and hydrophobic chromatography for MR1-1 purification
The conventional purification method (two anion-exchange columns followed by size exclusion chromatography) was used for large-scale purification of immunotoxins previously (Batra et al. 1990; Buchner et al. 1992; Mansfield et al. 1997a and 1997b). When the method was applied to MR1-1 at laboratory scale, it was found that the production yield was relative low (Kuan et al. 1999). To improve the method, several protein purification procedures including ammonium sulfate precipitation and hydrophobic interaction chromatography (HIC) were evaluated.
Ammonium sulfate was used to increase proteins’ hydrophobic binding to the HIC column. Ammonium sulfate is also known to cause protein “salting out,” so its effect on MR1-1 and impurity separation was examined. Various concentrations of ammonium sulfate were tested and results showed that MR1-1 remained soluble at the ammonium sulfate concentration below 1.5 M. Further increase of the salt concentration precipitated MR1-1 protein. At (NH4)2SO4 concentrations from 1.0 to 1.5, a significant amount of impurities were precipitated and could be separated away. MR1-1 could be completely precipitated at 2 M (NH4)2SO4 and the paste kept frozen for long-term storage. The precipitated protein was readily dissolved in a solution with less than 1.5 M (NH4)2SO4. MR1-1 cytotoxicity remained unchanged during the precipitation and dissolution process. The protein was also stable in the form of precipitation pellet during long-term frozen storage. This precipitation step provided a convenient and economical way for in-process hold and storage.
Several hydrophobic interaction chromatographic resins, including ether, butyl, and phenyl, were screened. The columns were loaded at 1 M (NH4)2SO4 at pH 7.4 and eluted with a linear gradient of (NH4)2SO4 from 1.0 to 0 M at pH 7.4 over 20 column volumes at 150 cm/h. Phenyl HP (GE HealthCare) provided the best separation of MR1-1 from other impurities. Separation at pH 7.0, 7.4, 8.0, and 8.5 were evaluated and pH 8.0 was found to be optimal (data not shown). Three (NH4)2SO4 concentrations, 1.0, 1.25, and 1.5 M, were evaluated for MR1-1 binding. Three times higher binding capacity of MR1-1 to Phenyl HP was observed when ammonium sulfate concentration was increased from 1 to 1.25 M. No further improvement was found at 1.5 M. No notable degradation of MR1-1 protein was observed in this solution for up to 1 week at 4 °C. Hence the MR1-1 protein was incubated in 1.25 M ammonium sulfate followed by centrifugation before loaded onto a Phenyl HP column.
A small-scale Phenyl HP chromatogram is shown in Fig. 3a. MR1-1 in 1.25 M ammonium sulfate was centrifuged at 10,000×g for 30 min. The supernatant containing 15 mg protein was adjusted to pH 8.0 and loaded to a 5 ml Phenyl HP (1.6 × 2.5 cm). The column was washed with1.25 M ammonium sulfate in buffer 20 mM Tris, pH 8.0 and eluted with a gradient from 1.25 M to 0 M ammonium sulfate in 20 mM Tris, pH 8.0. Two major peaks were eluted off the column. The first peak was MR1-1 (Fig. 3b lanes 3 to 5), and the second contained free heavy chain (VH) and heavy chain dimer (VH–VH, Fig. 3b lanes 6–10).

Intact MR1-1 (VH − VL) and VH (monomer and dimer) separation by Phenyl HP chromatography. a Phenyl HP chromatogram. Refolded MR1-1 solution, containing 15 mg of total protein in 1.25 M ammonium sulfate precipitation, was loaded to a 5-mL (1.6 × 2.5 cm) Phenyl HP column (from 0 to 30 min shown in the figure). The column was washed with 10 CV of 1.25 M ammonium sulfate in 20 mM Tris, pH 8.0 (30–40 min in the figure), and eluted with a 20 CV gradient (40–70 min) from 1.25 M (100 % B in the figure) to 0 M ammonium sulfate (0 % B in the figure). The gradient is shown in red. Absorbance at 280 nm is shown in blue. Fraction marks are shown in green. Peak 1 contained MR1-1 and peak 2 had mostly VH monomer and dimer. b SDS-PAGE showing separation of MR1-1 from impurities by Phenyl HP. Samples of Phenyl HP column fractions were taken and analyzed by non-reducing SDS-PAGE. Fraction numbers was shown below the lane number. Intact MR1-1 was eluted from the column (lanes 3-5) well separated from impurities that mainly were VH monomer and dimer
The Phenyl column provided a simple and efficient way to purify MR1-1. The product-containing peak could be easily collected when the effluent was monitored by UV absorbance at 280 nm. The product purity could be confirmed by SDS-PAGE analysis. Additionally, the step recovery was high because of the clean separation of MR1-1 from other impurities.
Comparison of MR1-1 purification methods at laboratory scale
The previously published MR1-1 purification method, similar to that of conventional dsFv-toxin, consists of two consecutive anion exchange steps and a size exclusion step. In the new method, we introduced an ammonium sulfate precipitation and a hydrophobic interaction step, and eliminated size exclusion and one anion exchange step. To compare the two methods, multiple purifications were performed from the same refolded MR1-1 starting material. The conventional method was tested with slight modification by substituting Source 15Q for Mono Q; 1.8 L (1.3 g total protein, 47.2 % purity, or 0.61 g of MR1-1) of refolded MR1-1 solution was loaded onto a Fast Flow Q column (5.0 × 14 cm, 275 ml) equilibrated with buffer A (20 mM Tris, pH 7.4, 1 mM EDTA). The column was washed with 5 CV of 0.1 M NaCl in buffer A, and eluted with a 10-CV gradient from 0.1 to 0.3 M NaCl in buffer A. Fractions containing MR1-1 were pooled and a total of 159 mg of protein was recovered (Fig. 4, lane 7). The pool was diluted five times in buffer A and loaded onto a Source 15Q (3.5 × 4.7 cm, 45 ml) column. Protein was eluted with a 30-CV gradient from 0.1 to 0.25 M NaCl in buffer A. A total of 70.8 mg protein was recovered in the fraction pool (Fig. 4, lane 8). The pool was concentrated and diafiltered against PBS (Fig. 4, lane 9) by tangential flow filtration, and subjected to Superdex S200 chromatography (2.6 × 62 cm, 329 ml). Fractions containing purified MR1-1 were pooled and sterile filtered. The purified bulk contained 46.3 mg of MR1-1. Purity was 98.7 % as determined by HPLC size exclusion chromatography analysis. The bulk was also analyzed by SDS-PAGE. A trace amount of VH-toxin impurity was observed on SDS-PAGE (Fig. 4, lane 10) that represented less than 2 % of the total protein by densitometry. MR1-1 and VH-toxin monomer were similar in size and charge, and thus similar chromatographic behaviors on ion-exchange and size exclusion columns. The two proteins eluted very close to each other in all three purification steps. SDS-PAGE analysis was required before fraction pooling at each step to ensure the VH-toxin monomer was separated away from the product. Stringent pooling had to be enforced and resulted in overall low recovery. Out of 610 mg of refolded MR1-1 in the starting material, only 46.3 mg of MR1-1 was recovered, representing a 7.6 % overall purification recovery, which is much higher than 2 % that observed by Lorimer et al. (1996), and within the range of 5–20 % reported by Kreitman (2009). Most of the losses were attributed to the poor separation of MR1-1 and VH-toxin monomer on the ion-exchange and size exclusion columns.

MR1-1 purification by conventional method: Fast Flow Q Sepharose, Source 15 Q, and size exclusion chromatography. Samples were taken from each step in the entire process including expression, refolding, and purification by conventional method and analyzed by SDS-PAGE. Lane 1 molecular weight standards; lane 2 VH whole cell lysate; lane 3 VL whole cell lysate; lane 4 VH inclusion bodies; Lane 5 VL inclusion bodies; lane 6 post refolding; lane 7 Q fast flow eluate; lane 8 Source 15 Q eluate; lane 9 post concentration and diafiltration by TFF; lane 10 size exclusion chromatography fraction pool; 5 μg of protein were loaded from lanes 4 to 10. Samples in lanes 2 to 5 were reduced, 6–10 were non-reduced
The new purification method was tested at the same scale with the same starting material. As described previously the method incorporated ammonium sulfate precipitation and hydrophobic interaction chromatography; 1.8 L refolded MR1-1 (1.30 g total protein with 47.2 % purity, or 0.61 g of MR1-1) was mixed with 1 L of 3.5 M ammonium sulfate resulting a final concentration of 1.25 M (NH4)2SO4. After centrifugation at 15,000×g for 1 h, the pellet was discarded and the supernatant, containing 465 mg of total protein, was loaded onto a Phenyl HP column (5 × 9 cm, 177 ml) pre-equilibrated with 1.25 M ammonium sulfate in 20 mM Tris buffer pH 8.0. After loading, column was washed with 10 CV of the same buffer, and eluted with a gradient from 1.25 M to 0 M (NH4)2SO4 in 20 mM Tris pH 8.0. The first peak that was collected contained 146.9 mg MR1-1 (Fig. 5 lane 8). Phenyl HP eluate was concentrated and diafiltered by TFF against 20 mM Tris pH 7.4 before loading onto a QFF column (2x10cm, 31 ml). The QFF column was washed with 10 CV of 100 mM NaCl in 20 mM Tris, pH 7.4, and eluted with 250 mM NaCl in 20 mM Tris pH 7.4. MR1-1 was collected as a single fraction; 129.1 mg of MR1-1 was recovered and the purity was 97.9 % by analytical HPLC size exclusion chromatography. The endotoxin level was further reduced from 1.26 to 0.34 EU/mg. The purification recovery was 21 %, nearly a 3-fold improvement over the conventional method.

MR1-1 purification by the new method: ammonium sulfate precipitation, Phenyl HP hydrophobic, and Fast Flow Q chromatography. Samples were taken from each step in the entire process including expression, refolding, and purification by the new method and analyzed by SDS-PAGE. Lane 1 molecular weight standards; lane 2 VH whole cell lysate; lane 3 VL whole cell lysate; lane 4 VH inclusion bodies; lane 5 VL inclusion bodies; lane 6 post refolding; lane 7 ammonium sulfate precipitation; lane 8 Phenyl HP eluate; lane 9 Q Fast Flow eluate, lane 10 post concentration and diafiltration by TFF; 5 μg of protein were loaded from lanes 4 to 10. Samples in lanes 2 to 5 were reduced, 6–10 were non-reduced
A direct comparison of the two methods in recovery, purity, and activity was listed in Table 1. Both products were tested by in vitro cytotoxicity assay and shown equally active. Both products exceeded 95 % purity. Endotoxin levels were below 0.5 EU/mg of protein. No microbial content was detected. While both methods produced quality product, the new method yielded almost three times more product. Additionally, the HIC provided such powerful product separation that one IEX column step and one SEC chromatograph were eliminated in the new method as compared to the previous method. The ammonium sulfate precipitation served as a holding point to isolate unit operations, as well as preliminary step to separate large amount of impurities. The significant yield increase and more streamlined process the new method brings substantial reduction in time and cost during clinical manufacturing.
Fermentation, refolding, and purification of HA22 for clinical development
Fermentations of HA22 heavy and light chain were conducted at 20-L scale. Fermentor (New Brunswick) batched with 12 L modified Superbroth with glucose and antibiotics was inoculated with the seed (5 % inoculum). The VL expression was induced by adding 1 mM IPTG at OD600 6.72. The fermentation was then allowed to continue for 3 h and the cell density reached OD600 of 15.2. Two hundred sixty-three grams of cells were harvested by centrifugation. The VH expression was induced by adding 1 mM IPTG at OD600 6.4. The fermentation was then allowed to continue for 3 h and the cell density reached OD60010. One hundred forty-three grams of cells were harvested by centrifugation. All cell pastes were stored in a ≤−70 °C freezer.
To prepare inclusion bodies for refolding, VL or VH cells were lysed by pressure homogenization as described in “Materials and methods”. Thirty-three grams of VL and 97.5 g of VH inclusion bodies were obtained and stored at ≤−70 °C.
For HA22 refolding, VL and VH inclusion bodies were each solubilized in 6 M guanidine–HCl, 100 mM Tris, pH 8, 2 mM EDTA to concentrations of 9.3 and 8.6 mg/mL, respectively. Both were then filtered through 0.45-μm (Millipore Millipak) filters. DTE (10 mg/mL) was added to each filtrate and they were continuously and gently stirred overnight at 2–8 °C. The following morning 400 mL of VL and 600 mL of VH solution were mixed gently. The mixture was then diluted rapidly 100-fold by mixing with 100 L of 100 mM Tris, pH 10.2, 2 mM EDTA, 500 mM arginine, and 0.9 mM oxidized glutathione. The mixing step was performed by pumping the two solutions together through a 1/2″ ID, 15-element SS static mixer at a 1:100 flow ratio. The mixed solution was stored at 2–8 °C for ∼46 h. The pH of the mixture was then adjusted to 7.54 with 6 M HCl. The pH-adjusted mixture was filtered through a 0.2-μm filter (Sartorius Sartobran PH), and then concentrated to 12 L using a 30-kD, 10 ft2 spiral TFF filter (Millipore). The concentrate was subjected to constant-volume diafiltration with 20 mM Tris, pH 7.4, 100 mM urea. The diafiltered retentate was stored at 2–8 °C for 3 days. Solid (NH4)2SO4 was then slowly added to the solution under constant agitation to obtain a concentration of 1.0 M, after which the solution was centrifuged at 12,300×g (average) for 30 min. The captured precipitate was discarded. Additional (NH4)2SO4 was added to the supernatant to increase the concentration to 2 M and the solution was re-centrifuged as described above to obtain 16 g of precipitate. This precipitate containing refolded HA22 was stored at ≤−70 °C.
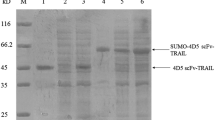
Purification of HA22 followed the process described in “Materials and methods”; 15.6 g of refolded HA22 ammonium sulfate precipitate was dissolved using a stir bar in a buffer containing 20 mM Tris–HCl, pH 8.0, 1 M ammonium sulfate. The dissolved protein solution was filtered with a 0.22-μm filter, equally divided into two parts, and subjected to the two-column back to back purification process. The elution fractions from the two Phenyl HP column (249 mL bed volume, 12.7 × 5 cm (h × ID)) runs were analyzed by non-reducing SDS-PAGE to determine the fraction pooling. The pooled HA22 solution was diluted with 20 mM Tris–HCl pH 7.4 buffer, equally divided into two parts, and loaded onto the QFF column (127 mL bed volume, 6.5 × 5 cm (h × ID)), back to back. The elution fractions from the two QFF column runs were analyzed by SDS-PAGE to determine the fraction pooling. The final pool contained 576 mg of purified HA22 as determined by Coomassie Plus assay. The purified protein was then processed by ultrafiltration and diafiltration to become the bulk drug substance for animal toxicology study (Fig. 6). The product was 99.5 % pure by HPLC-SEC and met all specifications for toxicology grade HA22.

HA22 purification by the new method: ammonium sulfate precipitation, Phenyl HP hydrophobic, and Fast Flow Q chromatography. Samples were taken from each step in the purification of HA22 toxicology lot and analyzed by SDS-PAGE. Lane 1 molecular weight standards; lane 2 Phenyl HP column load; lanes 3 and 4 Phenyl HP fraction pool; lane 5 Q Fast Flow fraction pool; lane 6 post concentration and diafiltration by TFF; lane 7 post 0.2 μm sterile filtration; 5 μg of protein were loaded in lane 2; 3 μg of protein were loaded from lanes 3 through 7. All samples were non-reduced
The process was scaled up to 80-L fermentation. Three manufacturing lots were produced to supply the materials for clinical trials. After inclusion body recovery and refolding, HA22 was purified through Phenyl HP and QFF chromatography and formulated by ultrafiltration and diafiltration. Each manufacturing lot yielded approximately 1–1.5 g of clinical grade HA22, with a recovery of 25–30 % from the purification process.
MR1-1 cGMP manufacturing
A MR1-1 cGMP manufacturing campaign was conducted successfully following the new purification method. Both light chain and heavy chain fermentation were conducted at 80-L scale. The expression was induced when the OD600 was greater than 4, with 1 mM IPTG for 2 h. Cells were harvested by centrifugation and lysed by high-pressure homogenization. Light chain and heavy chain inclusion bodies were recovered by tangential flow filtration. Refolding was scaled up to 200 L. An inline static mixer (Koflo) was utilized to provide effective mixing of denatured VH-toxin and VL solution into large quantity of refolding buffer. After refolding, proteins were precipitated with 2 M ammonium sulfate. This step led to a large volume reduction for convenient storage. Ammonium sulfate pellet containing 1,536 mg of total protein was re-dissolved in 1.25 M ammonium sulfate and purified by Phenyl HP followed by QFF column. Three hundred seventy-five milligrams of final product was produced. The overall purification recovery was 23 % from ammonium sulfate precipitate solubilization. The product was 98.9 % pure by HPLC-SEC and met all specifications for clinical grade MR1-1.
The novel purification process was demonstrated to be scalable for two dsFv immunotoxins. Clinical manufacturing was successfully conducted with improved yield. Product quality attributes such as purity, endotoxin, and residual host cell proteins were comparable or superior to those made from the conventional process. The HA22 and MR1-1 clinical lots were all ∼99 % in purity, contained <0.5 EU/mg endotoxin, and 10–16 ng/mg host cell proteins.
Discussion
We have successfully developed a novel clinical manufacturing process for dsFv-PE38 immunotoxins. Clinical supply of immunotoxins such as HA22, MR1-1, and other dsFv-PE38 molecules at relative large quantity is highly desirable (Wayne et al. 2010). However, it is a big challenge to increase the production yield with high purity at clinical grade. One of the major difficulties caused by that the molecular weight and charge of VH molecule (un-conjugated with VL) is very similar to the product VH–VL heterodimer. VH exists in significant amount after refolding and it was mixed with the product (Figs. 3 and 4). In vivo, VH alone may bind to the target CD22 (Harmsen and Haard 2007) with a more than two orders of magnitude lower affinity than that of VH–VL (Near et al. 1990). VH conjugated with toxin might cause additional toxicity and side effects in human clinical trials. Removing VH would be one of the key quality controls in this purification process design. Previously, the refolded immunotoxins (VH–VL), such as MR1-1 and BL22, were purified through two consecutive strong anion-exchange chromatographic steps with small resin size and large volume of gradient elution to increase the separation resolution. Then they were further purified through a size exclusion chromatographic step, which barely separate the VH from heterodimer (product) (Mansfield et al. 1997a and 1997b; Buchner et al. 1992; Kuan et al. 2000; Kreitman et al. 1999). This resulted in a poor recovery due to stringent pooling. Here, we report a new recovery and purification process that utilized ammonium sulfate precipitation followed by column chromatography with two notable mechanisms: hydrophobic interaction and anion exchange. Introduction of ammonium sulfate precipitation followed by HIC column chromatography were the two key factors for the process improvement especially separating VH from product VH–VL. Unlike the original process using two chromatography steps with the same mechanism of ion exchange, the new process is composed of three steps with three different purification mechanisms: precipitation, hydrophobic interaction, and ion exchange. Comparing to the conventional method, the new method is more efficient and streamlined, and most importantly, provided a significant yield increase (almost tripled, Table 1).
Two immunotoxins, MR1-1 and HA22, were successfully manufactured by using this method (in large scale) to supply clinical grade products. MR1‐1 and HA22 have been studied in phase 1 clinical trials for treatment of brain tumor [MR1‐1, at Duke University Medical Center (Shankar et al. 2006)] and in phase 1 and 2 trials for treatment of leukemia and lymphoma [HA22 a.k.a. CAT‐8015, at the National Institute of Health NCI‐07‐C‐0130 and NCI‐10‐C‐0067 (Kreitman 2012)]. As other dsFv-PE38 immunotoxin conjugates (Onda et al. 2011) would have structure and physiochemical properties similar to MR1-1 and HA22, we expect that this novel purification method could also be applied to the clinical production of those dsFv-toxin conjugates.
References
Archer GE, Sampson JH, Lorimer IAJ, McLendon RE, Kuan CT, Friedman AH, Friedman HS, Pastan IH, Bigner DD (1999) Regional treatment of epidermal growth factor receptor vIII-expressing neoplastic meningitis with a single-chain immunotoxin, MR-1. Clin Cancer Res 5:2646–2652
Bang S, Nagata S, Onda M, Kreitman RJ, Pastan I (2005) HA22 (R490A) is a recombinant immunotoxin with increased antitumor activity without an increase in animal toxicity. Clin Cancer Res 11:1545–1550
Batra JK, FitzGerald D, Gately M, Chaudhary VK, Pastan I (1990) Anti-Tac(Fv)-PE40, a single chain antibody Pseudomonas fusion protein directed at interleukin 2 receptor bearing cells. J Biol Chem 265:15198–15202
Beers R, Chowdhury C, Bigner D, Pastan I (2000) Immunotoxin with increased activity against epidermal growth factor receptor vIII-expressing cells produced by antibody phage display. Clin Cancer Res 6:2835–2843
Bird RE, Walker BW (1991) Single chain antibody variable regions. Trends Biotechnol 9:132–137
Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Withlow M (1988) Single-chain antigen binding proteins. Science 242:423–426
Bjorn MJ, Groetsema G, Scalapino L (1986) Antibody—Pseudomonas exotoxin A conjugates cytotoxic to human breast cancer cells in vitro. Cancer Res 46:3262–3267
Brinkmann U, Pai LH, FitzGerald DJ, Willingham M, Pastan I (1991) B3(Fv)-PE38KDEL, a single-chain immunotoxin that causes complete regression of a human carcinoma in mice. Proc Natl Acad Sci U S A 88:8616–8620
Buchner J, Pastan I, Brinkmann U (1992) A method for increasing the yield of properly folded recombinant fusion proteins: single-chain immunotoxins from renaturation of bacterial inclusion bodies. Analyt Biochem 205:263–270
Chaudhary VK, Queen C, Junghans RP, Waldmann TA, FitzGerald DJ, Pastan I (1989) A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature 339:394–397
FitzGerald DJ, Willingham MC, Pastan I (1988) Pseudomonas exotoxin—immunotoxins. Cancer Treatment Res 37:161–173
FitzGerald DJ, Wayne AS, Kreitman RJ, Pastan I (2011) Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res 71:6300–6309
Frankel AE (2002) Increased sophistication of immunotoxins. Clin Cancer Res 8:942–944
Frankel AE, Kreitman RJ, Sausville EA (2000) Targeted toxins. Clin Cancer Res 6:326–334
Harmsen MM, De Haard HJ (2007) Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol 77:13–22
Humphrey PA, Wong AJ, Vogelstein B, Friedman HS, Werner MH, Bigner DD, Bigner SH (1988) Amplification and expression of the epidermal growth factor receptor gene in human glioma xenografts. Cancer Res 48:2231–2238
Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, Ridge RJ, Bruccoleri EH, Crea R, Oppermann H (1988) Protein engineering of antibody binding sites: recovery of specific activity in an anti-dogoxigenin single-chain Fv analogue produced in Escherichia coli. Proc Nat Acad Sci (Wash) 85:5879–5883
Iglewski BH, Kabat D (1975) NAD-dependent inhibition of protein synthesis by Pseudomonas aeruginosa toxin. Proc Nat Acad Sci U S A 72:2284–2288
Kreitman RJ (2006) Immunotoxins for targeted cancer therapy. The AAPS Journal 8 (3) Article 63:E532–E551. doi:10.1208/aapsj080363
Kreitman RJ (2009) Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Cur Pharm Des 15:2652–2664
Kreitman RJ (2012) A phase 1/II study of CAT-8015 in adult relapsed or refractory B-cell non Hodgkin lymphoma and chronic lymphocytic leukemia. NCI-10_C-0067, NCT01086644. http://bethesdatrials.cancer.gov/clinical-research/search_detail.aspx?ProtocolID=NCI-10-C-0067 (2012) retrieved May 17, 2012
Kreitman RJ, Wang QC, FitzGerald DP, Pastan I (1999) Complete regression of human B-cell lymphoma xenografts in mice treated with recombinant anti-CD22 immunotoxin RFB4(dsFv)-PE38 at doses tolerated by cynomolgus monkeys. Int J Cancer 81:148–155
Kreitman RJ, Wilson WH, White JD, Stetler-Stevenson M, Jaffe ES, Giardina S, Waldmann TA, Pastan I (2000) Phase I trial of recombinant immnuotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol 18:1622–1636
Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, Pastan I (2001) Efficiency of the anti-CD22 recombinant immnuotoxin BL22 chemotherapy-resistant hairy-cell leukemia. N Engl J Med 345:241–247
Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I (2009) Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res 15:5274–5279
Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, Fitzgerald DJ, Lechleider R, Pastan I (2012) Phase I trial of anti-CD22 recombinant immunotoxin Moxetumomab Pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. doi:10.1200/JCO.2011.38.1756, epublish ahead of print
Kuan CT, Pastan I (1996) Improved antitumor activity of a recombinant anti-Lewis(y) immunotoxin not requiring proteolytic activation. Proc Natl Acad Sci U S A 93:974–978
Kuan CT, Reist CJ, Foulon CF, Lorimer IAJ, Archer G, Pegram CN, Pastan I, Zalutsky MR, Bigner DD (1999) 125I-labled anti-epidermal growth factor receptor-vIII single-chain Fv exhibits specific and high-level targeting of glioma xenografts. Clin Cancer Res 5:1539–1549
Kuan CT, Wikstrand CJ, Archer G, Beers R, Pastans T, Zalutsky M, Bigner DD (2000) Increased binding affinity enhances targeting of glioma zenografts by EGFRvIII-specific scFv. Int J Cancer 88:962–969
Lorimer IAJ, Wikstrand CJ, Batra SK, Bigner DO, Pastan I (1995) Immunotoxins that target an oncogenic mutant epidermal growth factor receptor expressed in human tumors. Clin Cancer Res 1:859–864
Lorimer IJ, Keppler-Hafkemeyer A, Beers RA, Pegram CN, Bigner DD, Pastan I (1996) Recombinant immunotoxins specific for a mutant epidermal growth factor receptor: targeting with a single chain antibody variable domain isolated by phage display. Proc Natl Acad Sci U S A 93:14815–14820
Mansfield E, Chiron MF, Amlot P, Pastan I, FitzGerald DJ (1997a) Recombinant RFB4 single-chain immunotoxin that is cytotoxic towards CD22-positive cells. Biochem Soc Trans 25:709–714
Mansfield E, Amlot P, Pastan I, FitzGerald DJ (1997b) Recombinant RFB4 immunotoxin exhibit potent cytotoxic activity for CD-22-bearing cells and tumors. Blood 90:2020–2026
Near RI, Ng SC, Mudgett-Hunter M, Hudson NW, Margolies MN, Seidman JG, Haber E, Jacobson MA (1990) Heavy and light chain contributions to antigen binding in an anti-digoxin chain recombinant antibody produced by transfection of cloned anti-digoxin antibody genes. Mol Immunol 27:901–909
Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ, Pastan I (2011) Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci U S A 108:5742–5747
Pai LH, Wittes R, Setser A, Willingham MC, Pastan I (1996) Treatment of advanced solid tumors with immnuotoxin LMB-1: an antibody linked to Pseudomonas exotoxin. Nat Med 2:350–353
Pastan I, Hassan R, FitzGerald DJ DJ, Kreitman RJ (2006) Immunotoxin therapy of cancer. Nat Rev Cancer 6:559–565
Pirker R, FitzGerald DJ, Hamilton TC, Ozols RF, Laird W, Frankel A, Willingham MC, Pastan I (1985) Characterization of immunotoxins active against ovarian cancer cells. J Clin Invest 76:1261–1267
Reisfeld RA, Gillies SD (1996) Recombinant antibody fusion proteins for cancer immunotherapy. Curr Top Microbiol Immunol 213:27–53
Reiter Y (2001) Recombinant immunotoxins is targeted cancer cell therapy. Adv Cancer Res 81:93–124
Reiter Y, Brinkmann U, Kreitman RJ, Jung SH, Lee B, Pastan I (1994) Stabilization of the Fv fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. Biochemistry 33:5451–5459
Shankar S, Vaidyanathan G, Kuan CT, Bigner DD, Zalutsky MR (2006) Antiepidermal growth factor variant III scFv fragment: effect of radioiodination method on tumor targeting and normal tissue clearance. Nucl Med Biol 33:101–110
Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, Steinberg SM, Stetler-Stevenson M, Fitzgerald DJ, Pastan I (2010) Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studies and phase I clinical trial. Clin Cancer Res 16:1894–1903
Wikstrand CJ, Hale LP, Batra SK, Hill MI, Humphrey PA, Kurpad SN, Mclendon RE, Moscatello D, Pegram CN, Reist CJ, Traweek ST, Wong AJ, Zalutsky MR, Bigner DD (1995) Monoclonal-antibodies against EGFRvIII are tumor-specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res 55:3140–3148
Wikstrand CJ, Reist CJ, Archer GE, Zalutsky MR, Bigner DD (1998) The class III variant of the epidermal growth factor receptor (EGFRvIII): characterization and utilization as an immunotherapeutic target. J Neuro Virol 4:148–158
Wikstrand CJ, Cokgor I, Sampson JH, Bigner DD (1999) Monoclonal antibody of human glioma: current status and future approaches. Cancer Metastasis Rev 18:451–464
Yamazaki H, Ohba Y, Tamaoki N, Shibuya M (1990) A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Jap J Cancer Res 81:773–779
Yokata T, Milenic DE, Withlow M, Schlom J (1992) Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res 52:3402–3408
Zhang Y, Chertov O, Zhang J, Hassan R, Pastan I (2011) Cytotoxic activity of immunotoxin SS1P is modulated by TACE-dependent mesothelin shedding. Cancer Res 71:5915–5922
Acknowledgment
The authors would like to thank Dr. Ira Pastan for the expression vectors and assay protocols. Mr. Joe Newland, Ray Rose, and Dr. Edward Wang contributed to the materials preparation for process development and cGMP production. Dr. Xiaoyi Yang and Gopalan Soman performed the bioactivity assay.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contracts N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mentioned of trade names, commercial products, or organizations imply endorsement by the US government.
Author information
Authors and Affiliations
Corresponding author
Additional information
Hua Jiang and Yueqing Xie contributed equally to this work.
Rights and permissions
About this article
Cite this article
Jiang, H., Xie, Y., Burnette, A. et al. Purification of clinical-grade disulfide stabilized antibody fragment variable—Pseudomonas exotoxin conjugate (dsFv-PE38) expressed in Escherichia coli . Appl Microbiol Biotechnol 97, 621–632 (2013). https://doi.org/10.1007/s00253-012-4319-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4319-2