
Abstract
3-Ketosteroid-Δ1-dehydrogenases (KstDs [EC 1.3.99.4]) catalyze the Δ1-dehydrogenation of steroids and are a class of important enzymes for steroid biotransformations. In this study, we cloned 12 putative KstD-encoding (kstd) genes from both fungal and Gram-positive microorganisms and attempted to overproduce the recombinant proteins in E. coli BL21(DE3). Five successful recombinant enzymes catalyzed the Δ1-desaturation of a variety of steroidal compounds such as 4-androstene-3,17-dione (AD), 9α-hydroxy-4-androstene-3,17-dione (9-OH-AD), hydrocortisone, cortisone, and cortexolone. However, the substrate specificity and catalytic efficiency of the enzymes differ depending on their sources. The purified KstD from Mycobacterium smegmatis mc2155 (MsKstD1) displayed high catalytic efficiency toward hydrocortisone, progesterone, and 9-OH-AD, where it had the highest affinity (K m 36.9 ± 4.6 μM) toward 9-OH-AD. On the other hand, the KstD from Rhodococcus erythropolis WY 1406 (ReKstD) exhibited high catalytic efficiency toward androst-4,9(11)-diene-3,17-dione (Diene), 21-acetoxy-pregna-4,9(11),16-triene-3,20-dione (Triene), and cortexolone, where in all three cases the K m values (12.3 to 17.8 μM) were 2.5–4-fold lower than that toward hydrocortisone (46.3 μM). For both enzymes, AD was a good substrate although ReKstD had a 3-fold higher affinity than MsKstD1. Reaction conditions were optimized for the biotransformation of AD or hydrocortisone in terms of pH, temperature, and effects of hydrogen peroxide, solvent, and electron acceptor. For the biotransformation of hydrocortisone with 20 g/L wet resting E. coli cells harboring MsKstD1 enzyme, the yield of prednisolone was about 90% within 3 h at the substrate concentration of 6 g/L, demonstrating the application potential of the newly cloned KstDs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
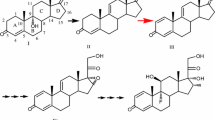
Steroids have important physiological functions with a wide range of clinical applications, and they are currently the second largest class of drugs after antibiotics (Donova and Egorova 2012). Among the steroidal drugs, many contain a double bond at the 1,2-position because they usually have enhanced potency and cause less drug-induced salt retention. For example, the anti-inflammatory activity of prednisolone acetate (PA) increased three to four times when an unsaturation was introduced into the 1,2-position of hydrocortisone acetate (HA) by Δ1-dehydrogenation (Fig. 1) (Zhang et al. 2013). Accordingly, Δ1-dehydrogenation of steroid compounds is an important reaction in the synthesis of medically useful steroidal molecules (Bhatti and Khera 2012; Donova 2007; Donova and Egorova 2012; Fernandes et al. 2003). Synthesis of these compounds may be achieved by chemical or biocatalytic methods. While the chemical methods require a complicated series of reaction steps due to the complex nature of the steroidal molecules (Chen et al. 2010; Jing et al. 2010; Marcos-Escribano et al. 2009), biotransformations of steroids are usually performed under mild conditions with high selectivity. Indeed, Δ1-dehydrogenation of 3-ketosteroids by actinobacteria has been used for the production of prednisolone and prednisteroids (Carballeira et al. 2009; Donova 2007; Donova and Egorova 2012).

3-Ketosteroid-Δ1-dehydrogenase (KstD)-catalyzed synthesis of prednisolone acetate from hydrocortisone acetate
When wild-type microbial cells are used, Δ1-dehydrogenation of 3-ketosteroids is usually accompanied by other reactions due to unwanted cellular enzymes in the microorganisms. In the biotransformation of pregna-4,9(11)-diene-17α,21-diol-3,20-dione 21-acetate and 17,21-diacetate by Nocardioides simplex VKM Ac-2033D, although the major Δ1-dehydrogenation products were identified as pregna-1,4,9(11)-triene-17α,21-diol-3,20-dione 21-acetate and pregna-1,4,9(11)-triene-17α,21-diol-3,20-dione 17,21-diacetate, respectively, the deacetylation and 20β-reduction products were also formed (Fokina and Donova 2003; Fokina et al. 2003). The microbial Δ1-dehydrogenation of 3-ketosteroids involves the enzyme 3-ketosteroid Δ1-dehydrogenase (KstD), which plays a crucial role in the early steps of steroid degradation by introducing a double bond between the C1-C2 atoms of the A ring of 3-ketosteroids. In order to improve the Δ1-dehydrogenation efficiency of 3-ketosteroids, two general strategies have been adopted. Genetic manipulations were performed with Mycobacterium neoaurum NwIB-01 and Arthrobacter simplex 156 to enhance the expression of KstD-encoding genes, yielding the recombinant strains with improved Δ1-dehydrogenation efficiency (Wei et al. 2014; Wei et al. 2010; Zhang et al. 2013). On the other hand, the kstd genes could be overexpressed in the well-developed host strains such as Escherichia coli, Bacillus subtilis, or Pichia pastoris to provide recombinant KstD enzymes for the desired biotransformation. However, these have not been well explored, considering highly effective KstD-catalyzed Δ1-dehydrogenation is in demand by the pharmaceutical steroid industry.
Up to now, some KstD enzymes have been identified in various microorganisms, e.g., Arthrobacter spp. (Choi et al. 1995a, Rhodococcus spp. (Morii et al. 1998), Pseudomonas spp. (Plesiat et al. 1991), Nocardia spp. (Itagaki et al. 1990a, b), and Mycobacterium spp. (Sukhodolskaya et al. 2007). The KstD enzyme from Nocardia corallina was purified and characterized by Itagaki and co-workers (Itagaki et al. 1990a, b), which triggered subsequent studies on the application of KstD in steroid biotransformations. The KstD enzyme of Pseudomonas testosteroni was found to contain a flavin adenine dinucleotide (FAD)-binding domain which is present in a number of related membrane proteins (Plesiat et al. 1991). Three KstDs, designated as KstD1, KstD2, and KstD3, had been characterized in Rhodococcus erythropolis SQ1 with activity toward different 3-ketosteroids, e.g., 4-androstene-3,17-dione (AD) and 9α-hydroxy-4-androstene-3,17-dione (9-OH-AD) (Knol et al. 2008; van der Geize et al. 2000, 2002). Subsequently, crystal structure and site-directed mutagenesis of KstD1 had revealed its catalytic mechanism, in which Tyr487 and Gly491 promote the keto-enol tautomerization and increase the acidity of the C2 hydrogen atoms of the substrate. This facilitates the abstraction of the axial β-hydrogen from C2 as a proton by Tyr318 with assistance of Tyr119, whereas the axial α-hydrogen from the C1 atom is transferred as a hydride ion to FAD (Rohman et al. 2013).
Most studies on KstD enzymes have been focused on the purification and elucidation of the possible physiological importance of the enzymes. We aim at developing a “toolkit” of KstD biocatalysts by characterizing potentially useful KstDs from newly cloned kstd genes of different origins in E. coli. In particular, two new KstD enzymes, MsKstD1 and ReKstD, were purified and characterized and the application potential of the former in the synthesis of prednisolone was explored.
Materials and methods
Reagents, strains, and media
All reagents were at least of analytical grade unless otherwise noted. 2,6-Dichlorophenloindophenol (DCPIP) and FAD were bought from Sigma. Testosterone was obtained from Acros Organics (NJ, USA). Cortexolone was purchased from Tokyo Chemical Industry Co., Ltd. 4-Androsten-3-one-5-ene-17-carboxylic acid (17-carboxylic acid) was bought from Adamas Reagent Co. Ltd. (Shanghai, China). Progesterone and phenazine methosulfate (PMS) were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Other steroids were donated by Zhejiang Xianju Junye Pharmaceutical Co., Ltd. (Zhejiang, China). All other chemicals were from commercial sources. Restriction endonucleases, T4 DNA ligases, and protein marker were obtained from Thermo Fisher Scientific. KOD DNA polymerase was purchased from Toyobo Life Science.
Mycobacterium neoaurum 2966 (NRRL B-3683) and M. neoaurum 2967 (NRRL B-3805) were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Germany). Mycobacterium smegmatis mc2155 (ATCC700084) was obtained from the American Type Culture Collection (ATCC, USA). Rhodococcus erythropolis WY 1406 strain (CGMCC 5096) was isolated from soil and is available from China General Microbiological Culture Collection Center (Beijing, China) (Wang et al. 2013).
Cloning of kstd genes and preparation of cell-free extracts
Nucleotide sequences of kstd genes were obtained from the National Center for Biotechnology Information GeneBank® using the kstd1 gene from R. erythropolis SQ1 as a query sequence (Rohman et al. 2013). Twelve putative kstd genes from different groups were selected in this study (Table 1). The five synthetic genes were cloned into plasmid pET21a(+) through codon optimization at http://www.prodoric.de/JCat (Grote et al. 2005). The other kstd genes were cloned from chromosomal DNA of M. smegmatis mc2155, M. neoaurum 2966, M. neoaurum 2967, and R. erythropolis WY 1406 with the primers shown in Table S1.
M . smegmatis mc2155,M. neoaurum 2966, M. neoaurum 2967, and R. erythropolis strains were cultivated in Luria-Bertani (LB) broth containing 1% (v/v) Tween 80 at 30 °C with shaking (200 rpm) for 16 h. Genomic DNAs were prepared by using the TianGen Bacteria DNA Kit DP302 (TianGen Biotech, Beijing) according to manufacturer’s instructions. The DNA fragment of Mskstd1 gene was obtained by PCR using KOD DNA polymerase. The following PCR program was used: 95 °C for 3 min followed by 30 cycles of 30 s at 95 °C, 30 s at 60 °C, 2 min at 68 °C, and then 68 °C for 10 min. The PCR product of Mskstd1 was cloned into NdeI/HindIII-digested pET28a(+). The plasmid, pET28a(+)-MsKstD1, was introduced into E. coli BL21 (DE3), resulting in recombinant strain pET28a(+)-MsKstD1-BL21. Similarly, other kstD genes were also cloned in pET28a(+) vector and transformed into E. coli BL21 (DE3).
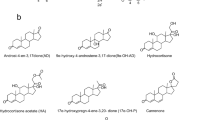
The recombinant strains were cultured at 37 °C in Terrific-Broth (TB) medium (200 mL) containing ampicillin (100 mg/L) or kanamycin (50 mg/L) until the optical density (OD) reached 1.0~2.0 at 600 nm. Subsequently, induction by the addition of 0.2 mM IPTG was performed for 20 h at 30 °C or for 24 h at 25 °C. Cells were separated by centrifuge at 4000×g for 10 min and washed twice with 100 mL 50 mM Tris-HCl buffer (pH 8.0). Protein production was analyzed by 0.1% SDS-12.5% polyacryamide gel electrophoresis. Subsequently, recombinant E. coli cells were resuspended in 50 mM Tris-HCl buffer (pH 8.0) and disrupted by high-pressure homogenizer. Cell debris was precipitated by centrifuge at 10,000×g for 30 min. The supernatants (cell-free extracts) were used for activity assays with a range of substrates as described below (Fig. 2).

Structures of the steroids tested in this study
Bioconversion of steroids with cell-free extract
A reaction mixture (2 mL) consisting of 2 mM PMS, 0.2 mL cell-free extracts, and 4 mg of substrate (Fig. 2; dissolved in 5% of dimethylsulfoxide (DMSO) or dimethylformamide (DMF) in the case of androst-4,9(11)-diene-3,17-dione (Diene)) in Tris-HCl buffer (50 mM, pH 8.0) was incubated at 30 °C. The E. coli BL21 (DE3) carrying the pET28a(+) plasmid was used as the control. Samples (200 μL) were withdrawn every hour and extracted with six volumes of ethyl acetate. The extracts were analyzed by thin-layer chromatography (TLC) and high-performance liquid chromatography (HPLC). TLC was performed on silica gel aluminum plates (Yantai Chemical Reagent Factory, China). The following solvent systems were used depending on the substrates: chloroform/methanol (25:1 v/v) for hydrocortisone, cortisone, and cortexolone; dichloromethane/methanol (20:1 v/v) for 11β,17-dihydroxy-6α-methyl-pregn-4-ene-3,20-dione (6α-Methyl) and hydrocortisone acetate (HA); petroleum ether/ethyl acetate (5:5 v/v) for 21-chloro-17-hydroxy-pregna-4,9(11)-diene-3,20-dione (21-Cl), anecortave, 17-carboxylic acid and testosterone; and petroleum ether/ethyl acetate (6:4 v/v) for AD, 21-acetoxy-pregna-4,9(11),16-triene-3,20-dinone (triene), diene, progesterone, pregna-4,9(11),16-triene-3,20-dione (PT), 9-OH-AD, and 17α-hydroxypregn-4-ene-3,20-dione (17α-OH-P). The compounds were visualized under UV light at 254 nm or by spraying with 20% H2SO4 and heating at 100 °C for about 5 min. HPLC analyses were carried out on Agilent 1200 system. For substrates hydrocortisone, 6α-Methyl, HA, cortisone, and cortexolone, an Agilent-C18 column (5 μm particles, 250 mm × 4.6 mm) was used with a mobile phase consisting of 30% acetonitrile and 70% water (v/v) at a flow rate of 0.6 mL/min. For other substrates a C18 column (Agilent, 5 μm particles, 150 mm × 4.6 mm) was used, and methanol and water (60:40 v/v) was used as the mobile phase at a flow rate of 0.6 mL/min. The UV absorbance was determined at 254 nm, and the column temperature was at 30 °C.
Purification of KstD enzymes
The His-tagged recombinant proteins were purified by Ni2+-chelating Sepharose affinity. All steps were carried out at 4 °C unless otherwise indicated. The cell pellet was suspended in 100 mL buffer A (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 30 mM imidazole). After supplementation with FAD (25 μM) and PMSF (1 mM), the cell suspension was lysed by high-pressure homogenizer and centrifuged at 10,000×g for 30 min. The supernatant was applied to the Ni2+-column followed by washing with buffer A. Elution was carried out using a linear imidazole gradient (30–500 mM) in buffer A. The fraction containing target protein was dialyzed for 12 h against Tris-HCl buffer (50 mM, pH 8.0) and then stored at −20 °C for further use. Protein concentrations were determined using the BCA Protein Assay Kit CW0014S (CWbiotech, Beijing), and protein purity was monitored by SDS-PAGE as described above.
Determination of the kinetic constants of purified KstD enzymes
The reaction mixture contained 1.5 mM PMS, 40 μM DCPIP, an appropriate amount of purified enzyme (5–15 μL), and 500 μM steroid substrate (20 μL solution in methanol) in 1 mL Tris-HCl buffer (50 mM, pH 7.0) (Knol et al. 2008). The reaction rates were determined by measuring the absorption of DCPIP at 600 nm (ε600nm = 18.7 × 103/cm/M) with multi-detection microplate reader at 30 °C. One unit of enzyme activity (U) is defined as the reduction of 1 μmol of DCPIP per minute.
The kinetic parameters of the KstD enzymes were determined by incubating the respective purified enzyme and varying concentrations of steroid substrate as described above. K m and Vmax values were obtained by fitting Michaelis-Menten equation to the data using the SigmaPlot program (version 12.0).
Optimal pH and temperatures of purified KstD enzymes
The optimal pH of the purified enzyme was determined at 30 °C in different buffers within a pH range of 5.0–10.0 (50 mM citric acid, 5.0–6.0; 50 mM sodium phosphate, 5.8–8.0; 50 mM Tris-HCl, 7.0–9.0; 50 mM Gly-NaOH, 9.0–10.0). The assay was started by addition of 500 μM AD into the reaction mixture. The optimal temperature was studied by carrying out the reaction in Tris-HCl buffer (50 mM, pH 8.0) at different temperatures ranging from 20 to 60 °C.
Effect of hydrogen peroxide on the activity of purified KstD enzymes
The effect of H2O2 was determined by supplementing different concentrations ranging from 0 to 5% (0, 0.05, 0.1, 0.2, 0.5, 1.0, 2.0, and 5.0% [v/v]) into the reaction mixture containing 1.5 mM PMS, 40 μM DCPIP, 5 μg purified enzyme, and 500 μM AD in Tris-HCl buffer (50 mM, pH 8.0). The activity was determined by measuring the absorption of DCPIP at 600 nm.
Optimization of process conditions for the bioconversion of hydrocortisone to prednisolone
The recombinant E. coli strain expressing MsKstD1 was cultured at 37 °C in TB medium containing kanamycin (50 mg/L) until the OD600 reached 1.0~2.0. Subsequently, induction with 0.2 mM IPTG was performed for 20 h at 30 °C. Cells were separated by centrifuge (4000×g) at late exponential phase (OD600, 8~10), and resuspended in 50 mM Tris-HCl (pH 8.0) for further use.
-
1.
Effects of DCPIP and PMS as electron acceptor: Tris-HCl buffer (1 mL, 50 mM, pH 8.0) consisting of 5 mM hydrocortisone, 5 μg enzyme (MsKstD1), and different concentrations of electron acceptors ranging from 0 to 6 mM (0, 1, 2, 5, and 6 mM) were incubated at 30 °C for 2 h in a little glass bottle (20 mL) with shaking at 200 rpm. The reaction mixtures were extracted with an equal volume of ethyl acetate and analyzed on HPLC as described above. The effect of PMS was further investigated at the concentrations ranging from 0 to 1.5 mM (0, 0.05, 0.1, 0.5, 1, and 1.5 mM). Samples (200 μL) were taken at 1-h interval for measuring the conversion of hydrocortisone by HPLC analysis.
-
2.
Effect of organic solvents and surfactant: Methanol, ethanol, isopropyl alcohol, t-butyl methyl ether, DMSO, DMF and tetrahydrofuran, and Tween 80 were used in this study. The reaction mixture containing 20 mg wet cells as described above, 2 mM hydrocortisone (suspended in 5% organic solvent or Tween 80) and 0.5 mM PMS in 1 mL of Tris-HCl buffer (pH 8.0, 50 mM) in 20-mL glass bottle was incubated at 30 °C for 1 h with shaking at 200 rpm. The reaction mixture was extracted with 2 mL ethyl acetate and evaporated to dryness. The residual was dissolved in methanol (2 mL), and the resulting solution was subjected to HPLC analysis to measure the conversion of hydrocortisone.
-
3.
Effect of biomass for Δ1-dehydrogenation of hydrocortisone: The bioconversion of hydrocortisone to prednisolone by recombinant whole cells was carried out in a 100-mL flask. The mixture containing 5 mM PMS and different amount of wet cells ranging from 1 to 100 g/L (1, 5, 10, 20, 50, and 100 g/L) in 20 mL 50 mM Tris-HCl (pH 8.0) buffer was incubated at 30 °C for 10 min, and 6 g/L hydrocortisone (dissolved in 5% v/v DMSO) was added into the reaction flask. The mixture was shaken at 200 rpm and 30 °C for 16 h. Samples (1 mL) were withdrawn at different time intervals and extracted three times each with a six volume of ethyl acetate. The ethyl acetate extracts were combined, and the solvent was removed in vacuum. The residue was dissolved in methanol and filtered. The filtrate was analyzed by HPLC to measure the amounts of hydrocortisone and prednisolone as described above. Each experiment was repeated three times.
Results
Δ1-Dehydrogenation of steroids by cell-free extracts of various KstD enzymes
Twelve putative kstd genes from different origins (Table 1) were selected, cloned, and overexpressed in E. coli BL21(DE3). Among these recombinant enzymes, five derived from a Streptomyces (SfKstD), Nocardia (NnKstD), Rhodococcus (ReKstD), and Mycobacterium (MsKstD1, MsKstD4) (see Table 1) were shown to have high 3-ketosteroid Δ1-dehydrogenase activity toward the steroidal compounds (Fig. 3). In general, these enzymes exhibited activity toward the steroids with C3-carbonyl group. MsKstD1 exhibited a high activity (>90% conversion) toward AD, hydrocortisone, Triene, progesterone, testosterone, 9-OH-AD, cortisone, and cortexolone (Fig. 3a), but low activity (<25%) toward 6α-Methyl, anecortave, and PT (Fig. 3b). The activity of MsKstD4 toward AD, Triene, progesterone, and testosterone was 40 to 60% higher than the other substrates. ReKstD showed high activities (>90% conversion) toward hydrocortisone, Triene, and 9-OH-AD, and moderate activity (about 80% conversion) toward AD, Diene, progesterone, testosterone, cortisone, cortexolone, and 17α-OH-P, but no activity was observed for 6α-Methyl. The conversions of these substrates with NnKstD were less than 40%. SfKstD showed a high activity (>90% conversion) toward Triene, progesterone, testosterone, and 9-OH-AD. Compared to other enzymes, SfKstD displayed the higher activity (40% conversion) toward 6α-Methyl.

Substrate profiles of cloned KstD enzymes using cell-free extracts of recombinant E. coli. a The substrates with higher than 80% conversion for MsKstD1. b The substrates with less than 80% conversion for MsKstD1. The values of the radar map indicate the conversion of substrates by recombinant E. coli expressing these genes
In general, the new KstD enzymes showed higher activities with steroids having a hydroxyl group (e.g., hydrocortisone) at C-21 than the corresponding acetic ester (HA). Most of the enzymes showed higher activity toward progesterone than those of testosterone, AD, and Diene, suggesting that an acetyl group at the C-17 position of the steroidal substrates is important for the activity. On the other hand, a lower activity toward PT may be due to the C=C structure at C9–C11 of the steroid substrates. Furthermore, introducing a hydroxyl group at the C17 position reduced the enzyme activity, e.g., 17α-OH-P. The presence of a functional group (hydroxyl or carbonyl group) or no substituent at the C11 position of the substrates (hydrocortisone, cortisone, and cortexolone) exerted almost no influence on the dehydrogenase activity of MsKstD1 and ReKstD, but not those of the SfKstD, NnKstD, and MsKstD4. The majority of enzymes were inactive toward steroidal substrates having a methyl substituent at the C6 position such as 6α-Methyl. As a whole, the KstD activity was dependent on the individual enzyme and the substrate structures. These enzymes showed no activity with steroids having a C3-hydroxyl group such as cholesterol and pregnenolone (data not shown).
Biochemical properties and kinetic parameters of purified MsKstD1 and ReKstD
To further study the catalytic properties of MsKstD1 and ReKstD, their purifications were performed by Ni2+ column chromatography. These proteins were purified to more than 90% homogeneity based on SDS-PAGE analysis (Fig. S1). The experimental molecular masses (M r ) of MsKstD1 and ReKstD were estimated to be 55.2 and 53.8 kDa, respectively. These values are in good agreement with the calculated values of 54,806 and 53,073 Da, respectively. The purified KstD proteins exhibited a bright yellow color and obvious flavoprotein absorptions at 275, 377, and 464 nm (data not shown). With the same purification procedure, NnKstD and MsKstD4 lost their activity upon purification. Due to low expression of SfKstD, this protein was not studied further.
The optimum temperatures of MsKstD1 and ReKstD activities were obtained at 25 and 30 °C (Fig. S2), respectively, and both their optimum pH values were around 8.0 (50 mM Tris-HCl) (Fig. S3). The enzymes were almost not active below pH 6.0. The purified proteins could be stored stably in 50 mM Tris-HCl buffer (pH 8.0) at 4 °C for at least 2 weeks.
It has been reported that molecular oxygen is used as the hydrogen acceptor in the enzymatic Δ1-dehydrogenation of steroids, generating hydrogen peroxide (H2O2) (Itagaki et al. 1990a, b). The effect of different concentrations of H2O2 on the enzyme activity was evaluated (Fig. S4). Compared to ReKstD, MsKstD1 exhibited better tolerance to H2O2. For MsKstD1, H2O2 exerted no significant effect on the enzyme activity at lower than 0.2% concentration, whereas at 0.5%, about 75% of the enzyme activity remained (Fig. S4a). In the case of ReKstD, the enzyme activity decreased much faster in the presence of H2O2 and less than 20% residual activity was detected at 0.5% of the oxidant (Fig. S4b).
The kinetic parameters of purified MsKstD1 and ReKstD were measured toward various steroids. The results are summarized in Table 2. Substrate affinity of MsKstD1 for 9-OH-AD (K m = 36.9 μM) was 4-fold higher than that of AD (K m = 138.5 μM). These K m values agree very well with those of MN-KstD1 from M. neoaurum ATCC25795 (Yao et al. 2014). However, ReKstD displayed similar affinities with 9-OH-AD and AD, and same as the K m values of KstD1 from R. erythropolis (van der Geize et al. 2002). Using AD as substrate, the K m values of KstDF from Aspergillus fumigatus, KS1DH from Arthrobacter simplex, and AcmBhis from Sterolibacterium denitrificans were 47, 71, and 100 μM, respectively (Chen et al. 2012; Chiang et al. 2008; Choi et al. 1995a, b).
MsKstD1 had the highest affinity toward 9-OH-AD (36.9 μM) and the highest reaction rate (k cat ) toward progesterone (127.8/s); the catalytic efficiency (k cat /K m) favored progesterone (1.9 × 106/s/M) as a substrate. ReKstD had the highest K m for cortexolone of 12.3 μM. The highest turnover rate per monomer (k cat ) was 50.2/s toward hydrocortisone, and the catalytic efficiency, k cat /K m, was calculated to be 1.7 × 106/s/M. The catalytic efficiency of MsKstD1 and ReKstD did not change significantly for substrates hydrocortisone, 21-Cl, HA, anecortave, progesterone, testosterone, cortisone, cortexolone, and 17α-OH-P (Table 2).
Optimization of process conditions for the bioconversion of hydrocortisone to prednisolone
The conversion of hydrocortisone to prednisolone is an important reaction due to its commercial interest. The Δ1-dehydrogenation reaction involves PMS or DCPIP as an electron acceptor. The results in Fig. 4a showed that PMS is more effective as an electron acceptor than DCPIP. In the presence of 0.05 mM PMS, the Δ1-dehydrogenation of hydrocortisone proceeded effectively (Fig. 4b). As the concentration of PMS increased by an order of magnitude (0.5 mM), the conversion rate increased by 10–20% after 2 h, reaching a maximum after 4 h and then plateaued. Further increase in PMS concentration did not benefit the transformation.

a Transformation of hydrocortisone by MsKstD1 with the different concentrations of phenazine methosulfate (PMS) and 2,6-dichlorophenloindophenol (DCPIP). b Time course of hydrocortisone conversion by MsKstD1 at low concentrations of PMS. The data were the mean values with standard deviation from triplicate experiments
The bioconversion of hydrocortisone to prednisolone by the resting cells harboring MsKstD1 enzyme was carried out in the presence of various co-solvents (Fig. 5). The highest yield (80%) of prednisolone was obtained when DMSO served as the co-solvent, followed by DMF and Tween 80 at about 70%.

The effects of co-solvent on the Δ1-dehydrogenation of hydrocortisone to prednisolone catalyzed by resting cells containing MsKstD1. The data were the mean values with SD from triplicate experiments. TBME tert-butyl methyl ether, DMSO dimethyl sulfoxide, DMF dimethyl formamide, THF, tetrahydrofuran
As a proof of concept, in a small-scale preparation run, the Δ1-dehydrogenation of hydrocortisone was performed with DMSO as the co-solvent at the substrate concentration of 6 g/L. The data characterizing the Δ1-dehydrogenation of hydrocortisone with 1–100 g/L of biomass are shown in Fig. 6. Transformation of hydrocortisone by 1 g/L wet cells produced only 40% prednisolone after 3 h. The yield of prednisolone by 20 g/L wet cells reached about 80% by the first 1 h, and the transformation was essentially completed in 3 h with a final yield of 90%. With increasing biomass, the yield increases sequentially. At the higher concentration (100 g/L), near 100% conversion was achieved in the first 1 h. In general, by the third hour, the yield of prednisolone reached 40% to near 100% (Fig. 6).

Transformation of hydrocortisone by resting cells of MsKstD1. The data were the mean values with SD from triplicate experiments
Discussion
The results of phylogenetic analysis (Fig. 7a) show that KstD sequences may be clustered into at least four distinct groups. The five active enzymes (SfKstD, NnKstD, ReKstD, MsKstD1, MsKstD4) are scattered among these different groups. ReKstD is highly similar to KstD1 from R. erythropolis SQ1 (97%) of which its structure has been analyzed (Rohman et al. 2013). MsKstD1, SfKstD, and NnKstD show about 40–45% amino acid sequence identity with the R. erythropolis SQ1 KstD1 protein. KstD1 is a flavoprotein with a conserved N-terminal FAD-dependent domain (Plesiat et al. 1991; Knol et al. 2008). Alignment of amino acid sequences of KstD1 from R. erythropolis SQ1 with those of other KstDs (Fig. 7b) shows that four residues (Y119, Y318, Y487, G491) essential for its activity in R. erythropolis SQ1 are conserved in these KstDs (Fig. 7b). The consensus FAD-binding sequences with the characteristic GSGX5-6AX2AX3GLX5EX5GGXXAXSG are also evident (de las Heras et al. 2012).

Phylogenetic tree and alignment of amino acid sequences of KstD. a Phylogenetic tree of 3-ketosteroid dehydrogenases. The scale length was set at 0.1. Mn Mycobacterium neoaurum ATCC 25795, Msmeg Mycobacterium smegmatis mc2155, Mtb Mycobacterium tuberculosis H37Rv, Nfa Nocardia farcinica IFM10152, Ro Rhodococcus jostii RHA1, SQ1 Rhodococcus erythropolis SQ1, 1815D Mycobacterium neoaurum VKM Ac-1815D, Psi Pimelobacter simplex, Nn Nocardia nova SH22a, Sa Salinispora arenicola, Afu Apergillus fumigatus Af293, Sfl Streptomyces flavovariabilis, Afl Apergillus flavus NRRL3357, Ao Apergillus oryzae RIB40. Asterisk indicates KstD enzymes studied in this study. b The sequence alignment of known KstD enzymes. SQ1 KstD from R. erythropolis SQ1 (PDB entry 4C3Y). Active site residues and residues involved in co-ordination of a FAD in SQ1 KstD are indicated by number sign. A conserved consensus sequence for FAD-binding region is indicated by asterisk
The purified MsKstD1 from M. smegmatis mc2155 catalyzes the Δ1-dehydrogenation of 3-ketosteroids with PMS or DCPIP as artificial electron acceptor. Different KstDs may utilize different electron acceptors. For example, DCPIP is an excellent electron acceptor for AcmB from S. denitrificans (Chiang et al. 2008), while PMS is highly efficient electron acceptor for KSTD from N. corallina (Itagaki et al. 1990a, b). The dehydrogenation of steroidal substrates is not only related with enzyme activity but also with the solubility of substrate. The co-solvents can increase the solubility of steroidal compounds in the aqueous reaction medium, thus significantly improving the transformation efficiency as seen by the conversion of hydrocortisone to prednisolone catalyzed by the MsKstD1 resting cells (Fig. 5).
The introduction of double bond into the 1,2-position of steroidal structure is one of the most important biotransformations of steroidal compounds. Although the KstD enzymes carrying out this reaction were studied with several bacteria (Chen et al. 2012; de las Heras et al. 2012; Sukhodolskaya et al. 2007) in the past two decades, these studies focused on limited steroidal substrates such as AD and 9-OH-AD. In this study, the newly identified KstDs from various strains were tested toward 16 substrates with diverse substituents on the steroidal nucleus, providing useful substrate profile information about these enzymes. These KstDs displayed activity toward steroids having a carbonyl group at C3 position. Among them, ReKstD and MsKstD1 exhibited high catalytic efficiency toward many steroidal substrates (hydrocortisone, Triene, progesterone, testosterone, 9-OH-AD, cortisone, etc.). In particular, MsKstD1 effectively catalyzed the Δ1-dehydrogenation of hydrocortisone to prednisolone with DMSO as the co-solvent at the substrate concentration of 6 g/L. Comparing the result to other microbial Δ1-dehydrogenation of hydrocortisone to prednisolone reported in the literature (Table 3), it can be seen that the recombinant cells pET28a(+)-MsKstD1-BL21 gave the highest time-space productivity of 1.83 g/(L h), a value highly competitive with that of Arthrobacter simplex ATCC 6946 (Vlahov et al. 1990). Thus, MsKstD1 may serve as a promising biocatalyst for the effective transformation of hydrocortisone to prednisolone.
Enzymatic Δ1-dehydrogenation is known to be a key process in steroid metabolism. The results from this study indicated that the recombinant KstDs showed different preferences to different substrates. Particularly, MsKstD1 is a useful biocatalyst for the one-step efficient transformation of hydrocortisone to prednisolone, advocating the possible utilization of the enzyme in large-scale production of prednisolone. Thus far, only one KstD crystal structure is available (Rohman et al. 2013), and the reaction mechanism is still unclear. Other enzymes such as 3-ketosteroid-Δ4-(5α) dehydrogenases are also known to catalyze the introduction of double bonds into steroids. Further studies are desired to understand the reaction mechanisms of double-bond formation by these enzymes, which may facilitate their utilization in biosynthesis of medically important steroidal compounds. It is anticipated that further research in this aspect and additional biocatalyst improvement by mutagenesis may facilitate the full potential of KstD in steroid production.
References
Adham NZ, El-Hady AA, Naim N (2003) Biochemical studies on the microbial Δ1-dehydrogenation of cortisol by Pseudomonas fluorescens. Process Biochem 38(6):897–902
Arinbasarova AGM, Akimenko VK, Koshcheyenko KA, Skryabin GK (1985) Redox reactions in hydrocortisone transformation by Arthrobacter globiformis cells. J Steroid Biochem 23:307–312
Bhatti HN, Khera RA (2012) Biological transformations of steroidal compounds: a review. Steroids 77(12):1267–1290
Bredehoft M, Baginski R, Parr MK, Thevis M, Schanzer W (2012) Investigations of the microbial transformation of cortisol to prednisolone in urine samples. J Steroid Biochem Mol Biol 129(1–2):54–60
Carballeira JD, Quezada MA, Hoyos P, Simeó Y, Hernaiz MJ, Alcantara AR, Sinisterra JV (2009) Microbial cells as catalysts for stereoselective red-ox reactions. Biotechnol Adv 27(6):686–714
Chen K, Liu C, Deng L, Xu G (2010) A practical Δ1-dehydrogenation of Δ4-3-keto-steroids with DDQ in the presence of TBDMSCl at room temperature. Steroids 75(7):513–516
Chen MM, Wang FQ, Lin LC, Yao K, Wei DZ (2012) Characterization and application of fusidane antibiotic biosynethsis enzyme 3-ketosteroid-Δ1-dehydrogenase in steroid transformation. Appl Microbiol Biotechnol 96(1):133–142
Chiang YR, Ismail W, Gallien S, Heintz D, Van Dorsselaer A, Fuchs G (2008) Cholest-4-en-3-one-Δ1-dehydrogenase, a flavoprotein catalyzing the second step in anoxic cholesterol metabolism. Appl Environ Microbiol 74(1):107–113
Choi KP, Molnár I, Yamashita M, Murooka Y (1995a) Purification and characterization of the 3-ketosteroid-Δ1-dehydrogenase of Arthrobacter simplex produced in Streptomyces liuidans. J Biochem 117:1043–1049
Choi KP, Murooka Y, Molnár I (1995b) Secretory overproduction of Arthrobacter simplex 3-ketosteroid Δ1-dehydrogenase by Streptomyces lividans with a multi-copy shuttle vector. Appl Microbiol Biotechnol 43:1044–1049
Donova MV (2007) Transformation of steroids by actinobacteria: a review. Appl Biochem Microbiol 43(1):1–14
Donova MV, Egorova OV (2012) Microbial steroid transformations: current state and prospects. Appl Microbiol Biotechnol 94(6):1423–1447
El-Hadi AA (2003) Production of prednisolone by Bacillus pumilus E601 cells incorporated in radiation induced poly(vinyl alcohol g-2 hydroxyethylmethacrylate) cryogels. Process Biochem 38(12):1653–1657
Fernandes P, Cruz A, Angelova B, Pinheiro HM, Cabral JMS (2003) Microbial conversion of steroid compounds: recent developments. Enzyme Microb Tech 32(6):688–705
Fokina VV, Donova MV (2003) 21-Acetoxy-pregna-4(5),9(11),16(17)- triene-21-ol-3,20-dione conversion by Nocardioides simplex VKM ac-2033D. J Steroid Biochem Mol Biol 87(4–5):319–325
Fokina VV, Sukhodolskaya GV, Baskunov BP, Turchin KF, Grinenko GS, Donova MV (2003) Microbial conversion of pregna-4,9(11)-diene-17α,21- diol-3,20-dione acetates by Nocardioides simplex VKM Ac-2033D. Steroids 68(5):415–421
van der Geize R, Hessels GI, van Gerwen R, Vrijbloed JW, van der Meijden P, Dijkhuizen L (2000) Targeted disruption of the KstD gene encoding a 3-ketosteroid Δ1-dehydrogenase isoenzyme of Rhodococcus erythropolis strain SQ1. Appl Environ Microbiol 66(5):2029–2036
van der Geize R, Hessels GI, Dijkhuizen L (2002) Molecular and functional characterization of the kstD2 gene of Rhodococcus erythropolis SQ1 encoding a second 3-ketosteroid Δ1-dehydrogenase isoenzyme. Microbiology 148(10):3285–3292
Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33:526–531
de las Heras LF, van der Geize R, Drzyzga O, Perera J, Navarro Llorens JM (2012) Molecular characterization of three 3-ketosteroid-Δ1-dehydrogenase isoenzymes of Rhodococcus ruber strain Chol-4. J Steroid Biochem Mol Biol 132(3–5):271–281
Itagaki E, Hatta T, Wakabayashi T, Suzuki K (1990a) Spectral properties of 3-ketosteroid-Δ1-dehydrogenase from Nocardia corallina. Biochim Biophys Acta 1040:281–286
Itagaki E, Wakabayashi T, Hatta T (1990b) Purification and characterization of 3-ketosteroid-Δ1-dehydrogenase from Nocardia corallina. Biochim Biophys Acta 1038:60–67
Jing Y, Xu CG, Ding K, Lin JR, Jin RH, Tian WS (2010) Protecting group effect on the 1,2-dehydrogenation of 19-hydroxysteroids: a highly efficient protocol for the synthesis of estrogens. Tetrahedron Lett 51(24):3242–3245
Kaul R, Mattiasson B (1986) Extractive bioconversion in aqueous two-phase systems. Appl Microbiol Biotechnol 24(4):259–265
Knol J, Bodewits K, Hessels GI, Dijkhuizen L, van der Geize R (2008) 3-Keto-5α-steroid Δ1-dehydrogenase from Rhodococcus erythropolis SQ1 and its orthologue in Mycobacterium tuberculosis H37Rv are highly specific enzymes that function in cholesterol catabolism. Biochem J 410(2):339–346
Marcos-Escribano A, Bermejo FA, Bonde-Larsen AL, Retuerto JI, Sierra IH (2009) 1,2-Dehydrogenation of steroidal 6-methylene derivatives. Synthesis of exemestane. Tetrahedron 65(36):7587–7590
Morii S, Fujii C, Miyoshi T, Iwami M, Itagaki E (1998) 3-Ketosteroid-Δ1-dehydrogenase of Rhodococcus rhodochrous: sequencing of the genomic DNA and hyperexpression, purification, and characterization of the recombinant enzyme. J Biochem 124(5):1026–1032
Plesiat P, Grandguillot M, Harayama S, Vragar S, Michel-Briand Y (1991) Cloning, sequencing, and expression of the Pseudomonas testosteroni gene encoding 3-oxosteroid Δ1-dehydrogenase. J Bacteriol 173(22):7219
Rohman A, van Oosterwijk N, Thunnissen AM, Dijkstra BW (2013) Crystal structure and site-directed mutagenesis of 3-ketosteroid Δ1-dehydrogenase from Rhodococcus erythropolis SQ1 explain its catalytic mechanism. J Biol Chem 288(49):35559–35568
Sukhodolskaya GV, Nikolayeva VM, Khomutov SM, Donova MV (2007) Steroid-1 -dehydrogenase of Mycobacterium sp. VKM Ac-1817D strain producing 9α-hydroxy-androst-4-ene-3,17-dione from sitosterol. Appl Microbiol Biotechnol 74(4):867–873
Vlahov R, Pramatarova V, Spassov G, Suchodolskaya GV, Koshcheenko KA (1990) Transformation of microcrystalline hydrocortisone by free and immobilized cells of Arthrobacter simplex. Appl Microbiol Biotechnol 33(2):172–175
Wang Y, Li J, Wu Q, Zhu D (2013) Microbial stereospecific reduction of 3-quinuclidinone with newly isolated Nocardia sp. and Rhodococcus erythropolis. J Mol Catal B Enzym 88:14–19
Wei W, Wang FQ, Fan SY, Wei DZ (2010) Inactivation and augmentation of the primary 3-ketosteroid-Δ1-dehydrogenase in Mycobacterium neoaurum NwIB-01: biotransformation of soybean phytosterols to 4-androstene- 3,17-dione or 1,4-androstadiene-3,17-dione. Appl Environ Microbiol 76(13):4578–4582
Wei W, Fan SY, Wang FQ, Wei DZ (2014) Accumulation of androstadiene-dione by overexpression of heterologous 3-ketosteroid Δ1-dehydrogenase in Mycobacterium neoaurum NwIB-01. World J Microb Biot 30(7):1947–1954
Yao K, Xu LQ, Wang FQ, Wei DZ (2014) Characterization and engineering of 3-ketosteroid-Δ1-dehydrogenase and 3-ketosteroid-9α-hydroxylase in Mycobacterium neoaurum ATCC 25795 to produce 9α-hydroxy-4-androstene-3,17-dione through the catabolism of sterols. Metab Eng 24:181–191
Zhang H, Tian Y, Wang J, Li Y, Wang H, Mao S, Liu X, Wang C, Bie S, Lu F (2013) Construction of engineered Arthrobacter simplex with improved performance for cortisone acetate biotransformation. Appl Microbiol Biotechnol 97(21):9503–9514
Acknowledgements
This work was financially supported by the STS Program of Chinese Academy of Sciences (KFJ-SW-STS-164) and the National High Technology Research and Development Program (“863”Program) of China (Nos. 02011AA02A211). The authors thank the sponsorship from Zhejiang Xianju Junye Pharmaceutical Co., Ltd. (Zhejiang, China), and greatly appreciate Professor Peter Lau at the same institute for his help and constructive suggestions on this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
This article does not contain any studies with human participants or animals performed by the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 354 kb)
Rights and permissions
About this article

Cite this article
Wang, X., Feng, J., Zhang, D. et al. Characterization of new recombinant 3-ketosteroid-Δ1-dehydrogenases for the biotransformation of steroids. Appl Microbiol Biotechnol 101, 6049–6060 (2017). https://doi.org/10.1007/s00253-017-8378-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8378-2