
Abstract
The opportunistic human pathogen Pseudomonas aeruginosa produces an extracellular polysaccharide called alginate. This is especially relevant in pulmonary infection of cystic fibrosis patients where it protects the bacteria from the hosts’ immune system and the diffusion of antibiotics. Here a connection between the stability of a proposed alginate polymerisation/secretion complex and the regulation of the operon encoding these proteins was assessed. Experimental evidence was provided for a periplasmic multiprotein complex composed of AlgX, AlgK, and the regulatory protein MucD. Disruption of the alginate machinery in a mucoid strain, either by removal, or over production of various essential proteins resulted in an at least 2-fold increase in transcription of a lacZ reporter under the control of the algD promoter. Instability of the complex was indicated by an increase in secretion of alginate degradation products. This increase in transcription was found to be dependent on the negative regulatory protein MucD. Surprisingly, over production of MucD leads to a 3.3-fold increase in transcription from the alginate promoter and a 1.7-fold increase in the levels of alginate produced, suggesting an additional positive regulatory role for MucD in mucoid strains. Overall, this study provided experimental evidence for the proposed periplasmic multiprotein complex and established a link of a constituent of this complex, MucD, to transcriptional regulation of alginate biosynthesis genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pseudomonas aeruginosa is a clinically important opportunistic human pathogen, and its ability to produce a thick extracellular matrix predominantly composed of alginate significantly contributes to its pathogenicity. This is of particular relevance in cystic fibrosis (CF) patients where P. aeruginosa pulmonary infections are the leading cause of both morbidity and mortality (Hay et al. 2010a).
The CF lung provides a unique environment to the pathogen which induces the overproduction of alginate by the bacteria, contributing to the clogging of the lung, while protecting the bacteria from the host immune response and antibiotic treatment (Pier et al. 2001; Simpson et al. 1988, 1989; Slack and Nichols 1981; Song et al. 2003). This switch to a mucoid phenotype is widely recognised as a poor prognosis indicator for patients, after which eradication of the infection is extremely unlikely. The exact mechanisms responsible for this switch are unclear but appear to involve a complex arrangement of transcriptional regulation, post-translational regulation and the mutation of hyper-mutable regions of the genome (Rehm 2010).
The 12 genes encoding the core alginate biosynthesis machinery are located in a single operon. AlgD and AlgA are involved in precursor synthesis; AlgI, AlgJ and AlgF are involved in acetylation of alginate (Franklin and Ohman 2002); AlgG for epimerization (Franklin et al. 1994); AlgL for cleavage (regulating the length of the polymer or degrading misguided alginate) (Schiller et al. 1993); AlgE is an outer membrane export porin (Rehm et al. 1994; Hay et al. 2010b); Alg8 is involved in polymerisation (Remminghorst and Rehm 2006a; Remminghorst et al. 2009); and Alg44 is involved in post-translational regulation (Remminghorst and Rehm 2006a, b; Hay et al. 2009). The remaining two proteins, AlgK and AlgX, have unclear functions. These proteins are essential for the production of intact alginate (loss of AlgK or AlgX results in the secretion of short alginate degradation products) and are thought to play some sort of structural or protective role, guiding the alginate polymer through the periplasmic space (Jain and Ohman 1998; Robles-Price et al. 2004; Gutsche et al. 2006). Recently, it has been suggested that AlgK may interact with the outer membrane protein AlgE (Keiski et al. 2010). Interestingly, the purification of AlgX from P. aeruginosa resulted in the co-purification of the regulatory protein MucD which demonstrated the first interaction of proteins involved in alginate biosynthesis (Gutsche et al. 2006). MucD is a periplasmic protease involved in the regulation of alginate biosynthesis. It has been suggested that proteins predicted to be involved in polymerization–export of alginate form a complex spanning from the inner to the outer membrane with a bridging periplasmic protein scaffold (Fig. 1) (Rehm 2010).

Schematic representation of predicted alginate biosynthesis complex and regulatory network. All interactions in the presented biosynthesis machinery are hypothetical except AlgX and MucD and AlgK and AlgX as indicated by stars. Plus and minus symbols indicate the effect the respective protein has on the subsequent protein in the pathway (not its overall regulatory role). Adapted from Hay et al. (2010a)
The alginate operon is under the tight control of a promoter upstream of algD, and transcription is initiated from this promoter via an alternate sigma factor AlgU (σ22, AlgT). The algU gene is itself located in a partially auto-regulated operon called the “switch” loci containing the genes algU, mucA, mucB, mucC and mucD (Ramsey and Wozniak 2005; Firoved and Deretic 2003; Deretic et al. 1994; Chitnis and Ohman 1993). This region is a common site of mutations in clinical (mucoid) isolates, one study found that 80% of all clinical isolates contained mutations in this region (Boucher et al. 1997). MucA, an intramembrane anti-sigma factor which sequesters AlgU at the membrane, is at the apex of a regulated intramembrane proteolysis (RIP) cascade (Schurr et al. 1996; Mathee et al. 1997; Wood and Ohman 2009). Several steps of this proteolytic cascade had been elucidated: MucB binds to the periplasmic side of MucA protecting it from proteolysis (Martin et al. 1993; Mathee et al. 1997). AlgW (Escherichia coli DegS homologue) is a periplasmic protease which is activated by the C-terminus of particular misfolded proteins (Qiu et al. 2007) and cleaves the C-terminus of MucA which is subsequently cleaved on the cytosolic side by the intramembrane protease YaeL (MucP, PA3649) leading to the release of AlgU (Cezairliyan and Sauer 2009; Wood and Ohman 2009). MucD, a homologue to the E. coli periplasmic serine protease DegP/HtrA, appears to be playing a role antagonistic to that of AlgW degrading misfolded proteins which would otherwise activate AlgW (Wood and Ohman 2006, 2009; Boucher et al. 1996; Qiu et al. 2007). MucD also appears to be involved in response to stresses such as excessive heat or response to reactive oxygen species as MucD mutants showed increased sensitivity to H2O2 and heat killing (Boucher et al. 1996).
Here evidence for the existence of a periplasmic multiprotein complex was provided. Furthermore, based on the apparent interaction between the regulatory protein, MucD, and other proteins of the alginate biosynthesis machinery, the relationship between the stability of the proposed alginate biosynthesis multiprotein complex and the transcriptional regulation of the alginate biosynthesis operon was investigated. Instability of the complex was achieved by removing or over producing proposed members of the complex. The activation of the alginate promoter in the presence and absence of MucD was assessed.
Materials and methods
Bacterial strains and growth conditions
The bacterial strains, plasmids and oligonucleotides used in the present study are listed in Supplementary Table 1. E. coli strains were grown in LB medium at 37°C. E. coli strains S17-1 or SM10 was used for conjugative transfer of the suicide plasmids derived from pEX100T, the flipase encoding plasmid pFLP2 and the φCTX-based integration vector mini-CTX-Palg-lacZ. Where required, antibiotics were used at the following concentrations: ampicillin 100 μg/ml, gentamicin 10 μg/ml and streptomycin 30 μg/ml. P. aeruginosa strains were grown in LB or PI(A) medium (Pseudomonas isolation [agar] medium—20 g of peptone, 10 g of K2SO4, 1.4 g MgCl2, 0.025 g of triclosan and 20 ml of glycerol per litre) at 37°C. Where required, antibiotic concentrations used for P. aeruginosa strains were as follows: gentamicin 300 μg/ml carbenicillin 300 μg/ml and tetracycline 200 μg/ml. All chemicals were purchased from Merck KGaA (Darmstadt, Germany) and Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated.
Isolation, analysis and manipulation of DNA
General cloning procedures were performed as described previously (Sambrook et al. 1989). pBBR1MCS-5, pTZ110 and pHERD-derived plasmids were transferred to P. aeruginosa strains via electroporation as previously described (Choi et al. 2006). DNA primers, dNTPs, Taq and Platinum Pfx polymerases were purchased from Sigma-Aldrich. DNA sequences of plasmid constructs were confirmed by DNA sequencing.
Construction and confirmation of deletion mutants
The P. aeruginosa deletion mutants for genes alg8, alg44, algX and algE were performed using the suicide plasmids described previously (Remminghorst and Rehm 2006a, c; Gutsche et al. 2006; Hay et al. 2010b). Suicide vectors for the construction of deletion mutants in the genes mucD, algU and rpoN were constructed as follows. Two regions upstream and downstream of the target gene were amplified using Pfx polymerase resulting in the fragments mucDN (comprising bases −2 to 406 relative to the designated mucD coding region followed by a BamHI site) and mucDC (bases 1,008 to 1,410 preceded by a BamHI), algUN (bases −164 to 260 relative to the first start codon flanked by a ScaI site and BamHI site) and algUC (bases 797 to 1,217 flanked by a BamHI site and ScaI site) and rpoNN (bases −15 to 389 flanked by a EcoRV site and BamHI site) and rpoNC (bases 1,118 to 1,503 flanked by a BamHI site and EcoRV site). Both PCR products were hydrolyzed by using BamHI and inserted into the vector pGEM-T Easy (Promega, Madison, WI, USA). Vector pPS856 (Hoang et al. 1998) was hydrolyzed with BamHI and the fragment containing the aacC1 gene (encoding gentamicin acetyltransferase) flanked by two FRT (Flp recombinase target) sites was inserted into the BamHI site between the two regions of the target gene. The fragments comprising the gentamicin cassette flanked by the upstream and downstream regions of the genes of interest were then released by hydrolysis with SmaI (for mucD), EcoRI (for rpoN) or ScaI (for algU) and inserted into SmaI site of vector pEX100T (Hoang et al. 1998), resulting in plasmids pEX100T:ΔmucDΩGm, pEX100T:ΔalgUΩGm and pEX100T:ΔrpoNΩGm.
These suicide plasmids were transferred into P. aeruginosa and transconjugants were selected on mineral salt medium (Schlegel et al. 1961) containing gentamicin and 5% (wt/vol) sucrose. Cells growing on this selective medium should have emerged from double crossover events. Gene replacement was confirmed after subculture of cells on PIA medium containing gentamicin and using PCR with primers upstream and downstream of the homologous regions.
The Flp recombinase encoding vector pFLP2 (Hoang et al. 1998) was transferred into P. aeruginosa ΩGm strains and grown on PIA containing carbenicillin for 12 h. The pFLP2 vector was later cured from the strain by growth on PIA medium containing 5% (wt/vol) sucrose. Gentamicin- and carbenicillin-sensitive cells were analysed by PCR for loss of the gentamicin-resistant cassette. Strains with mutations in multiple genes were constructed in a stepwise manner, deleting the genes sequentially in the order they are named in the strain.
To confirm that construction of the deletion mutants did not result in any polar effects, deletion mutants were complemented in trans with a plasmid containing the open reading frame of the deleted gene. Plasmids for the complementation of the alg8, alg44, algX and algE deletion mutants (pBBR1MCS-5:alg8, pBBR1MCS-5:alg44, pBBR1MCS-5:algX and pBBR1MCS-5:algE) are described in previous studies (Remminghorst and Rehm 2006a, c; Hay et al. 2010b; Gutsche et al. 2006). Plasmids for the complementation of the mucD, algU and rpoN deletion mutants were constructed as follows: the mucD, algU and rpoN open reading frames were amplified using the primers mucD-F-HiSDNd and mucD-R-SacI; algU-F-HiSDNd and algU-R-BamHI; and rpoN-F-HiSDNd and rpoN-R-BamHI, respectively. The fragments were hydrolysed with HindIII and SacI (mucD) and HindIII and BamHI (algU and rpoN) and ligated into corresponding sites in the broad host range plasmid pBBR1MCS-5 (Kovach et al. 1995), resulting in the plasmids pBBR1MCS-5:mucD, pBBR1MCS-5:algU and pBBR1MCS-5:rpoN.
Activation of the alginate promoter
Activation of the alginate promoter in deletion mutants was assessed using the plasmid pTZ110:Palg, and this plasmid contains lacZ under the control of the algD promoter. The promoter region, located at −854 to 1 bp relative to the algD open reading frame, was amplified using the primers Palg-F-HiNo and Palg-R-Ba. The product was hydrolysed with HindIII and BamHI and ligated into the HindIII and BamHI sites of the plasmid pTZ110 (Schweizer and Chuanchuen 2001). This plasmid was then transferred to the P. aeruginosa strain of interest.
Additionally, to assess the effect of artificially increasing the copy number of various genes (in trans) involved in alginate biosynthesis, strains were constructed with the same transcriptional reporter (lacZ) fusion described above integrated into the genome. These strains were created using an integration proficient φCTX-based plasmid. The promoter region was amplified as described above and ligated into the HindIII and BamHI sites of the plasmid mini-CTX-lacZ (Becher and Schweizer 2000), resulting in the plasmid mini-CTX:Palg-lacZ. This plasmid was then transferred to the P. aeruginosa strain of interest and subsequently plated on PIA media containing tetracycline to select for colonies that have undergone recombination with the plasmid and subsequent integration of the alginate promoter reporter along with the tetracycline resistance gene. Integration at the attB site was confirmed by PCR using the primers Pser-down and Pser-up.
Activity of these transcriptional reporter promoter fusions was assessed as follows: strains were grown on PI(A) agar plates containing 200 μg/ml carbenicillin for 48 h. A representative sample of the bacterial lawn was scraped from the agar surface, washed three times in TBS (pH 7.8) and resuspended to an OD600 of approximately 0.2–0.3. The β-galactosidase activity of this was measured using a modified Miller method (Zhang and Bremer 1995; Miller 1972b). Briefly, 20 μl of the cells was added to 80 μl of permeabilisation solution (0.8 mg/ml hexadecyltrimethylammonium bromide, 0.4 mg/ml sodium deoxycholate, 5.4 μl/ml β-mercaptoethanol, 100 mM Na2HPO4, 20 mM KCl, 2 mM MgSO4) and incubated at 30°C for 15 min. Six hundred microlitres of the substrate solution (1 mg/ml o-nitrophenyl-β-d-galactoside, 2.7 μl/ml β-mercaptoethanol, 60 mM Na2HPO4, 40 mM NaH2PO4). After a yellow colour has developed, the reactions are stopped with the addition of 700 μl stop solution (1 M Na2CO3) and time recorded. The OD420 was recorded and Miller units calculated (Miller 1972a).
Construction of a conditional non-mucoid PDO300 strain
PDO300 is an isogenic derivative of PAO1 with a non-functional mucA gene (muc22A allele). As the deletion mutants of mucD in the PDO300 and PDO300ΔalgX background were seemingly not possible, a strain with the WT mucA gene under the control of the araBAD promoter (PBAD) was constructed. The mucA gene was amplified using the primers mucA-F-SDNd and mucA-R and ligated into the pGEM-T Easy cloning vector (Promega, WI, USA). The resulting plasmid was hydrolysed with NcoI and SalI and ligated into the corresponding sites on the arabinose-inducible vector pHERD26T (Qiu et al. 2008a), resulting in pHERD26T:mucA. A 4,274-bp fragment containing the araC gene and mucA under the control of the PBAD promoter was hydrolysed from the plasmid pHERD26T:mucA with NheI, and the resulting 5′ overhangs were filled in with T4 DNA polymerase (blunted). This fragment was hydrolysed with SalI and the 1,576-bp araC-PBAD-MCS containing fragment was ligated into a SalI and SmaI hydrolysed integration proficient vector mini-CTX2 (Hoang et al. 2000), resulting in mini-CTX2PBAD:mucA.
mini-CTX2PBAD:mucA was transferred by conjugation to the strains PDO300 and PDO300ΔalgX. Transconjugates were selected for on tetracycline containing media and integration confirmed as described above, resulting in the strains PDO300-CTX2PBAD:mucA and PDO300ΔalgX-CTX2PBAD:mucA.
Alginate production assays
Alginate was harvested and purified as described previously (Hay et al. 2009). Uronic acid content was assessed through a modification of the Blumenkrantz and Asboe-Hansen (1973) method described previously using 100% alginic acid from brown algae (Sigma-Aldrich) as a standard (Hay et al. 2009).
Free/dialysable uronic acids (alginate degradation products) were measured in the supernatant of 2 ml of overnight cultures. Briefly, total uronic acid content of the supernatant was determined, the supernatants were filtered with Amicon Ultra-0.5 (Millipore) centrifugal filters (nominal molecular weight cut-off 10 kDa) and the flow through was collected (containing free uronic acids and short length alginate degradation products) and the uronic acid content determined.
Purification of MucD/AlgX and pull-down experiments
A C-terminal translational fusion or MucD to a hexahistadine tag was constructed: mucD was amplified with the primers mucD-F-HiSDNd and mucD-R-6×His-BamHI. This was hydrolysed with HindIII and BamHI and ligated into corresponding sites in the plasmid pBBR1MCS-5, resulting in pBBR1MCS-5:mucDHis. This plasmid was introduced into the P. aeruginosa strain PAO1ΔmucD and PAO1ΔmucDΔalgX and the E. coli strain Rosetta 2 (Novagen). The P. aeruginosa strains were grown in 500 ml of rich media (32 g l−1 tryptone, 20 g l−1 yeast extract, 5 g l−1 NaCl) at 37°C for 24 h. Rosetta 2 strains were grown in 500 ml of LB at 37°C until an OD600 of 0.6 was reached, after which expression was induced with the addition of IPTG to a final concentration of 1 mM. Cells were harvested and washed three times in one volume of TBS (pH 7.8) and suspended in 1/10th volume of lysis buffer (150 mM NaCl, 100 mM Tris–HCl, 0.2% Triton X-100, pH 8.0) with 1 mg ml−1 lysozyme and 1 mg ml−1 DNase. This was incubated for 20 min at 4°C with shaking and subsequently lysed by sonication. Insoluble cell debris was removed by centrifugation at 16,000×g for 20 min at 4°C. His-tagged MucD was purified from the lysate using TALON™ DynaBeads® (Invitrogen).
C terminally strep-tagged AlgX was purified as follows: The plasmid pBBR1MCS-5:algXStrep (Gutsche et al. 2006) was introduced into P. aeruginosa strains PDO300ΔalgX, PAO1ΔalgX, PAO1ΔmucDΔalgX and E. coli strain Rosetta 2. Cell lysates were prepared as described above. Supernatants were subjected to affinity purification with a Strep-Tactin® Superflow™ 1 ml Column (Novagen) according to the manufacturer’s instructions.
Where the protease activity was to be assessed, purifications were completed in the absence of any protease inhibitors.
Analysis of proteins
Protein concentrations were determined using the Quant-iT™ Protein Assay Kit (Invitrogen). Proteins were separated by SDS-PAGE on 10% acrylamide gels. Bands of interest were identified by tryptic peptide fingerprinting using matrix-assisted laser desorption ionisation-time of flight/mass spectrometry (MALDI-TOF/MS) by the Centre for Protein Research at the University of Otago.
Analysis of protease activity
The protease activity of various protein extracts was assayed using Universal protease substrate (resorufin-labelled casein) (Roche) according to the manufacturer’s instructions. Fifty micrograms of total protein from the purified MucDHis containing fractions was assayed with the addition of buffer or 150 μg of BSA, purified AlgX containing fractions or total membrane fractions from mucD deletion mutants. These were incubated for 120 min and the amount of free resorufin was then assessed by measuring the absorbance at 574 nm.
Gel filtration chromatography
One milligram of total protein in 500 μl was loaded on to a Superdex 200 10/300GL column. Two column volumes of lysis buffer (with 0.02% Triton X-100 instead of 2%) was passed through the collum at 0.5 ml min−1. The absorbance at 280 nm was monitored. Fractions were collected in 0.5 ml steps and subsequently assessed by SDS-PAGE.
Results
Generation and characterisation of algX/mucD mutants
To better understand any interaction between MucD and AlgX, we attempted to generate a set of deletion mutants for these two genes in both non-mucoid (PAO1) and mucoid (PDO300) parent strains. Disruption of mucD in PAO1 led to a mucoid phenotype (PAO1ΔmucD) with alginate levels similar to that of the mucoid strain PDO300 (Table 1). The non-mucoid phenotype could be restored by providing the mucD gene in trans. This is consistent with previous findings (Yorgey et al. 2001; Wood and Ohman 2006; Boucher et al. 1996). Disruption of algX in the non-mucoid strain did not visibly alter the phenotype of the parent. Disruption of both algX and mucD in the non-mucoid parent (PAO1ΔmucDΔalgX) resulted in a non-mucoid strain, though short, dialysable, uronic acid-containing molecules, i.e. alginate degradation products, could be detected in the culture supernatant. As would be expected, providing algX in trans to the double mutant resulted in a mucoid phenotype whereas providing mucD alone or both mucD and algX resulted in a non-mucoid phenotype (Table 1). It should be noted that both PAO1ΔmucD and more dramatically PAO1ΔmucDΔalgX showed impaired growth characteristics, growing slower and reaching cell densities about 0.73 and 0.38 times less than that of wild type, respectively. These strains also appeared to be more susceptible to lysis during washing with TBS. This growth could be restored to wild-type levels when complemented with mucD or mucD and algX in trans. All other strains had similar growth rates (data not shown).
Disruption of algX in the mucoid parent (PDO300ΔalgX) resulted, as previously described (Robles-Price et al. 2004; Gutsche et al. 2006), in a non-mucoid phenotype with the secretion of free uronic acids. Alginate production could be restored by providing algX in trans (Table 1).
Intriguingly, although multiple attempts were made to disrupt mucD in the mucoid strain PDO300 and its isogenic ΔalgX strain, no mutants could be generated. PDO300 is an isogenic derivative of PAO1 generated through the replacement of mucA with the defective mucA22 allele (from the clinical isolate FRD1) containing a single base pair deletion in a string of guanine residues which results a premature stop codon and a truncated MucA missing 48 residues from its periplasmic C-terminus (Mathee et al. 1999). In order to address this issue, we attempted to mimic the PAO1 environment by providing PDO300 with a conditionally expressed functional WT mucA and attempt to disrupt mucD in this strain. Accordingly, the full-length mucA (under the control of the arabinose-inducible pBAD promoter) was integrated into the genome via the CTX2 vector, which resulted in strains PDO300-CTX2PBAD:mucA and PDO300ΔalgX-CTX2PBAD:mucA. PDO300-CTX2PBAD:mucA showed a mucoid colony morphology in the absence of arabinose and a non-mucoid colony morphology in the presence of 0.5% arabinose (data not shown). Attempts were made to knock out mucD in these strains in the presence of various concentrations of arabinose with no success. Further attempts to disrupt mucD in another mucoid strain, the clinical isolate FRD1, also proved unsuccessful.
Experimental evidence for periplasmic multiprotein complex composed of AlgK, AlgX and MucD
To provide experimental evidence for the longtime proposed periplasmic multiprotein complex and its implications to regulation of alginate biosynthesis, further analysis of protein–protein interactions was conducted. Hexahistadine-tagged MucD was purified from strains PAO1ΔmucD(pBBR1MCS-5:mucD6xHis) and PAO1ΔmucDΔalgX(pBBR1MCS-5:mucD6xHis) using TALON Dynabeads (Fig. 2a). One primary band corresponding to full-length mature MucD and three truncations of MucD were observed and identified by MALDI-TOF/MS. Both of these extracts showed similar levels of protease activity as measured using resorufin-labelled casein. Strep II-tagged AlgX was enriched from PDO300ΔalgX(pBBR1MCS-5:algXstrep), PAO1ΔalgX(pBBR1MCS-5:algXstrep) and PAO1ΔmucDΔalgX(pBBR1MCS-5:algXstrep) cell lysates using Strep-Tactin superflow columns. Several co-eluting proteins could be detected and were identified by MALDI-TOF/MS (Fig. 2). In purified fractions from PDO300ΔalgX(pBBR1MCS-5:algXstrep), three bands were identified as the essential alginate biosynthesis protein AlgK; one at 49.5 kDa corresponding to the mature full-length protein; and two truncations of AlgK, one at 45.6 kDa and one at 43.7 kDa, and the previously described co-eluting protein MucD could be identified in two protein bands 47.8 kDa (full-length) and 43.7 kDa (truncated) (Fig. 2a). Several other biotin-containing proteins were present (data not shown). In purified fractions from PAO1ΔalgX(pBBR1MCS-5:algXstrep), only AlgX and MucD could be identified with AlgX being in much lower quantities than when purified from PDO300ΔalgX. From PAO1ΔmucDΔalgX(pBBR1MCS-5:algXstrep), only very small amounts of strep-tagged AlgX and full-length AlgK could be identified (Fig. 2a).

a Purification of strep-tagged AlgX and hexahistadine-tagged MucD from various P. aeruginosa strains. SDS-PAGE gel shows AlgX and co-eluting proteins. b Gel filtration chromatography (OD280nm) of the strep-tagged AlgX purification from P. aeruginosa PDO300- ΔalgX(pBBR1MCS-5:algXStrep). Inset SDS-PAGE of peak of interest. All protein identities were confirmed by MALDI-TOF/MS
A purified AlgX fraction from PDO300ΔalgX(pBBR1MCS-5:algXstrep) was separated by gel filtration chromatography. One major peak could be detected at approximately 14 min (approximately 70 kDa) with a lower molecular weight shoulder. SDS-PAGE and subsequent MALDI-TOF/MS of the protein bands showed that this peak was composed of full-length strep-tagged AlgX and the two shorter truncations of AlgK. The shoulder was composed of strep-tagged AlgX alone (Fig. 2b).
As MucD is a serine protease, we assessed the influence of AlgX, the AlgX–AlgK complex or envelope fractions containing members of the alginate biosynthesis machinery had on the protease activity of MucD. MucD was purified from PAO1ΔmucDΔalgX(pBBR1MCS-5:mucDHis). Approximately 3 times molar excess of AlgX purified from E. coli Rosetta 2 (pBBR1MCS-5:algXStrep) or the AlgX–AlgK complex purified from PAO1ΔmucDΔalgX(pBBR1MCS-5:algXStrep) was added to the purified MucD. Neither AlgX nor the AlgX–AlgK complex had any significant effect on protease activity when compared to BSA as control. This was repeated with crude envelope fractions from various strains, and again no effect on protease activity could be detected (Supplementary Table 2).
Loss of members of the alginate polymerase machinery affects the activation of the alginate promoter
Due to the previously described interaction between AlgX, a proposed member of the alginate synthesis/secretion machinery, and MucD, a regulatory protein involved in the transcriptional regulation of alginate biosynthesis (Gutsche et al. 2006), we assessed whether the loss of various members of the alginate biosynthesis machinery would have an effect on the levels of expression from the alginate promoter.
The plasmid pTZ110:Palg was constructed which contains the lacZ gene under the control of the promoter upstream of the alginate operon. This plasmid was introduced into various strains with disruptions in genes involved in the synthesis or secretion of alginate. The β-galactosidate activity and levels of alginate production were assessed. In the mucoid strain PDO300, disruption of alg8 and alg44 led to 2.5- and 4-fold reductions in alginate promoter activity, respectively, whereas disruption of algX or algE resulted in a 2.2-fold increase in alginate promoter activity (Table 2). As previously demonstrated, these mutants did not produce alginate, although dialysable uronic acids could be detected in the ΔalgX and ΔalgE mutants.
Disruption of these same genes had no effect on the minimal levels of transcription and alginate production observed in the non-mucoid strain PAO1. Interestingly, although the mucoid strain PAO1ΔmucD produced alginate at levels equivalent to PDO300, the activity of the alginate promoter was about 6.5 times less in the PAO1ΔmucD strain. Disruption of both mucD and algX in PAO1 resulted in a 5-fold increase in promoter activity over PAO1ΔmucD (Table 2).
Artificially increasing the levels of alginate biosynthesis proteins leads to increased transcription from the alginate promoter when AlgX and MucD are present
To further assess the influence the alginate biosynthesis machinery has on the levels of transcription from the alginate promoter, various proteins essential for alginate biosynthesis were overproduced. To do this, the lacZ alginate promoter reporter fusion was integrated into the chromosome via the integration proficient mini-CTX2 vector. This allowed us to artificially increase the copy number of genes of interest by providing them on plasmids. It should be noted that the chromosomal promoter lacZ reporter reported significantly lower β-galactosidase activity than the plasmid born reporter (pTZ110:Palg), but the relative levels of transcription and alginate production were similar in the strains PDO300CTXPalglacZ, PDO300ΔalgXCTXPalglacZ, PAO1CTXPalglacZ, PAO1ΔmucDCTXPalglacZ and PAO1ΔmucDΔalgXCTXPalglacZ to those carrying the pTZ110:Palg (Fig. 3).

Alginate promoter activity (as measured by β-galactosidase activity) (a, c, e and g) and alginate production (b, d, f and h) of strains overproducing various proteins involved in alginate production. PDO300Palg is PDO300CTXPalglacZ, PAO1Palg is PAO1CTXPalglacZ, PDO300ΔalgXPalg is PDO300ΔalgXCTXPalglacZ and PAO1ΔmucDPalg is PAO1ΔmucDCTXPalglacZ
Overexpression of algX, algE, alg44 and alg8 in PDO300CTXPalglacZ resulted in significant increases in transcription from the alginate promoter (3-, 3-, 2- and 2-fold, respectively) (Fig. 2a). Levels of alginate production in these strains did not tightly correlate with promoter activity, but all strains did produce elevated levels of alginate (Fig. 3b). Levels of dialysable uronic acids were about 10-fold higher in PDO300 strains containing multiple copies of algX, alg44 and algE, whereas dialysable uronic acid levels were only slightly elevated when multiple copies of alg8 were present (Supplementary Table 3). Overexpression of algX, algE, alg44 or alg8 in PAO1 neither induced transcription from the alginate promoter nor the production of alginate or dialysable uronic acids (data not shown and Supplementary Table 3).
Overexpression of algE, alg44 and alg8 in PDO300ΔalgX resulted in no significant increases in transcription from the alginate promoter (when compared to PDO300ΔalgX). Complementation by overexpression of algX resulted in a slight increase in transcription levels (Fig. 3c). As expected, only expression of algX in PDO300ΔalgX could restore alginate production (Fig. 3d). Apart from the complemented strain, which showed 33% dialysable uronic acid, all strains showed similar levels of uronic acids in the culture supernatant to PDO300ΔalgX(pBBR1MCS-5) with 100% of it being dialysable (Supplementary Table 3).
Contrary to PDO300, overexpression of algX, algE, alg44 and alg8 in mucoid strain PAO1ΔmucD had no effect on the level of promoter activity or the level of alginate production (Fig. 2e, f). Free uronic acid constituted 9.91% of the total uronic acids in the culture supernatant of the PAO1ΔmucD(pBBR1MCS-5) strain and increased to 17.2%, 16.4% and 22.3% when multiple copies of algX, alg44 or algE, respectively were present (Supplementary Table 3).
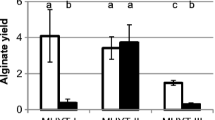
MucD can act both as a negative and positive regulator
As discussed above, disruption of mucD in PAO1 resulted in a mucoid phenotype and increased alginate promoter activity. Overexpression of mucD in PAO1 did not have an impact on the level of transcription from the alginate promoter act or on the levels of alginate production. Complementation of PAO1ΔmucD with mucD in trans resulted in a near complete reduction in promoter activity and alginate production (Fig. 4a, b). This does not seem to be mirrored in PDO300. Surprisingly, overexpression of mucD (thought to be a negative regulator) in PDO300 resulted in the most marked increase in promoter transcription observed (3.3-fold) (Fig. 4a, b). As the only difference between PDO300 and PAO1 is the defective mucA22 allele, it would seem that MucD is playing a different, positive regulatory role in the absence of full-length MucA.

Alginate promoter activity (as measured by β-galactosidase activity) (a) and alginate production (b) strains overproducing MucD. PDO300Palg is PDO300CTXPalglacZ, PAO1Palg is PAO1CTXPalglacZ, PDO300ΔalgXPalg is PDO300ΔalgXCTXPalglacZ and PAO1ΔmucDPalg is PAO1ΔmucDCTXPalglacZ
Increased levels of transcription associated with aberrations to the stoichiometry of the alginate biosynthesis proteins require AlgU
Since PDO300 has a truncated (and seemingly non-functional) MucA, we hypothesised that changes in alginate promoter activity in PDO300-derived strains may be via a route independent of the conventional MucA–AlgU, anti-sigma factor, complex. Within the alginate promoter region, there is a binding site for an alternative sigma factor, RpoN. Under certain conditions, RpoN has been shown to be required for transcription from the alginate promoter, while inhibiting transcription under other conditions (Boucher et al. 2000). Thus, the genes encoding the sigma factors AlgU and RpoN were disrupted in several strains to assess which sigma factor is responsible for the changes in the activation of the alginate promoter.
Disruption of algU in all strains assessed resulted in a loss of alginate production as well as reduction of alginate promoter activity to levels similar to those of PAO1, indicating that the increases in alginate promoter transcription observed in the ΔalgX and ΔmucD mutants ultimately require the AlgU sigma factor (Supplementary Table 4). Furthermore, overproduction AlgX, Alg8, Alg44, AlgE and MucD in PDO300ΔalgU did not result in increased levels of transcription seen in WT PDO300.
Disruption of rpoN produced a slight reduction in both alginate promoter activity and levels of alginate produced in PDO300. PAO1ΔmucDΔrpoN had no change in promoter activation nor in the levels of alginate produced. Artificially changing the levels of AlgX, Alg8, Alg44, AlgE or MucD had no significant effect on the increased levels of transcription associated with them (Supplementary Table 4).
Discussion
For decades, an alginate polymerisation/secretion multiprotein complex anchored in the cytoplasmic membrane had been proposed (Fig. 1) (Rehm 2010). So far, only one protein–protein interaction between AlgX and MucD had been experimentally shown (Gutsche et al. 2006). To provide experimental evidence for this multiprotein complex, it was investigated whether the AlgX–MucD complex interacts with further subunits. Here pull-down experiments using affinity chromatography were conducted. This led to identification of AlgK interacting with AlgX which interacts with MucD (Fig. 2). This is the first direct interaction demonstrated between essential alginate biosynthesis proteins. AlgK is a lipoprotein of unclear function which is encoded in the alginate operon and essential for the mucoid phenotype and the production of full-length alginate (Jain and Ohman 1998). AlgK is associated with the outer membrane, and there is some evidence to suggest that AlgK is involved in the localisation of AlgE to the outer membrane (Keiski et al. 2010). When AlgX was purified from PDO300ΔalgX(pBBR1MCS-5:algXStrep), AlgK was predominately present in a truncated 45.6-kDa version with small amounts of full-length AlgK and MucD. However, when purified in the absence of MucD (i.e. from PAO1ΔmucDΔalgX(pBBR1MCS-5:algXStrep)), AlgK was only present in the full-length form. This could indicate that the proteolytic activity of MucD is cleaving AlgK, though the reason for this seems unclear. The fact MucD was present in far lower quantities than either AlgX or AlgK and that no MucD containing complex could be detected in the gel filtration chromatography suggested that the interaction between MucD and AlgX/AlgK is weak or transient. MucD could be co-purified with AlgX from the non-mucoid PAO1ΔalgX(pBBR1MCS-5:algXStrep), suggesting that the AlgX– MucD interaction is not dependent on AlgK. Similarly, copurification of AlgK from the mucoid strain PAO1ΔmucDΔalgX(pBBR1MCS-5:algXStrep), i.e. in the absence of MucD, suggests that the AlgX–AlgK interaction is not dependent on MucD. Additionally, AlgX or AlgK does not appear to have an effect on the protease activity of MucD.
One possible explanation could be that the interaction of AlgX/AlgK with MucD might cause sequestration of MucD at the alginate polymerisation/secretion complex, making it unavailable for its regulatory role (Fig. 1). External stresses could cause instabilities in the complex and the release of MucD from the complex where it could exert its positive regulatory role increasing transcription of the alginate operon (possibly restoring the multiprotein complex).
The presence of the regulatory protein, MucD, in the periplasmic multiprotein complex suggested a link between the assembly of the multiprotein complex and regulation of alginate biosynthesis. Regulation of alginate biosynthesis is a complex process involving a combination of transcriptional regulation, post-translational regulation and the mutation of “hyper-mutable” regions of the genome. This regulatory network involves both globally acting regulators and alginate-specific regulators. Recently, several of the steps of a RIP cascade involved in activating transcription of the alginate operon in response to cell wall stresses have been elucidated. At least five proteases have been shown to be involved in the proteolysis of MucA, and all but MucD have been shown to positively influence alginate production in non-mucod strains (Wood and Ohman 2006, 2009; Wood et al. 2006; Qiu et al. 2007, 2008b) (Fig. 1). MucD is thought to repress the activation of alginate production by degrading misfolded proteins that would otherwise activate the protease AlgW, though this relationship is unclear as it has recently been demonstrated that mucD mutants remain mucoid in the absence of AlgW but are dependent on the MucP protease (Qiu et al. 2007; Damron and Yu 2011).
Here it was shown that there is a connection between the stability of the proposed alginate biosynthetic complex and the level of transcription from the alginate promoter. Removing or increasing the copies of various members of the proposed complex results in instability of the complex as indicated by the secretion of short dialysable uronic acids, presumably the products of alginate degradation by AlgL (Robles-Price et al. 2004). In PDO300, overproduction of the various subunits resulted in an at least 2-fold increase in the level of transcription from the alginate promoter as well as increased levels of alginate biosynthesis (Fig. 2). The different levels of increased promoter activity observed with the different subunits could be due to the relative effect each subunit has on the stability of the complex. It appears that this activation is dependent on MucD, as overproduction of these same proteins in the mucoid PAO1ΔmucD had no significant effect on the levels of transcription from the alginate promoter or the levels of alginate production. It is possible that AlgX is also required as no increase in transcription was observed in PDO300ΔalgX strains overproducing these proteins, but any effect may be masked by the already elevated levels of transcription observed in this strain.
PDO300 is an isogenic derivative of PAO1 with a truncated, seemingly non-functional MucA anti-sigma factor (Mathee et al. 1999), yet the increases in transcription associated with instability of the alginate biosynthesis complex appeared to be dependent on the sigma factor AlgU. This suggested that any communication between the alginate secretion complex and the transcriptional machinery still involves AlgU. As the truncation in MucA is in the periplasmic C-terminus, it is possible that the truncated MucA present in PDO300 (and many clinical isolates) is still able to bind/sequester the cytosolic AlgU, but may be more prone to proteolysis by the RIP cascade. Instability of the secretion complex may speed up the proteolysis of MucA, possibly via MucD, and thus increase the levels of transcription. It should also be noted that instability of the complex (as assessed here) is not sufficient to induce expression of the alginate biosynthesis genes or a mucoid phenotype in the non-mucoid strain PAO1 containing full-length MucA.
Interestingly, though our results regarding the disruption of mucD in PAO1 are consistent with the proposed negative regulatory role of MucD, our results regarding the overproduction of MucD seem to suggest a positive regulatory role, at least in the absence of full-length MucA. Overproduction of MucD in PDO300 resulted in a 3.3-fold increase in the levels of transcription from the alginate promoter and a 1.7-fold increase in the levels of alginate biosynthesis (Fig. 3). This positive regulatory role would seem to be dependent on AlgX, as overproduction of MucD in PDO300ΔalgX does not result in an increased transcription, though this could be due to the already elevated transcription levels in this strain. Overproduction of MucD in PAO1 has neither effect on the levels of transcription nor the levels of alginate biosynthesis. This suggests that in the presence of full-length MucA, MucD acts as a negative regulator; however, in situations where MucA is truncated (i.e. PDO300), MucD may be acting as a positive regulator. This is complicated by the fact that it was not apparently possible to generate a mucD-deficient strain in the mucA22 strains PDO300 and FRD1. It cannot be ruled out that overproduction of MucD aids in the degradation of MucA when it is already truncated (and thus not “protected” by MucB) due to the muc22A mutation. Frame shift mutations in mucA resulting in truncations, such as the mucA22 mutation, are by far the most common mutations observed in clinical mucoid isolates (Ciofu et al. 2008; Boucher et al. 1997). This inability to generate the ΔmucD mutant could suggest that MucD may be essential for the survival in mucA22-based mucoid strains. This is strengthened by the finding that PAO1ΔmucD and PAO1ΔmucDΔalgX showed impaired growth and were prone to cell lysis. Also, ΔmucD mutants have previously been demonstrated to be more susceptible to heat and reactive oxygen species (Boucher et al. 1996).
Intriguingly, though PAO1ΔmucD produced alginate at levels equivalent to PDO300, the alginate promoter activity was about 6.5 times less than in PDO300. This could indicate that the increase in alginate production observed with the loss of mucD may, at least to some extent, occur at a post-transcriptional level.
Overall, in this study, further evidence for a periplasmic multiprotein complex involved in alginate biosynthesis and its transcriptional regulation was obtained.
References
Becher A, Schweizer HP (2000) Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 29(5):948–950, 952
Blumenkrantz N, Asboe-Hansen G (1973) New method for quantitative determination of uronic acids. Anal Biochem 54(2):484–489
Boucher JC, Martinez-Salazar J, Schurr MJ, Mudd MH, Yu H, Deretic V (1996) Two distinct loci affecting conversion to mucoidy in Pseudomonas aeruginosa in cystic fibrosis encode homologs of the serine protease HtrA. J Bacteriol 178(2):511–523
Boucher JC, Yu H, Mudd MH, Deretic V (1997) Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect Immun 65(9):3838–3846
Boucher JC, Schurr MJ, Deretic V (2000) Dual regulation of mucoidy in Pseudomonas aeruginosa and sigma factor antagonism. Mol Microbiol 36(2):341–351
Cezairliyan BO, Sauer RT (2009) Control of Pseudomonas aeruginosa AlgW protease cleavage of MucA by peptide signals and MucB. Mol Microbiol 72(2):368–379
Chitnis CE, Ohman DE (1993) Genetic analysis of the alginate biosynthetic gene cluster of Pseudomonas aeruginosa shows evidence of an operonic structure. Mol Microbiol 8(3):583–593
Choi KH, Kumar A, Schweizer HP (2006) A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64(3):391–397
Ciofu O, Lee B, Johannesson M, Hermansen NO, Meyer P, Hoiby N (2008) Investigation of the algT operon sequence in mucoid and non-mucoid Pseudomonas aeruginosa isolates from 115 Scandinavian patients with cystic fibrosis and in 88 in vitro non-mucoid revertants. Microbiology 154(Pt 1):103–113
Damron FH, Yu HD (2011) Pseudomonas aeruginosa MucD regulates the alginate pathway through activation of MucA degradation via MucP proteolytic activity. J Bacteriol 193(1):286–291
Deretic V, Schurr MJ, Boucher JC, Martin DW (1994) Conversion of Pseudomonas aeruginosa to mucoidy in cystic fibrosis: environmental stress and regulation of bacterial virulence by alternative sigma factors. J Bacteriol 176(10):2773–2780
Firoved AM, Deretic V (2003) Microarray analysis of global gene expression in mucoid Pseudomonas aeruginosa. J Bacteriol 185(3):1071–1081
Franklin MJ, Ohman DE (2002) Mutant analysis and cellular localization of the AlgI, AlgJ, and AlgF proteins required for O acetylation of alginate in Pseudomonas aeruginosa. J Bacteriol 184(11):3000–3007
Franklin MJ, Chitnis CE, Gacesa P, Sonesson A, White DC, Ohman DE (1994) Pseudomonas aeruginosa AlgG is a polymer level alginate C5-mannuronan epimerase. J Bacteriol 176(7):1821–1830
Gutsche J, Remminghorst U, Rehm BH (2006) Biochemical analysis of alginate biosynthesis protein AlgX from Pseudomonas aeruginosa: purification of an AlgX–MucD (AlgY) protein complex. Biochimie 88(3–4):245–251
Hay ID, Remminghorst U, Rehm BH (2009) MucR, a novel membrane-associated regulator of alginate biosynthesis in Pseudomonas aeruginosa. Appl Environ Microbiol 75(4):1110–1120
Hay ID, Rehman ZU, Ghafoor A, Rehm BHA (2010a) Bacterial biosynthesis of alginates. J Chem Technol Biotechnol 85(6):752–759
Hay ID, Rehman ZU, Rehm BH (2010b) Membrane topology of the outer membrane protein AlgE which is required for alginate production in Pseudomonas aeruginosa. Appl Environ Microbiol 76(6):1806–1812
Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212(1):77–86
Hoang TT, Kutchma AJ, Becher A, Schweizer HP (2000) Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43(1):59–72
Jain S, Ohman DE (1998) Deletion of algK in mucoid Pseudomonas aeruginosa blocks alginate polymer formation and results in uronic acid secretion. J Bacteriol 180(3):634–641
Keiski CL, Harwich M, Jain S, Neculai AM, Yip P, Robinson H, Whitney JC, Riley L, Burrows LL, Ohman DE, Howell PL (2010) AlgK is a TPR-containing protein and the periplasmic component of a novel exopolysaccharide secretin. Structure 18(2):265–273
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM 2nd, Peterson KM (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166(1):175–176
Martin DW, Schurr MJ, Mudd MH, Deretic V (1993) Differentiation of Pseudomonas aeruginosa into the alginate-producing form: inactivation of mucB causes conversion to mucoidy. Mol Microbiol 9(3):497–506
Mathee K, McPherson CJ, Ohman DE (1997) Posttranslational control of the algT (algU)-encoded sigma22 for expression of the alginate regulon in Pseudomonas aeruginosa and localization of its antagonist proteins MucA and MucB (AlgN). J Bacteriol 179(11):3711–3720
Mathee K, Sternberg C, Ciofu O, Jensen P, Campbell J, Givskov M, Ohman DE, Hoiby N, Molin S, Kharazmi A (1999) Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiol 145:1349–1357
Miller JH (1972a) Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Miller JH (1972b) Experiments in molecular genetics. Cold Spring Harbor Press, Cold Spring Harbor
Pier GB, Coleman F, Grout M, Franklin M, Ohman DE (2001) Role of alginate O acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect Immun 69(3):1895–1901
Qiu D, Eisinger VM, Rowen DW, Yu HD (2007) Regulated proteolysis controls mucoid conversion in Pseudomonas aeruginosa. Proc Natl Acad Sci USA 104(19):8107–8112
Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD (2008a) PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74(23):7422–7426
Qiu D, Eisinger VM, Head NE, Pier GB, Yu HD (2008b) ClpXP proteases positively regulate alginate overexpression and mucoid conversion in Pseudomonas aeruginosa. Microbiology 154(Pt 7):2119–2130
Ramsey DM, Wozniak DJ (2005) Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol Microbiol 56(2):309–322
Rehm BH (2010) Bacterial polymers: biosynthesis, modifications and applications. Nat Rev Microbiol 8(8):578–592
Rehm BH, Boheim G, Tommassen J, Winkler UK (1994) Overexpression of algE in Escherichia coli: subcellular localization, purification, and ion channel properties. J Bacteriol 176:5639–5647
Remminghorst U, Rehm BH (2006a) Alg44, a unique protein required for alginate biosynthesis in Pseudomonas aeruginosa. FEBS Lett 580(16):3883–3888
Remminghorst U, Rehm BH (2006b) Bacterial alginates: from biosynthesis to applications. Biotechnol Lett 28(21):1701–1712
Remminghorst U, Rehm BHA (2006c) In vitro alginate polymerization and the functional role of Alg8 in alginate production by Pseudomonas aeruginosa. Appl Environ Microbiol 72(1):298–305
Remminghorst U, Hay ID, Rehm BH (2009) Molecular characterization of Alg8, a putative glycosyltransferase, involved in alginate polymerisation. J Biotechnol 140(3–4):176–183
Robles-Price A, Wong TY, Sletta H, Valla S, Schiller NL (2004) AlgX is a periplasmic protein required for alginate biosynthesis in Pseudomonas aeruginosa. J Bacteriol 186(21):7369–7377
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Plainview
Schiller NL, Monday SR, Boyd CM, Keen NT, Ohman DE (1993) Characterization of the Pseudomonas aeruginosa alginate lyase gene (algL): cloning, sequencing, and expression in Escherichia coli. J Bacteriol 175(15):4780–4789
Schlegel HG, Kaltwasser H, Gottschalk G (1961) [A submersion method for culture of hydrogen-oxidizing bacteria: growth physiological studies] article in German. Arch Mikrobiol 38:209–222
Schurr MJ, Yu H, Martinez-Salazar JM, Boucher JC, Deretic V (1996) Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J Bacteriol 178(16):4997–5004
Schweizer HP, Chuanchuen R (2001) Small broad-host-range lacZ operon fusion vector with low background activity. Biotechniques 31(6):1258, 1260, 1262
Simpson JA, Smith SE, Dean RT (1988) Alginate inhibition of the uptake of Pseudomonas aeruginosa by macrophages. J Gen Microbiol 134(1):29–36
Simpson JA, Smith SE, Dean RT (1989) Scavenging by alginate of free radicals released by macrophages. Free Radic Biol Med 6(4):347–353
Slack MP, Nichols WW (1981) The penetration of antibiotics through sodium alginate and through the exopolysaccharide of a mucoid strain of Pseudomonas aeruginosa. Lancet 2(8245):502–503
Song Z, Wu H, Ciofu O, Kong KF, Hoiby N, Rygaard J, Kharazmi A, Mathee K (2003) Pseudomonas aeruginosa alginate is refractory to Th1 immune response and impedes host immune clearance in a mouse model of acute lung infection. J Med Microbiol 52(Pt 9):731–740
Wood LF, Ohman DE (2006) Independent regulation of MucD, an HtrA-like protease in Pseudomonas aeruginosa, and the role of its proteolytic motif in alginate gene regulation. J Bacteriol 188(8):3134–3137
Wood LF, Ohman DE (2009) Use of cell wall stress to characterize sigma 22 (AlgT/U) activation by regulated proteolysis and its regulon in Pseudomonas aeruginosa. Mol Microbiol 72(1):183–201
Wood LF, Leech AJ, Ohman DE (2006) Cell wall-inhibitory antibiotics activate the alginate biosynthesis operon in Pseudomonas aeruginosa: roles of sigma (AlgT) and the AlgW and Prc proteases. Mol Microbiol 62(2):412–426
Yorgey P, Rahme LG, Tan MW, Ausubel FM (2001) The roles of mucD and alginate in the virulence of Pseudomonas aeruginosa in plants, nematodes and mice. Mol Microbiol 41(5):1063–1076
Zhang X, Bremer H (1995) Control of the Escherichia coli rrnB P1 promoter strength by ppGpp. J Biol Chem 270(19):11181–11189
Acknowledgements
This study was supported by research grants to B.H.A.R. from Massey University. I.D.H was funded by a Massey University Doctoral scholarship.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 214 kb)
Rights and permissions
About this article
Cite this article
Hay, I.D., Schmidt, O., Filitcheva, J. et al. Identification of a periplasmic AlgK–AlgX–MucD multiprotein complex in Pseudomonas aeruginosa involved in biosynthesis and regulation of alginate. Appl Microbiol Biotechnol 93, 215–227 (2012). https://doi.org/10.1007/s00253-011-3430-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3430-0