
Abstract
Microorganisms play important roles in the tobacco aging process. However, microbial communities on flue-cured tobacco leaves (FCTL) remain largely unknown. In this study, the total microbial genomic DNA of unaged and aging FCTL from Zimbabwe were isolated using a culture-independent method, and the bacterial communities were investigated through analyzing two 16S rRNA gene libraries. Eighty-four and 65 operational taxonomic units were obtained from the libraries of the unaged and aging FCTL, respectively. The following genera were represented more than 4% in both libraries (aging and unaged library): Sphingomonas (4.84%, 4.18%), Stenotrophomonas (4.84%, 5.23%), Erwinia (5.81%, 4.88%), Pantoea (19.35%, 18.47%), and Pseudomonas (21.29%, 24.04%). The dominant species varied between the two libraries. Specifically, several dominant species in unaged FCTL including Pseudomonas fulva, Pseudomonas sp. (AM909658), Klebsiella sp. (HM584796), and Pantoea sp. (AY501386) were not identified in aging FCTL, while several dominant species in aging FCTL such as Pantoea sp. (GU566350), Pseudomonas sp. (EF157292), and Buttiauxella izardii were not found in unaged FCTL. The phylogenetic analysis showed that bacteria from unaged and aging FCTL were divided into two clades, and two unique subclades were identified in aging FCTL. Our results revealed for the first time the bacterial diversities on Zimbabwe tobacco, and provided a basis for clarifying the roles of bacteria in aging process of FCTL.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tobacco is economically the most important nonfood crop worldwide. The flue-cured tobacco leaves (FCTL) is one of the most important type of tobacco in the world, including China. Cured but unaged tobacco leaves have a sharp, disagreeable odor and an undesirable aroma, and produce harsh, irritating smoke. In the industrial production, a further process called fermentation or aging is typically applied to improve the quality of the FCTL (Guo et al. 2004). Aging greatly improves the aroma and color, reduces irritating smoke, and improves the overall tobacco quality (Peng et al. 2009). Tobacco fermentation is a very complicated process, and it has been linked to the enzymatic actions of bacteria, fungi, and other chemical interactions within the leaves (Jensen and Parmele 1950). Tobacco fermentation can be divided into artificial and natural fermentation. Compared to natural aging, artificial fermentation can shorten the fermentation period and reduce cost (Guo et al. 2004). However, because little is known about the number and type of microorganisms appropriate for conducive aging, it has been difficult to control artificial fermentation (Yang et al. 2008). Tamayo and Cancho (1953) were the first to inoculate tobacco leaves with microorganisms to improve the aroma. In recent years, increasing reports showed these microorganisms on FCTL can accelerate the aging process and improve the quality of tobacco leaves through its growth activities (Han and Ye 1997; Yang et al. 2008; Huang et al. 2010a).
The conventional media and culture condition are not appropriate for the growth of most microorganisms in nature, and more than 85% microbes cannot be obtained in pure culture (Amann et al. 1995). The development of molecular biology techniques has provided a useful method to study these uncultured microorganisms. Culture-independent 16S rRNA gene sequence analysis has been widely used to identify microbial diversities in soil (Williamson et al. 2003), air (Tringe et al. 2008), animal gut (Hill et al. 2005), and other environments. Recently, Huang et al. (2010b) and Zhao et al. (2007) analyzed the bacterial diversities of several FCTLs using the culture-independent method. The analyzed tobacco leaves include several widely cultivated in China such as K326, Zhongyan 100, NC89, and Zhongyan 101. However, microbial communities in several other important tobacco leaves, such as Zimbabwe and Brazil FCTL, are not fully understood. Zimbabwe has natural conditions for the production of high-quality FCTL, and Zimbabwe tobacco leaves are the main materials for producing the high-grade cigarette. Understanding the microbial communities of Zimbabwe tobacco leaves would be very important for effectively controlling the tobacco aging process and improving the quality of FCTL. In this study, the total microbial genomic DNAs were isolated from the unaged and aging Zimbabwe FCTL, and the bacterial communities of these two samples were analyzed and compared by their 16S rRNA clone libraries. Moreover, the phylogenetic trees of bacteria in unaged and aging FCTL were constructed based on the 16S rRNA sequences.
Materials and methods
Sampling of tobacco
Unaged and aging Zimbabwe tobacco leaves were sampled from Hongyun Honghe Tobacco (Group) Co, Ltd. (Kunming, China), and the grade of samples are L1OT. Unaged tobacco leaves and those aged for 24 months were sampled and stored at −20°C for use.
DNA extraction from the microbial community of tobacco
Sixty grams of tobacco leaves were divided into three equal parts and placed in three flasks with 250 mL sterilized 0.1 M phosphate buffer (pH 7.0) for 30 min, respectively. Later, the tobacco leaves were washed with a sonicator for 10 min, and the microorganisms were collected by centrifugation at 10,000×g for 30 min. The microbial genomic DNA was extracted according to our recently described protocol (Huang et al. 2010b). The genomic DNA isolated from three samples were mixed and used as template for 16S rRNA gene amplification.
Amplification of the bacterial 16S rRNA genes
Primers 799f (5′-GGTAGTCCACGCCGTAAACGATG-3′; position 781 through 799 according to Escherichia coli number) and 1492r (5′-GGTTACCTTGTTACGACTT-3′; position 1492 through 1,510 according to E. coli number) were selected to amplify the 16S rRNA of bacteria. These primers are specific to bacteria and have a low affinity for chloroplast DNA (Jurkevitch et al. 2000). The PCR amplification was done according to the conditions described by Sun et al. (2008), and the band size of approximately 715 bp was purified.
Construction of the 16S rRNA clone library
The purified PCR products were ligated into the pMD18-T vector (Takara, Japan) and transformed into competent cells (E. coli DH5α) to construct the 16S rRNA clone library. In order to identify the bacterial diversity on unaged and aging tobacco samples, two 16S rRNA clone libraries were respectively constructed. Inserts of 16S rRNA genes from recombinant clones were re-amplified using universal primers M13 and RV. Products of PCR amplification were purified and sequenced with a 3730-nucleotide sequencer (ABI, USA).
Analysis of the 16S rRNA sequence data
Chimeric sequences were identified using the B2C2 program (Gontcharova et al. 2010). The resulting sequences were fully aligned with ClustalX (Thompson et al. 1997). On the basis of the alignment, a distance matrix was constructed by using the DNADIST program from PHYLIP (ver. 3.66, Felsenstein 1989) with the default parameters. The program DOTUR with the furthest neighbor algorithm was used to group sequences into operational taxonomical units (OTUs) or phylotypes that represented the number of 16S rRNA sequence similarity groupings (Schloss et al. 2005). A 97% cutoff value was used so that sequences with more than 97% similarity were considered the same. A sequence was selected randomly from each OTU and compared with those available sequences in GenBank using the BLAST program to determine their approximate phylogenetic affiliation and sequence similarities (Altschul et al. 1990). The diversity of the clone libraries was investigated by rarefaction analysis, and the rarefaction curves were calculated using the RarefactWin program (Holland 2004).
Phylogenetic analysis
Nucleotide sequences were aligned initially using ClustalX (Thompson et al. 1997) and then manually adjusted. Distance matrices and phylogenetic trees were calculated according to the Kimura two-parameter model (Kimura 1980) and the neighbor-joining (Saitou and Nei 1987) algorithm using the MEGA (version 4.0) software packages (Tamura et al. 2007). One thousand bootstraps were performed to assign confidence levels to the nodes in the trees.
Nucleotide sequence accession numbers
The nucleotide sequence data reported in this paper were deposited in GenBank with accession numbers JF820125-JF820273.
Results
Statistical analysis of the clone library
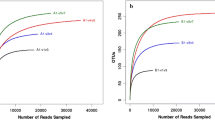
According to the Phylipwx program, the 303 16S rRNA clones from the unaged FCTL were grouped into 87 OTUs, and the 316 clones from the aging FCTL sample were grouped into 66 OTUs. Through the BLAST analysis, three OTUs containing 16 clones in unaged FCTL and one OTU containing 6 clones in aging FCTL belonged to the tobacco chloroplast. The calculated rarefaction curve (Fig. 1) showed that the two samples are close to their respective OTU saturation curve, indicating that the clone libraries already included most of the dominant bacteria species on tobacco samples.

Rarefaction curve of bacterial 16S rRNA clone library of the unaged and aging Zimbabwe FCTL
Analyses of bacterial communities and dominant species on unaged and aging FCTL
As shown in Table 1, the two samples contained a large diversity of bacteria, which were divided mainly into Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. Twenty-two genera including Pseudomonas, Bacillus, Rhizobium, Sphingomonas, Achromobacter, Methylobacteriun, Stenotrophomonas, and Buttiauxella were detected in both the unaged and aging FCTL; these genera contained 49 OTUs (81.88% clone ratio) and 37 OTUs (81.61% clone ratio) in unaged and aging FCTL, respectively. Eight genera including Roseomonas, Novosphingobium, and Xanthomonas were only detected in the unaged FCTL and these genera contained 10 OTUs and 5.57% of the total clone library. In contrast, 16 genera including Agrobacterium, Curtobacterium, Arthrobacter, Flavobacterium, and Actinomyces were only detected in aging FCTL, which consisted of 19 OTUs and 11.94% of the total clone library.
In both samples, the genera with greater than 4% of the clone libraries were Sphingomonas, Stenotrophomonas, Erwinia, Pantoea, and Pseudomonas, and the most dominant genera were Pantoea and Pseudomonas, representing 18.47–24.04% of the clone libraries. The main OTUs in the genus Pantoea included Pantoea vagans and Pantoea sp. (GU566350), and those in the genus Pseudomonas included Pseudomonas putida, Pseudomonas fulva, Pseudomonas sp. (AM909658), and Pseudomonas sp. (EF157292). The OTUs of Pseudomonas decreased significantly from 11 to 3 after aging treatment. Moreover, the percentages of Buttiauxella and Klebsiella differed significantly between the two samples: Buttiauxella was represented by 0.35% in unaged FCTL and by 7.10% in aging FCTL; Klebsiella was represented by 6.62% in unaged FCTL and by 0.65% in aging FCTL.
The dominant species (i.e., those present in greater than 3% of the libraries) were also variable between the two samples (Table 2). P. vagans and Pseudomonas sp. (AM909658) were both present by more than 10% in unaged FCTL, while Pantoea sp. (GU566350) and Pseudomonas sp. (EF157292) were represented by more than 10% in aging FCTL. P. vagans decreased from 14.63% to 6.77% after aging treatment, while P. putida increased from 2.79% to 9.03%. Erwinia sp. (EU336938) and Stenotrophomonas maltophilia were equally dominant in these two samples. Moreover, several dominant species such as P. fulva, Pseudomonas sp. (AM909658), Klebsiella sp. (HM584796), Acinetobacter sp. (HM137026), and Pantoea sp. (AY501386) were only detected in unaged FCTL, while several dominant species such as Pantoea sp. (GU566350), Pseudomonas sp. (EF157292), and Buttiauxella izardii were only detected in aging FCTL.
Phylogenetic analysis of the bacterial community on tobacco leaves
Phylogenetic trees (Fig. 2) were constructed based on the 16S rRNA sequences of the 84 OTUs and 65 OTUs from unaged and aging FCTL, respectively. Two trees of bacteria from unaged (Fig. 2a) and aging FCTL (Fig. 2b) shared similar shape, and those bacteria were clustered into two clades (A and B). In unaged FCTL, clade A contained 77 OTUs (97.21% clones) and were divided into two subclades (A1 and A2), and subclade A1 consisted of two clusters (A11 and A12, Fig. 2a). Cluster A11 (gamma-proteobacteria) contained 51 OTUs (81.88% clones), which included 13 genera, and the dominant genera were Stenotrophomonas, Pantoea, and Pseudomonas. Cluster A12 (beta-proteobacteria) contained five OTUs (3.48% clones) and included Achromobacter, Herbaspirillum, and Massilia. Subclade A2 (alpha-proteobacter) contained 21 OTUs (11.85% clones) and included Aurantimonas, Methylobacterium, Rhizobium, and Rhodobacter.


The phylogenetic tree of bacteria in Zimbabwe FCTL based on 16S rRNA sequences. a Unaged FCTL, b aging FCTL. OTU sequences to the most closely related sequences obtained from GenBank. Bootstrap values are 1,000 replicates
In the aging FCTL, clade A contained 44 OTUs (occupies 88.39% clones) and were divided into three subclades (A1, A2, and A3), subclade A1 included three clusters (A11, A12, and A13; Fig. 2b). Cluster A11 (gamma-proteobacteria) contained 17 OTUs (68.39% clones), which included 11 genera and the dominant ones were Buttiauxella, Pantoea, and Pseudomonas. Cluster A12 (beta-proteobacteria) contained three OTUs (3.87% clones) and included Achromobacter and Massilia. Subclade A2 (alpha-proteobacter) includes 20 OTUs (14.84% clones) and included Aurantimonas, Methylobacterium, Rhizobium, and Rhodobacter. Subclade A3 contained two OTUs, epsilon-proteobacteria and delta-proteobacteria with one clone each. Interestingly, subclade A3 and cluster A13 were unique in aging FCTL, and the member of cluster A13 belongs to uncultured bacteria.
Similarly, clade B could be further divided into two subclades (B1 and B2). In the unaged sample, subclade B1 contained 3 OTUs (1.05% clones) in genera Aerococcus, Bacillus, and one uncultured bacterium which has 93% similarity to Paenibacillus sp. (HM099647); subclade B2 included two clusters Actinobacteria and Bacteroidetes, containing 4 OTUs (1.74% clones) in three genera Microbacterium, Chryseobacterium, and Sphingobacteria. More bacteria OTUs were identified in aging sample: subclade B1 contained 8 OTUs (4.84% clones) in five genera Saccharibacillus, Paenibacillus, Bacillus, Enterococcus, and Moryella; subclade B2 also included two clusters Actinobacteria and Bacteroidetes, containing 13 OTUs (6.77% clones) in genera Microbacteri, Curtobacterium, Arthrobacter, Chryseobacterium, Flavobacterium, Pedobacter, Sphingobacterium, and an uncultured bacterium.
Discussion
In this study, we systematically analyzed the bacterial communities in the aging and unaged Zimbabwe FCTL using culture-independent method. Compared to our previous report (Huang et al. 2010b), the bacterial communities in Zimbabwe FCTL were different from those in tobacco K326 (grade C3F): 84 and 65 OTUs were detected from the unaged and aging Zimbabwe FCTL, respectively, while 50 and 42 OTUs were obtained from the unaged and aging FCTL (K326). Although the OTU numbers of bacteria in these two tobacco samples showed similar trends, their bacterial species and dominant genera were quite different. Bacillus spp. and Pseudomonas spp. were the two dominant genera in FCTL (K326), while the most dominant genera were Pantoea and Pseudomonas in Zimbabwe FCTL. These differences of bacterial communities might be related to the tobacco varieties, and further analysis could help understand effect of bacterial diversities in the aging process of FCTL.
From the phylogenetic analysis (Fig. 2), the OTUs and the clone ratios of bacteria in clade A decreased, while these in clade B increased significantly after aging process. Tobacco leaves lose water gradually during aging process, which could impact the bacterial communities in FCTL, and the physiological activities of these bacteria would in turn contribute to bioconversion of macromolecular compounds (e.g., starch, cellulose, and protein) and improving the quality of tobacco leaves. Different from FCTL of K326 tobacco leaves where beta-proteobacteria were only detected in the unaged FCTL, these bacteria were found in both aging and unaged Zimbabwe FCTL. In addition, the clone ratio of beta-proteobacteria increased through the aging, except for Achromobacter detected in tobacco K326. Other genera such as Massilia and Herbaspirillum were only found in Zimbabwe FCTL. At present, the effects of these beta-proteobacteria in the tobacco aging process were unknown.
Alpha-proteobacteria increased during aging of Zimbabwe FCTL, similar to those of tobacco K326 (Huang et al. 2010b), except that more alpha-proteobacteria bacteria were detected in Zimbabwe FCTL, such as Roseomonas, Novosphingobium, Aurantimonas, Agrobacterium, and Bosea. Moreover, in Zimbabwe FCTL, gamma-proteobacteria had an absolute advantage over other bacterium groups in both the number of OTU and clone. Different from the increase of OTU in alpha-proteobacteria, the number of OTU for gamma-proteobacteria decreased from 51 to 17 after aging treatment, and the dominant bacteria were B. izardii, Pseudomonas sp., Pantoea sp., S. maltophilia, Erwinia sp., P. vagans, and P. putida.
In Zimbabwe FCTL, Pseudomonas and Pantoea were the most dominant genera with significant changes during the aging process (Table 2). The clone ratio of P. vagans decreased by 7.86% after aging treatment, while P. putida increased by 6.27%. Similarly, P. fulva, Pseudomonas sp. (AM909658) and Klebsiella sp were the dominant species in unaged FCTL, while Pantoea sp. (GU56350), B. izardii and Pseudomonas sp. (EF157292) were more prevalent in aging FCTL. P. putida and its close relatives are capable of degrading nicotine in tobacco leaves (Newton et al. 1977; Civilini et al. 1997; Li et al. 2010). The prevalence of other species such as Klebsiella sp., Acinetobacter sp., and B. izardii also showed obvious change during the aging process. An increasing number of reports (Ruan et al. 2005; Chen et al. 2008; Li et al. 2010) have showed Pseudomonas could speed the aging process and improve the quality of tobacco leaves, while the role of other dominant bacteria in the aging process of tobacco remain unknown.
Recently, the microbial community structures on leaves of three varieties Zhongyan 100, NC89, and Zhongyan 101 were studied by denaturing gradient gel electrophoresis (DGGE) technique (Zhao et al. 2007). Five dominant 16S rDNA DGGE bands were sequenced, and they were found most similar to two cultured microbial species Bacteriovorax sp. EPC3, Bacillus megaterium, and three uncultured microbial species, respectively. The microbial community structure and dynamics on Italian Toscano cigar were also investigated by culture-based and culture-independent approaches (Di Giacomo et al. 2007); they found that Staphylococcaceae (Jeotgalicoccus and Staphylococcus) and Lactobacillales (Aerococcus, Lactobacillus, and Weissella), Bacillus spp., and Actinomycetales (Corynebacterium and Yania) were the most commonly detected bacteria at the early, middle, and late periods of fermentation, respectively. More recently, Huang et al. (2010b) reported that the dominant microbial population was Pseudomonas and Bacillus in tobacco K326 by 16S rRNA RFLP technology. These results showed that the dominant microbial population might be closely related to the tobacco varieties.
In this study, a large number of bacterial OTUs were identified on unaged and aging FCTL from Zimbabwe through direct sequencing. Although some microflora might be missed due to the limitation of the method, our results revealed for the first time the bacterial diversities on unaged and aging Zimbabwe FCTL. This study identified many uncultured bacteria in unaged and aging FCTL. Overall, more uncultured bacteria species were found in the aging Zimbabwe FCTL than in those of K326 FCTL (Huang et al. 2010b). These uncultured bacteria likely contribute to flue-cured tobacco aging process.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Chen CM, Li XM, Yang JK, Gong XW, Li X, Zhang KQ (2008) Isolation of nicotine-degrading bacterium Pseudomonas sp. Nic22, and its potential application in tobacco processing. Int Biodeter Biodegr 62:226–231
Civilini M, Domenis C, Sebastianutto N, Bertoldi M (1997) Nicotine decontamination of tobacco agro-industrial waste and its degradation by micro-organisms. Waste Manag Res 15:349–358
Di Giacomo M, Paolino M, Silvestro D, Vigliotta G, Imperi F, Visca P, Alifano P, Parente D (2007) Microbial community structure and dynamics of dark fire-cured tobacco fermentation. Appl Environ Microbiol 73:825–837
Felsenstein J (1989) PHYLIP—phylogeny of inference package (version 3.2). Cladistics 5:164–166
Gontcharova V, Youn E, Wolcott RD, Hollister EB, Gentry TJ, Dowd SE (2010) Black box chimera check (B2C2): a windows-based software for batch depletion of chimeras from bacterial 16S rRNA gene datasets. Open Microbiol J 4:47–52
Guo X, Jin B, Chen G (2004) Tobacco fermentation. Tob Sci Technol 11:7–9 (Chinese)
Han J, Ye B (1997) Microorganism activity in tobacco fermentation and application. Chinese Tob Sci 1:13–14 (Chinese)
Hill JE, Hemmingsen SM, Goldade BG, Dumonceaux TJ, Klassen J, Zijlstra RT, Goh SH, Van Kessel AG (2005) Comparison of ileum microflora of pigs fed corn-, wheat-, or barley-based diets by chaperonin-60 sequencing and quantitative PCR. Appl Environ Microbiol 71:867–875
Holland SM (2004) Analytic rarefaction. http://www.uga.edu/∼strata/software/. Accessed 9 July 2007
Huang JW, Duan YQ, Zhe W, Wang MF, Zhang KQ, Yang JK (2010a) Improvement of flue-cured tobacco quality with Bacillus pumilus. Tob Sci Technol 8:61–64 (Chinese)
Huang JW, Yang JK, DuanYQ GuW, Gong XW, Zhe W, Can Su, Zhang KQ (2010b) Bacterial diversities on unaged and aging flue-cured tobacco leaves estimated by 16S rRNA sequence analysis. Appl Microbiol Biotechnol 88:553–562
Jensen C, Parmele H (1950) Fermentation of cigar-type tobacco. Ind Eng Chem 42:519–522
Jurkevitch E, Minz D, Ramati B, Barel G (2000) Prey range characterization, ribotyping, and diversity of soil and rhizosphere Bdellovibrio spp. isolated on phytopathogenic bacteria. Appl Environ Microbiol 66:2365–2371
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Li HJ, Li XM, Duan YQ, Zhang KQ, Yang JK (2010) Biotransformation of nicotine by microbiology: the case of Pseudomans spp. Appl Microbiol Biotechnol 86:11–17
Newton RP, Geiss VL. Jewell JN, Gravely LE (1977) Process for reduction of nicotine content of tobacco by microbial treatment. US Patent 4,037,609
Peng Y, Yang T, Liu M, Wang G, Li X (2009) Technical methods of accelerating tobacco fermentation. J Hebei Agri Sci 13:62–64 (Chinese)
Ruan AD, Min H, Peng X, Huang Z (2005) Isolation and characterization of Pseudomonas sp. strain HF-1, capable of degrading nicotine. Res Microbiol 156:700–706
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Sun L, Qiu F, hang X, Dai X, Dong X, Dong X, Song W (2008) Endophytic bacterial diversity in rice (Oryza sativa L.) roots estimated by 16S rDNA sequence analysis. Microb Ecol 55:415–424
Tamayo A, Cancho F (1953) Microbiology of the fermentation of Spanish tobacco. Int Congr Microbiol 6:48–50
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tringe SG, Zhang T, Liu X, Yu Y, Lee WH, Yap J, Yao F, Suan ST, Ing SK, Haynes M, Rohwer F, Wei CL, Tan P, Bristow J, Rubin EM, Ruan Y (2008) The airborne metagenome in an indoor urban environment. PLoS One 3(4):e1862
Williamson KE, Wommack KE, Radosevich M (2003) Sampling natural viral communities from soil for culture-independent analyses. Appl Environ Microbiol 69:6628–6633
Yang JK, Duan YQ, Chen CM, Li QH, Huang JQ, Zhang KQ (2008) Identification and phylogenesis analysis of cultivable microorganisms on tobacco leaf surface during aging. Tob Sci Technol 11:51–55 (Chinese)
Zhao MQ, Wang BX, Li FX, Qiu LY, Li FF, Wang SM, Cui JK (2007) Analysis of bacterial communities on aging flue-cured tobacco leaves by 16S rDNA PCR-DGGE technology. Appl Microbiol Biotechnol 73:1435–1440
Acknowledgments
We are grateful to Prof. Jianping Xu of the Dept. Biology, McMaster University, Canada, for valuable comments and critical discussions. This work was supported by the National Natural Science Foundation of China (approved no. 31060012), the West Light Foundation of the Chinese Academy of Sciences (to Jinkui Yang), and by projects from the Department of Science and Technology of Yunnan Province (approved nos. 2006GG22 and 2009CI052).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
List of GenBank accession numbers for individual nucleotide sequence (DOC 89 kb)
Rights and permissions
About this article
Cite this article
Su, C., Gu, W., Zhe, W. et al. Diversity and phylogeny of bacteria on Zimbabwe tobacco leaves estimated by 16S rRNA sequence analysis. Appl Microbiol Biotechnol 92, 1033–1044 (2011). https://doi.org/10.1007/s00253-011-3367-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3367-3