
Abstract
Flue-cured tobacco leaves (FCTL) contain abundant bacteria, and these bacteria play very important roles in the tobacco aging process. However, bacterial communities on aging FCTL are not fully understood. In this study, the total microbial genome DNA of unaged and aging flue-cured tobacco K326 were isolated using a culture-independent method, and the bacterial communities were investigated by restriction fragment length polymorphism analysis. Comparison of the number of operational taxonomic units (OTUs) between the cloned libraries from the unaged and aging FCTL showed that the microbial communities between the two groups were different. Fifty and 42 OTUs were obtained from 300 positive clones in unaged and aging FCTL, respectively. Twenty-seven species of bacteria exist in both the unaged and aging FCTL, Bacillus spp. and Pseudomonas spp. were two dominant genera in FCTL. However, 23 bacterial species were only identified from the unaged FCTL, while 15 species were only identified from the aging FCTL. Interestingly, more uncultured bacteria species were found in aging FCTL than in unaged FCTL.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tobacco is economically the most important nonfood crop worldwide. Because of its disagreeable odor and undesirable aroma, unaged flue-cured tobacco is not suitable as cigarette products. A further process called fermentation or aging for about 2 years is typically applied to improve the quality of the flue-cured tobacco. Aging greatly improves the aroma and other qualities desirable in smoking products. This process has been linked to the enzymatic actions of bacteria, fungi, and other chemical interactions within the leaves (Jensen and Parmele 1950). Many microorganisms on aging tobacco leaves likely play important roles in the aging process. For example, bacteria have been found as the agents initiating the fermentation process in bulks of cigar leaf tobacco (Jensen and Parmele 1950). Increasing reports have supported the microbial theory of fermentation as applied to the preparation of tobacco leaves for cigar-making purposes (Tamayo and Cancho 1953; Di Giacomo et al. 2007; Zhao et al. 2007; Yang et al. 2008; Gong et al. 2009).
Appropriate microorganisms and fermentation conditions are necessary in the aging process of tobacco leaves to improve quality and aroma of tobacco leaves (Zhao et al. 2007; Chen et al. 2008). Tamayo and Cancho (1953) were the first to inoculate tobacco leaves with microorganisms to improve the aroma. Later, thermophiles including eight strains of Bacillus subtilis, five strains of Bacillus coagulans, four strains of Bacillus megaterium, and three strains of Bacillus circulans were isolated from broadleaf tobacco. Some of these strains in pure single culture or mixed cultures were employed to enrich the normal thermophilic microbial flora of “sweating” tobacco (English et al. 1967). However, those studies employed traditional culture-dependent methods that might have caused an underestimate of microflora diversity. This is because only a fraction of the bacteria known in natural habitats can be cultivated on laboratory media (Amann et al. 1995; Head et al. 1998; Hugenholtz et al. 1998; Zhao et al. 2007).
In recent years, new technologies developed in molecular biology have overcome the limitations of culturing process for studying microbial diversities. These techniques not only can analyze the culturable microorganisms but also the unculturable ones. Culture-independent 16S rRNA gene sequence analysis has been employed to study bacterial communities in environmental samples without prior cultivation (Amann et al. 1995; Head et al. 1998; Hugenholtz et al. 1998). This method has revealed a significantly broader diversity of 16S rRNA gene sequence types than culture-based studies (Amann et al. 1995; Head et al. 1998; Hugenholtz et al. 1998; Sun et al. 2008). The combination of phylogenetic sequence analysis with restriction fragment length polymorphism (RFLP) of PCR-amplified bacterial 16S rRNA genes has become a powerful tool to investigate bacterial communities (Amann et al. 1995; Head et al. 1998).
Knowledge about the microbial communities in tobacco leaves of unaged and aging flue-cured tobacco leaves (FCTL) will provide important basis for artificial regulation of the aging and fermentation processes. However, there is currently only a limited knowledge about the microorganisms on FCTL, especially the uncultured microorganisms. Recently, Zhao et al. (2007) analyzed the bacterial communities on aging FCTL of zhongyan 100, NC89, and zhongyan 101 by 16S rRNA PCR-DGGE technology, and five dominant 16S rRNA DGGE bands were sequenced, which were found most similar to two cultured microbial species Bacteriovorax sp. EPC3, B. megaterium, and three uncultured microbial species, respectively. However, DGGE analysis tends to reveal only the dominant microflora, which is a serious deficiency because minor organisms may make important biological contributions to an ecosystem as other studies indicated (Yang et al. 2001). In this study, tobacco K326, one of the widely planted tobacco species in China, was chosen. The total microbial genomic DNAs were isolated from the unaged and aging FCTL K326, and the bacterial communities of these two samples were analyzed by 16S rRNA amplification and RFLP analysis. Moreover, the phylogenetic trees of bacteria in unaged and aging FCTL were constructed based on the 16S rRNA sequences.
Materials and methods
Sampling of leaf
Tobacco K326 (grade C3F) was collected from Hongyun Honghe Tobacco (Group) Co, Ltd. (Kunming, China). Tobacco samples were placed at room temperature ranging from 4.7°C to 23.7°C to age and were selected for sampling after 0 (unaged FCTL) and 24 months (aging FCTL). The two samples were immediately stored at −20°C and analyzed within a month.
DNA extraction from the microbial community of tobacco
The modified SDS-CTAB procedure (Zhou et al. 1996, 1997; Jackson et al. 1997) was used to isolate total bacterial genomic DNA from the surface of tobacco leaves. Twenty grams of leaves were collected from a composite sample, which were divided into three equal parts and placed in three flasks with 330 mL sterilized 0.1 M phosphate buffer (pH 7.0) for 30 min, respectively. Later, the tobacco leaves were washed with a sonicator for 10 min to get most of the bacteria on leaf surfaces suspended in the phosphate buffer. The bacteria were then collected by centrifugation at 10,000×g for 30 min. The bacteria were then suspended in 2 mL DNA extracting solution (0.1 M N2HPO4, 0.1 M EDTA, 0.1 M Tris-base, 1.5 M NaCl) and 80 μL proteinase K. This mixture solution was incubated in a 37°C water bath for 60 min, with mixing by gently inverting the tube every 20 min. Then, 1 mL 20% SDS and 60 μL β-mercaptoethanol were added. The mixture was further incubated in a 60°C water bath for 120 min, again with mixing by gently inverting the tube every 20 min. During this time, the sample was frozen with liquid nitrogen every 40 min. Centrifugation was used to remove the precipitate at 5,000×g for 10 min. The supernatant was mixed with an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1). After centrifugation at 8,000×g for 2 min, the supernatant was transferred to a new tube, DNA was precipitated by adding 0.6 times volume of isoamyl alcohol (−20°C) to the supernatant and isolated by centrifugation at 13,000×g for 10 min. The pellet was then washed with 200 μL of 70% ethanol and centrifuged at 13,000×g for 5 min. The DNA pellet was dried under vacuum and then suspended in 40 μL of TE buffer (pH 8.0). The genomic DNA isolated from three samples was mixed and used as template for 16S rRNA gene amplification.
Amplification of the bacterial 16S rRNA genes
The pair of primers 799f (5′-GGTAGTCCACGCCGTAAACGATG-3′; position 781 through 799 according to Escherichia coli number) and 1492r (5′-GGTTACCTTGTTACGACTT-3′; position 1492 through 1510 according to E. coli number) were selected to amplify the 16S rRNA of bacteria. The pair of primers 799f and 1492r could not amplify chloroplast DNA. In order to remove the possible inhibitors of PCR, 1/10–1/50 dilution of template DNA was used for PCR amplification of 16S rRNA sequences. The PCR amplification was done according to the conditions described by Sun et al. (2008), and the band size of approximately 735 bp was purified.
Construction of the 16S rRNA clone library
The purified PCR products were ligated into the pMD18-T vector (Takara, Japan) and transformed into competent cells (E. coli DH5α) to construct 16S rRNA clone library. In order to identify the diversity of bacteria on unaged and aging FCTL, two 16S rRNA clone libraries were respectively constructed. Three hundred clones containing the inserts of correct size were verified by colony PCR. All of the positive clones were screened by RFLP.
RFLP analysis of 16S rRNA
Inserts of 16S rRNA genes from recombinant clones were re-amplified with vector primers M13 (5′-CGACGTTGTAAAACGACGGCCAGT-3′) and RV (5′-GGAAAC AGCTATGACCATGATTAC-3′). The amplifications were subjected to RFLP analysis by separate enzymatic digestions with HhaI (Takara, Japan) and MspI (Takara, Japan) endonucleases following the manufacturer's instructions, and the digested DNA fragments were electrophoresed in 3% agarose gels. After staining with ethidium bromide, the gels were photographed using Genesnap from SynGene (USA), and scanning image analysis was performed with GeneTools from SynGene, with manual adjustments as required.
Estimation of the size of clone library
An operational taxonomic unit (OTU) was defined as a group of clones that had identical banding patterns (unique RFLP pattern) obtained from two independent digestions with different restriction enzymes. The diversity of the clone libraries was investigated by rarefaction analysis. Rarefaction curves were calculated using the RarefactWin program (Holland 2004).
DNA sequencing and phylogenetic analysis
One to three representative clones for each RFLP pattern was sequenced with the M13 and RV primers. Sequencing was done on an automated ABI 3730 sequencer (Applied Biosystems, USA). The resulting sequences were compared with those available in GenBank using the BLAST program to determine their approximate phylogenetic affiliation and sequence similarities (Altschul et al. 1990). Chimeric sequences were identified using the CHECK-CHIMERA program of the Ribosomal Database Project (Maidak et al. 1997).
Nucleotide sequences were initially aligned using Clustal X and then manually adjusted. Distance matrices and phylogenetic trees were calculated according to the Kimura two-parameter model (Kimura 1980) and neighbor-joining (Saitou et al. 1987) algorithms using the MEGA (version 4.0) software packages (Tamura et al. 2007). One thousand bootstraps were performed to assign confidence levels to the nodes in the trees. A clade was defined as a main branch in the phylogenetic tree, and a cluster was referred to as a sub-branch within a clade.
Nucleotide sequence accession numbers
The nucleotide sequence data reported in this paper were deposited in GenBank with accession numbers GQ128386, GQ128387, GQ128391–GQ128405, GQ128407–GQ128431, and GU722211–GU722260.
Results
Statistical analysis of the clone library and RFLP
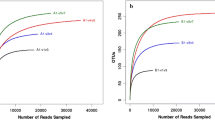
According to the RFLP patterns, 300 16S rRNA clones from unaged FCTL were grouped into 50 OTUs, and 300 clones from aging FCTL were grouped into 42 OTUs. The calculated rarefaction curve is shown in Fig. 1.

Rarefaction curve of bacterial 16S rRNA clone library of the unaged and aging FCTL K326
The 50 OTUs from the unaged FCTL consisted of four groups of bacteria: alpha-proteobacteria, gamma-proteobacteria, firmicutes, and high G + C Gram-positive bacteria (Table 1). Beta-proteobacteria were not isolated from the unaged FCTL, and two subclades of proteobacteria contained 191 clones, represented by 34 OTUs, accounted for 63.7% of the clone library. In the proteobacteria, the dominant species were related to Pseudomonas spp. and Enterobacter spp. Meanwhile, 105 clones were related to firmicutes, and this group was represented by 15 OTUs and accounted for 35% of the clone library. In the firmicutes, the dominant species were related to B. subtilis (FJ527658, EU880528), B. pumilus (EF488975), and B. circulans (AB271747). Moreover, the high G + C Gram-positive bacteria included only 1 OTU within the uncultured Micrococcus sp. (GU086431), accounting for 1.33% of the clone library (Table 1).
The 42 OTUs from the aging FCTL consisted of five groups of bacteria: alpha-proteobacteria, beta-proteobacteria, gamma-proteobacteria, firmicutes, and high G + C Gram-positive bacteria (Table 1). Alpha-, beta-, and gamma-proteobacteria were subclades of the proteobacteria, which made up the largest fraction of the clone library. Three groups of proteobacteria included 160 clones, represented by 28 OTUs, and accounted for 53.33% of the clone library. In the proteobacteria, the dominant species were related to Pseudomonas spp. Meanwhile, 136 clones were related to firmicutes, the second most abundant fraction, this group was represented by 13 OTUs and accounted for 45.33% of the clone library. In the firmicutes, the dominant species were related to Bacillus spp. Similarly, the high G + C Gram-positive bacteria included only 1 OTU within the uncultured Mycobacterium sp., accounting for 1.33% of the clone library (Table 1).
Phylogenetic analysis of the bacterial community on tobacco leaves
Phylogenetic trees (Fig. 2) was constructed based on the 16S rRNA sequences of the 50 OTUs and 42 OTUs from unaged and aging FCTL, respectively.


16S rRNA-based dendrogram showing the phylogenetic relationship of unaged (a) and aging FCTL (b). OTU sequences to the most closely related sequences obtained from GenBank and RDP database. Bootstrap values (n = 1,000 replicates) of ≥50% are reported as percentages. The scale bar represents the number of changes per nucleotide position. Archaebacterium Halococcus morrhuae (X00662) was used as the outgroup. Accession numbers are given at the end of each sequence. Alpha, alpha-proteobacteria; Beta, beta-proteobacteria; Gamma, gamma-proteobacteria; High, high G + C Gram-positive bacteria
Two phylogenetic trees of bacteria from unaged (Fig. 2a) and aging FCTL (Fig. 2b) shared similar shape, and those bacteria were divided into four clades (clade A, B, C, and D) (Fig. 2). Clade A, B, C, and D corresponded to alpha- and gamma-proteobacteria, firmicutes, and high G + C Gram-positive bacteria, respectively. Clade A consisted of two well-supported clusters (cluster 1 and 2), and most bacteria in this group were related to gamma-proteobacteria, with more OTUs identified in unaged FCTL (Fig. 2a) than that in aging FCTL (Fig. 2b). Enterobacter spp., one of the dominant species in cluster 1 of unaged FCTL, were not found in aging FCTL. However, Pseudomonas spp., a dominant species in cluster 2, was found in both samples. There were other differences between the two samples. For example, Xanthomonas spp. disappeared after aging for 24 months, but uncultured Acinetobacter spp. emerged in clade A of aging FCTL. Achromobacter xylosoxidans, a member of beta-proteobacteria also emerged in aging FCTL. Clade B consisted of two well-supported clusters (cluster 3 and 4), and these bacteria were related to alpha-proteobacteria. Within this cluster, similar OTUs were identified in both sample, though more OTUs and clones of Sphingomonas spp. were found in aging FCTL than in unaged FCTL. Clade C also consisted of two well-supported clusters (cluster 5 and 6), and they were related to firmicutes. However, more OTUs were identified from unaged FCTL, and several bacteria [such as Anoxybacillus sp. (GU166747) and Staphylococcus sp. (GU086429)] disappeared after 24 months of aging. Bacillus spp. were the dominant organisms of clade C, cluster 6. Micrococcus sp. and uncultured Mycobacterium sp. formed clade D in the unaged and aging FCTL, respectively. They both belonged to the high G + C Gram-positive bacteria.
Discussion
In the analysis of library, when the rarefaction curve became flat or reached a plateau, the size of the clone library would be considered representative of the diversity in the original sample (Guan et al. 2008). In this study, these rarefaction curves did become flat (Fig. 1), indicating that the size of our clone library was sufficiently large for the study and that these clone libraries included most of the dominant bacteria species on the FCTL.
Comparison of the number of OTUs between the cloned libraries from the unaged and aging FCTL showed that the microbial community in each group was different. We found that 23 species of bacteria were only identified from the unaged FCTL (Table 1). These bacteria likely failed to adapt to the FCTL aging environment. Most of these species belonged to gamma-proteobacteria, and Enterobacter is one of the most dominant genera, including Enterobacter oryzae (EF488760), Enterobacter cloacae (GQ426323), Enterobacter sakazakii (EF088366), and Enterobacter sp. (EF471901). On the contrary, 15 species of bacteria were only identified from the aging FCTL (Table 1), and these bacteria were likely enriched during the aging process and could contribute to the aging of FCTL. These bacteria were divided in alpha-, beta-, gamma-proteobacteria, and firmicutes, including Sphingomonas sp. (AB288313), A. xylosoxidans (FM163487), Aerococcus sp. (FJ405325), Bacillus amyloliquefaciens (EU855192), Bacillus licheniformis (AY017347), and uncultured bacteria (e.g., EU071494, FJ626756, EF511440, AB460075, and DQ346448). Moreover, the number of clones in FCTL also showed obvious changes after aging. The clones of gamma-proteobacteria were decreased by 14%, but the clones of firmicutes were increased by 10.33% and the clones of Bacillus spp. were increased from 31.6% to 42%, a result similar to previous reports (English et al. 1967), and suggested that these Bacillus species played an important role in improving the quality of tobacco leaves during aging process.
Recently, Zhao et al. (2007) reported that there were certain similarities of bacterial communities (similarity up to 63%) in tobacco leaves among the three varieties. In our study, 27 species of bacteria exist in both of unaged and aging FCTL (Table 1). Bacillus and Pseudomonas were two dominant genera in tobacco leaves. In these species, some bacteria had known functions. For example, Bacillus spp. can enhance the development of a desirable aroma and improve the smoking qualities of the tobacco (English et al. 1967). B. subtilis and B. circulans, enriched either alone or together, caused a more rapid appearance of a pleasing aroma in Pennsylvania “Wrapper B” filler tobacco (English et al. 1967). Bacillus thuringiensis is part of the natural microflora in the stored-tobacco environment, and this species might be involved in controlling pests in storage products (Kaelin et al. 1994). Similarly, another dominant bacterial species, Pseudomonas spp., can degrade nicotine and improve the quality of tobacco leaves (Chen et al. 2008; Ruan et al. 2005; Wang et al. 2005; Li et al. 2010).
The unculturable bacteria species in the two groups of tobacco leaves were also different. There were 45 clones of unculturable bacteria in the unaged FCTL, and these can be grouped into 9 OTUs. In contrast, 68 clones of unculturable bacteria were found in aging FCTL, and they can be grouped into 13 OTUs (Table 1). Among these, three OTUs were only identified from unaged FCTL, including uncultured Citrobacter sp. (GQ417492), uncultured bacterium (FJ795213), and uncultured soil bacterium (DQ248282). Meanwhile, seven OTUs were only identified from aging FCTL, including uncultured Sphingomonas sp. (FJ192268), uncultured Pseudomonas sp. (EU071494), uncultured Acinetobacter sp. (FJ192770, FJ193182), uncultured bacteria (GU272316, AB460075), and uncultured Mycobacterium sp. (EU071480). However, six OTUs were found in both of samples, including uncultured Pseudomonas sp. (EU341207), uncultured Escherichia sp. (EU175925), uncultured Acinetobacter sp. (FJ387638), uncultured Ralstonia sp. (FJ193264), and uncultured soil bacteria (FJ184323, DQ248243). These uncultured bacteria might contribute significantly to the tobacco leaf aging process and would be of great interest to isolate and culture them in the lab in order to understand their metabolic activities and potential applications to the cigarette industry.
Previous studies showed that unaged and early stage of aging FCTL contained abundant bacteria, and the numbers and types of bacteria decreased gradually during aging process (e.g., Qiu et al. 2000; Zhao et al. 2000; Zhu et al. 2001; Yang et al. 2008). Recently, Zhao et al. (2007) reported that the number and the intensity of DGGE bands were higher in tobacco leaves of 0 to 6 months aging than 9 to 12 months aging. In this study, more OTUs were also identified on unaged FCTL. Although some microflora might be missed due to the limitation of the method, our results revealed for the first time the bacterial diversities on unaged and aging FCTL (tobacco K326) by 16S rRNA RFLP technology. Our results provided a basis for clarifying the roles of microorganisms in FCTL and for establishing the condition to artificially enhance aging and fermentation.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Chen CM, Li XM, Yang JK, Gong XW, Li X, Zhang KQ (2008) Isolation of nicotine-degrading bacterium Pseudomonas sp. Nic22, and its potential application in tobacco processing. Int Biodeter Biodegr 62:226–231
Di Giacomo M, Paolino M, Silvestro D, Vigliotta G, Imperi F, Visca P, Alifano P, Parente D (2007) Microbial community structure and dynamics of dark fire-cured tobacco fermentation. Appl Environ Microbiol 73:825–837
English CF, Bell EJ, Berger AJ (1967) Isolation of thermophiles from broadleaf tobacco and effect of pure culture inoculation on cigar aroma and mildness. Appl Microbiol 15:117–119
Gong XW, Yang JK, Duan YQ, Dong JY, Zhe W, Wang L, Li QH, Zhang KQ (2009) Isolation and characterization of Rhodococcus sp. Y22 and its potential application to tobacco processing. Res Microbiol 160:200–204
Guan T, Wu J, Zhi X, Tang S, Xu L, Li W, Zhang L (2008) Actinobacterial diversity of a sediment sample from Xiaoerkule Lake (Chinese). Acta Microbiol Sin 48:851–856
Head IM, Saunders JR, Pickup RW (1998) Microbial evolution, diversity, and ecology, a decade of ribosomal RNA analysis of uncultivated microorganisms. Microb Ecol 35:1–21
Holland SM (2004) Analytic rarefaction. http://www.uga.edu/~strata/software/. Accessed 09 July 2007
Hugenholtz P, Goebel MB, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Jackson CR, Harper JP, Willoughby D, Roden EE, Churchill PF (1997) A simple, efficient method for the separation of humic substances and DNA from environmental samples. Appl Environ Microbiol 63:4993–4995
Jensen CO, Parmele HB (1950) Fermentation of cigar-type tobacco. Ind Eng Chem 42:519–522
Kaelin P, Morel P, Gadani F (1994) Isolation of Bacillus thuringiensis from stored tobacco and Lasioderma serricorne (F.). Appl Environ Microbiol 60:19–25
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Li HJ, Li XM, Duan YQ, Zhang KQ, Yang JK (2010) Biotransformation of nicotine by microorganism: the case of Pseudomonas spp. Appl Microbiol Biotechnol 86:11–17
Maidak BL, Olsen GJ, Larsen N, Overbeek R, McCaughey MJ, Woese CR (1997) The RDP ribosomal database project. Nucleic Acids Res 25:109–110
Qiu LY, Zhao MQ, Yu XM, Qi WC, Zhang WQ (2000) Isolation and identification of microflora on tobacco leaves during the natural fermentation of flue-cured tobacco (Chinese). Tob Sci Technol 3:14–17
Ruan AD, Min H, Peng XH, Huang Z (2005) Isolation and characterization of Pseudomonas sp. strain HF-1, capable of degrading nicotine. Res Microbiol 156:700–706
Saitou N, Nei M, Lerman LS (1987) The neighbor-joining method, a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sun L, Qiu F, Zhang X, Dai X, Dong X, Song W (2008) Endophytic bacteria diversity in rice (Oryzative L.) roots estimated by 16S rDNA sequence analysis. Microb Ecol 55:415–424
Tamayo AI, Cancho FG (1953) Microbiology of the fermentation of Spanish tobacco. Int Congr Microbiol 6:48–50
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Wang SN, Xu P, Tang HZ, Meng J, Liu XL, Ma CQ (2005) “Green” route to 6-hydroxy-3-succinoyl-pyridine from (S)-nicotine of tobacco waste by whole cells of a Pseudomonas sp. Environ Sci Technol 39:6877–6880
Yang CH, Crowley DE, Borneman J, Keen NT (2001) Microbial phyllosphere populations are more complex than previously realized. Proc Natl Acad Sci USA 8:3889–3894
Yang JK, Duan YQ, Chen CM, Li QH, Huang JQ, Zhang KQ (2008) Identification and phylogenesis analysis of cultivable microorganisms on tobacco leaf surface during aging (Chinese). Tob Sci Technol 11:51–55
Zhao M, Qiu L, Zhang W, Qi W, Yue X (2000) Study on the changes of biological activity of flue-cured tobacco leaves during aging (Chinese). Journal of Central China Agricultural University 19:537–542
Zhao MQ, Wang BX, Li FX, Qiu LY, Li FF, Wang SM, Cui JK (2007) Analysis of bacterial communities on aging flue-cured tobacco leaves by 16S rDNA PCR-DGGE technology. Appl Microbiol Biotechnol 73:1435–1440
Zhou JZ, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62:316–322
Zhou JZ, Davery E, Figure JB (1997) Phylogenetic diversity of a bacterial community determined from Siberian tundra soil DNA. Microbiology 143:3913–3919
Zhu DH, Chen R, Chen ZG, Zhou YF, Han JF, Jin BF, Song RB, Yu J (2001) The relationship between microorganisms and enzyme activities in flue-cured tobacco during aging and fermentation (Chinese). Acta Tob Sin 7:26–30
Acknowledgements
We are grateful to Prof. Jianping Xu of the Dept. Biology, McMaster University, for valuable comments and critical discussions. This work was supported by projects from the Department of Science and Technology of Yunnan Province (approved nos. 2006GG22 and 2009CI052).
Author information
Authors and Affiliations
Corresponding author
Additional information
Jingwen Huang and Jinkui Yang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Huang, J., Yang, J., Duan, Y. et al. Bacterial diversities on unaged and aging flue-cured tobacco leaves estimated by 16S rRNA sequence analysis. Appl Microbiol Biotechnol 88, 553–562 (2010). https://doi.org/10.1007/s00253-010-2763-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2763-4