
Abstract
A gene (Tx-est1) encoding a thermostable feruloyl-esterase was isolated from the genome of the Gram-positive hemicellulolytic thermophilic bacterium Thermobacillus xylanilyticus. This gene contains an open reading frame of 1,020 bp encoding a protein with molecular mass of 37.4 kDa, similar to feruloyl-esterases from cellulolytic bacteria and fungi. The recombinant enzyme Tx-Est1 was expressed and produced in Escherichia coli. Tx-Est1 contains the conserved putative lipase residues Ser 202, Asp 287, and His 322 which act as catalytic triad in its C-terminus part. Purified Tx-Est1 was active against phenolic acid derivatives and stable at high temperatures. Optimal activity was observed at 65 °C and the optimal pH was around 8.5. The kinetic parameters of the esterase were determined on various substrates. The enzyme displayed activity against methyl esters of hydrocinnamic acids and feruloylated arabino-xylotetraose, exhibiting high specificity and affinity for the latter. Our results showed that Tx-Est1 is a thermostable feruloyl-esterase which could be useful to hydrolyze arabinoxylans from graminaceous plant cell walls as the enzyme is able to release phenolic acids from a lignocellulose biomass.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Arabinoxylans are a predominant form of hemicellulose (Aman and Nordkvist 1983). These polymers consist of β-1,4-linked chains in which xylose residues are periodically substituted by arabinose, glucuronic acid, feruloyl, coumaroyl, or acetyl ester groups. In graminaceous plants, some phenolic compounds such as ferulic acid can form diferulic bridges, providing cross-linkages between the heteroxylans chains and between heteroxylans and lignin (Saulnier and Thibault 1999; Ishii 1997). The presence of such structures can reduce accessibility to polysaccharides (hemicelluloses and cellulose) and limit their enzymatic fractionation (Ishii 1997). Due to the heterogeneity and complexity of xylans, their complete enzymatic hydrolysis necessitates the combined action of several families of glycoside hydrolases, such as β-1,4 endoxylanases, arabinofuranosidases, glucuronidases, β-1,4 xylosidases and carbohydrate esterases (Shallom and Shoham 2003). Feruloyl- or cinnamoyl-esterases hydrolyze the ester bonds between phenolic acid compounds and the sugar residues in the arabinoxylans side chain. These enzymes could play an important role in plant cell wall hydrolysis as ester-linked hydroxycinnamic acid derivatives are responsible for the covalent linkages between xylan chains. Breakdown of the ester linkage makes the plant cell wall more accessible to enzyme attack and allows better solubilization of arabinoxylans by endoxylanases or other enzymes (Faulds et al. 2006; Faulds et al. 2004; Topakas et al. 2004; Panagiotou et al. 2007). Feruloyl-esterases have applications in biotechnological processes such as biorefining, pharmaceutical, and food industries (Koseki et al. 2009a; Faulds 2010). Although several chemical treatments, such as alkaline hydrolysis, are able to solubilize ester-linked phenolic compounds from a lignocellulose biomass, the use of feruloyl-esterase would be more environmentally friendly. To this issue, efficient biocatalysts are needed to increase xylan degradation within the plant cell biomass. However, thermostable feruloyl-esterase remained scarce (Huang et al. 2010; Abokitse et al. 2010; Donaghy et al. 2000). Thermophilic organisms are of special interest as their enzymes are active and stable at high temperatures (Turner et al. 2007). Thermobacillus xylanilyticus is a strict aerobic hemicellulolytic thermophilic spore-forming bacterium (Touzel et al. 2000) that is able to breakdown plant cell wall polysaccharides by producing enzymes such as glycoside hydrolases. Three thermostable enzymes, including two endoxylanases (GH10 and GH11) and an arabinofuranosidase (GH51), were obtained from this bacterium (Debeche et al. 2000; Samain et al. 1997; Connerton et al. 1999). These enzymes exhibited efficient lignocellulose biomass fractionation (Beaugrand et al. 2004a; Benamrouche et al. 2002; Remond et al. 2008). Therefore, T. xylanilyticus represents an interesting potential source of thermostable enzymes. In this paper, we report the biochemical characterization of a single domain thermostable carbohydrate esterase. This is the first feruloyl-esterase to be identified from T. xylanilyticus. The recombinant enzyme was purified and characterized on various synthetic substrates as well as on various lignocelluloses.
Materials and methods
Bacterial strains and growth conditions
T. xylanilyticus strain XE has been deposited in the Collection Nationale de Cultures de Microorganismes (France) under the number CNCM I-1017 and was grown in basal medium supplemented with 10% CO2 as previously described (Touzel et al. 2000). E. coli strains were grown aerobically at 37 °C in Luria–Bertani (LB) medium (Difco). E. coli transformants were selected by adding 50 μg.ml−1 of kanamycin (Sigma-Aldrich) to the LB medium.
DNA purification
Genome DNA from non-sporulated T. xylanilyticus culture was prepared with the QiaAmpDNA mini kit, according to the manufacturer’s recommendations. Plasmid DNA was purified from E. coli culture with the QIaprep Spin Miniprep kit (Qiagen, France). The concentrations and purities of the genome and plasmid DNA were determined by gel agarose electrophoresis and spectrophotometry (A260 and A280).
Isolation of Tx-est1 esterase gene by degenerate PCR and genome walking
Sequences of genes encoding esterase family 1 were obtained from the GenBank database. The block maker program (http://blocks.fhcrc.org/blocks/blockmkr/make_blocks.html) and CODEHOP (Rose et al. 1998) were used to generate blocks of similar amino acids and degenerate primers from four proteins from Bacillus clausii KSM-K16 (BAD64277), Bacillus halodurans C-125 (BAB05159), Bacillus licheniformis DSM-13 (AAU40191), and Geobacillus kaustophilus HTA426 (BAD75150). These primers are listed in Table 1 and corresponded to the highly conserved amino acids RRKWYHPEG and WTYWQPDIP. They led to the amplification of a 400-bp product.
Amplifications were performed with a two-step polymerase chain reaction (PCR) program: an initial denaturation at 94 °C for 10 min followed by five cycles consisting of a denaturation step at 94 °C for 30 s, an annealing step at 57 °C for 1 min, and an elongation step at 72 °C for 30 s. Forty cycles consisting of a denaturation step at 94 °C for 30 s, an annealing step at 50 °C for 1 min, and an elongation step at 72 °C for 30 s, were then performed. The reaction was terminated with a final elongation step of 7 min at 72 °C. After amplification, all PCR products were analyzed by agarose gel electrophoresis.
Nucleotide sequences of the entire gene encoding the esterase were obtained by applying genome-walking procedures given in the Universal Genome walker kit (Clontech, Takara Bio, France) and using four libraries constructed with genome DNA from T. xylanilyticus as template. The entire Tx-est1 gene was then isolated by successive amplifications and sequencing steps with esterase-specific primers (Table 1).
The presence of the putative esterase gene in the genome of T. xylanilyticus was verified by designing two specific primers (Est-fw and Est-rev) and amplifying the entire gene by PCR.
Cloning of the esterase genes in E. coli
The generally used cloning and transformation techniques were applied. The putative esterase entire gene and truncated esterases were amplified by PCR using two specific primers that allowed the amplification of a PCR fragment flanked by the cloning sites SacI (N-terminal) and HindIII (C-terminal) (Table 1). The resulting PCR product was cloned into SacI/HindIII digested pET28b vector (Merck, Germany). This produced the recombinant plasmid pET28b-6×His-Tx-Est1 that encoded an additional 36 amino acids N-terminal sequence including a 6×His-tag. Plasmids were used to transform electrocompetent E. coli JM109 (DE3) cells which were plated onto LB agar medium with kanamycin (50 μg.ml−1).
Identification of the catalytic domain by site-directed mutagenesis
Mutants of Tx-Est1 were created to confirm the identity of the active site residues. Mutants Ser202Ala, Asp287Ala, Asp285Ala, and His322Leu were generated by using the Quickchange site-directed mutagenesis kit (Agilent, France) and the primers listed in Table 1. The nucleotides modified by mutagenesis are indicated in upper case. Mutations were confirmed by sequence analysis of both DNA strands. Mutated plasmids were used to transform E. coli strains as described above.
Heterologous expression and purification of the 6×His-tagged-Tx-Est1 recombinant and mutants
E. coli JM109 DE3 cells were used to express the 6×His-Tx-Est1 recombinant protein and mutants. E. coli JM109DE3 cells were grown at 37 °C with agitation until the OD600 was 0.3–0.5. Then isopropyl-beta-d-thiogalactopyranoside (IPTG) (1.5 mM) was added and the culture was incubated for a further 16 h at 37 °C.
The 6×His-Tx-Est1 recombinant protein was purified from E. coli cell extracts by nickel-based-resin affinity chromatography (Profinity IMAC resin Bio-Rad, France) after cell disruption using a French press. The 6×His-Tx-Est1 recombinant protein was eluted with 100 mM imidazole and dialyzed against 50 mM NaH2PO4, pH 8 using a regenerated cellulose membrane (Spectra/Por; Spectrum Lab, The Netherlands) with a molecular mass cutoff between 6 and 8 kDa.
Protein determination
Protein concentration was determined by measuring the absorbance of the pure enzyme solution at 280 nm. The experimental extinction coefficient of the pure esterase at pH 8.5 was 40,073 M−1.cm−1.
Recombinant E. coli cytoplasmic proteins and purified recombinant proteins were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (12% acrylamide) according to the general instructions. The molecular weight of the recombinant protein was estimated with molecular weight standards (ColorPlus Prestained Protein Marker, New England Biolabs, UK). Protein bands were visualized by Coomassie blue staining.
The identity of the recombinant protein was confirmed by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI–TOF-MS) analyses. The MALDI-TOF-MS profiles were compared with the theoretical trypsin digestion of the enzyme and the percentage coverage and mascot score were deduced. Analyses were performed at the proteomic analysis facilities at INRA (Clermont-Ferrand/Theix, France). Its experimental molecular weight was determined by matrix-assisted laser desorption/ionization–liquid chromatography mass spectrometry (MALDI–LC-MS).
DNA sequencing and analysis
Purified PCR products and plasmids from the E. coli transformants, purified as described above, were used for automated sequencing. Sequences of both strands were determined with universal M13 forward and reverse primers or with the specific primers listed in Table 1 (Millegen Labège, France).
Sequence analyses were performed at the National Center for Biotechnology Information (NCBI) and the EBI websites (http/www.ncbi.nlm.nih.gov/ and http://www.ebi.ac.uk, respectively). Phylogenetic analyses were based on neighbor-joining analysis after alignment with ClustalW. Evolutionary distances were calculated using the Prodist matrix. Bootstrap analyses were performed with 500 replicates. Analyses were conducted using the facilities at the phylogeny.fr website (Dereeper et al. 2008).
Determination of esterase activities
Esterase activities were assayed against various substrates dissolved in sodium phosphate buffer (50 mM NaH2PO4, pH 8.5) and tested for 5 min to measure the initial rates of reaction.
Acetyl-esterase activity was measured by determining the rate of hydrolysis of p-nitrophenyl-acetate (Sigma-Aldrich) to p-nitrophenol (pNP) at 50 °C for 5 min. The reaction mixture contained 0.8 ml of sodium phosphate buffer, 0.1 ml of 20 mM pNP-acetate, and 0.1 ml of appropriately diluted enzyme. The increased absorbance at 405 nm was measured with a recording spectrophotometer (Uvikon 933). The extinction coefficient for pNP under these assay conditions was 12.8 × 103 M−1.cm−1.
Feruloyl-esterase activity was tested against feruloylated arabino-xylotetraose (FAX4), produced and purified as previously described (Remond et al. 2008), and methyl ferulate (MF), from Apin Chemicals (UK). Activity was assayed by analyzing the ferulic acid released from FAX4 and MF according to previously described procedures (Ralet et al. 1994). The extinction coefficients of ferulic acid, methyl ferulate, and FAX4 in the condition assays were 3,271 M−1.cm−1; 6,196 M−1.cm−1; and 10,500 M−1.cm−1, respectively. Activity against methyl-sinapinate (MS) and methyl-p-coumarate (MpC) was assayed as described above by analyzing the sinapic and p-coumaric acids released. The extinction coefficients of methyl-sinapinate, methyl-p-coumarate, sinapic and p-coumaric acids were 10,576 M−1.cm−1, 6,912 M−1.cm−1, 4,180 M−1.cm−1 and 4,025 M−1.cm−1, respectively. In all cases, substrate autohydrolysis was evaluated by incubating MF, MS, MpC, and pNP-acetate at 50 °C in pH 8.5 sodium phosphate buffer during 5 min.
One unit of enzyme activity was defined as the amount of enzyme required to produce 1 μmole of product per min in the standard conditions assay.
Determination of optimal pH and temperature, pH stability, and thermostability
Optimal pH was determined by measuring feruloyl-esterase activity against MF in universal buffer (with pH between 3 and 10) for 5 min at 50 °C. Stability at pH 8.5 was determined by analyzing residual enzyme activity after incubating the enzyme in NaH2PO4 buffer for 48 h at 20 °C or 50 °C or for several weeks at 4 °C. Optimal temperature was also estimated by measuring the enzyme activity at various temperatures (20–80 °C) in phosphate buffer at pH 8.5. Thermal stability was determined by measuring the residual activity at 15-and 60-min intervals at temperatures of 50, 55, 60, 65, 70, 75 °C for 24 h. The remaining activity was assayed under standard conditions as described above.
Determination of kinetic parameters: K m and k cat
Kinetic parameters against methyl esters of hydroxycinnamic acid (methyl ferulate MF, methyl-p-coumarate MpC, and methyl-sinapinate MS) and FAX4 were determined for the initial rates of reaction by incubating the enzyme with these substrates at 50 °C pH 8.5 for 10 min. The substrate concentrations ranged from 10 μM to 2.5 mM. K m and k cat values were calculated using Lineweaver–Burk plots after monitoring the release of ferulic, sinapic, p-coumaric acids by high-performance liquid chromatography (HPLC) (waters) as described in Beaugrand et al. (2004b). Kinetic parameters against pNP-acetate were determined spectrophotometrically as described for the specific activity assay, with substrate concentrations ranging from 10 μM to 5 mM.
Effect of inhibitors and mutants activity
The effects of two inhibitors, phenylmethylsulphonyl fluoride (PMSF) and diethylpyrocarbonate (DEPC) were determined as described above with 200 μM MF as substrate and with 1.5, 3, and 30 μg of enzyme. The concentrations of inhibitor added to the reaction volume ranged from 1 to 5 mM. Mutant activities were estimated as previously described, but the reactions were performed with the Ser202Ala, Asp287Ala, and His322Leu mutants and without enzyme dilution. The residual activity represented the percentage of activity remaining, under the tested conditions, in relation to the activity of the wild-type esterase or the maximal activity without inhibitors at 50 °C.
Determination of esterase activity against lignocellulose substrates
Destarched wheat bran and wheat straw were provided by ARD (Pomacle, France). Maize bran was provided by Limagrain (Ennezat, France) and destarched as previously described (Beaugrand et al. 2004b). Ester-linked phenolic acids, referred to as alkali-extractable phenolic acids, were released from 40 mg of substrate using 2 M NaOH with constant stirring at 35 °C as described by Beaugrand et al. (2004b). Prior to enzyme hydrolysis, the lignocellulose substrates (800 mg) were hydrated in 20 ml of sodium phosphate buffer (50 mM NaH2PO4, pH 8.5) for 16 h at 50 °C. Esterase was added (4 IU per gram of substrate) and the reaction occurred under constant stirring for 24 h at 50 °C. Synergistic effects between GH11 xylanase from =T. xylanilyticus (Tx-Xyl11) and Tx-Est1 were investigated under the same conditions, using 280 IU of Tx-Xyl11. Negative controls without enzyme were performed under the same conditions. All experiments were conducted in duplicate.
After a 24-h incubation, the supernatants were separated from the residual biomass by centrifuging (5,000 × g for 10 min), and the reaction was stopped by boiling at 100 °C for 10 min. The supernatant was acidified to pH 1–2 with 6 M HCl, then mixed with 3,4,5,trimethoxy-trans-cinnamic acid as internal standard. The phenolic acid content was quantified by HPLC as described previously and expressed as micrograms of ferulic, p-coumaric or diferulic acids released per gram of lignocellulose substrate.
The results were compared by applying statistical analyses (Student’s test). Significant differences were obtained with p ≤ 0.05.
Nucleotide sequence accession numbers
The nucleotide sequence data for the Tx-Est1 are available at Genbank under the accession number GU592999.
Results
Tx-Est1 a new feruloyl-esterase from T. xylanilyticus
Degenerate PCR combined with genome-walking procedures were used to identify a xylanolytic enzyme from the T. xylanilyticus genome and an open reading frame of 1,020 bp. In order to confirm that T. xylanilyticus actually possessed this open reading frame (ORF), the corresponding gene was isolated by PCR using genome DNA as template and specific primers. The ORF was successfully amplified in the genome of the bacterium as a DNA fragment of approximately 1 kb was amplified. Southern blot analysis indicated that this ORF was present as a single copy in the genome (data not shown).
The ORF was named Tx-Est1 and encoded a 340-amino acid protein with a predicted molecular mass of 37.7 kDa and an isoelectric point of 5.04. No signal sequence was predicted in the N-terminus part of the protein which suggests that Tx-Est1 is an intracellular protein.
The deduced amino acid sequence was used to perform a BLAST search at the NCBI database. This search revealed a high similarity to the well-characterized feruloyl-esterase domains of the bifunctional enzyme XynY from Clostridium thermocellum (CAA58242 52%) and XynB from Ruminococcus albus (BAB3949 50%). Similarities of around 45% were observed with other feruloyl-esterases from rumen bacteria (Cellulosilyticum ruminicola ACZ98598, Prevotella ruminicola ACN78954, and Ruminococcus flavefaciens CAB93667). Surprisingly, Tx-Est1 presented 43% similarity with feruloyl-esterase from the rumen fungus Orpinomyces sp. PC-2 (AAF70241) whereas no similarity was observed with other well-characterized fungal feruloyl-esterases. All these proteins belong to the carbohydrate esterase family 1 in the carbohydrate-active enzymes database (CAZY) classification (http://www.cazy.org).
The phylogenetic analysis shown in Fig. 1 indicates that Tx-Est1 is closely related to XynB (R. albus) and Xyn Y (C. thermocellum). Both proteins cluster with the feruloyl-esterase domain of XynZ (C. thermocellum, AAA23286) and FaeA from C. ruminicola. Except for FaeA from Orpinomyces sp., Tx-Est1 is relatively distant from the other fungal feruloyl-esterases such as FaeB from Neurospora crassa (CAC05587) or cinnamoyl esterase from Penicillium funiculosum (CAC14144).

Phylogenetic relationships between Tx-Est1 esterase and other characterized bacterial and fungal feruloyl-esterases belonging to the CE1 family. Sequence names are shown in the tree. The bootstrap value is based on 500 replicates. Accession numbers of used proteins are XynY from C. thermocellum (CAA58242), XynB from R. albus (BAB3949), Xyn1 from Ruminococcus sp. (CAA90271), bifunctionnalGH10/Fae1 from P. ruminicola (ACN78954), XynE R. flavefaciens (CAB93667), FaeA Orpinomyces sp. PC-2 (AAF70241), XynZ from C. thermocellum (AAA23286), FaeA from C. ruminicola (ACZ98598), FaeB from N. crassa (CAC05587), and cinnamoyl esterase from P. funiculosum (CAC14144)
Multiple alignment of the entire protein with the most closely related esterase sequences (>40% similarity) indicated that Tx-Est1 contained, in its C-terminal part, the conserved putative lipase triad residues Ser202, Asp287, and His322. The putative catalytic nucleophile Ser 202 belongs to the Gly-X-Ser-X-Gly lipase pentapeptide consensus. The putative oxyanion hole residues in the N-terminal part of the protein were conserved between the different proteins. In Tx-Est1 these residues are probably constituted by the consensus “Gly-Val-Gly-Gly-Asp” at position 91-95. The different amino acid sequence analyses indicated that Tx-Est1 could be classified as a feruloyl-esterase protein.
Overexpression and purification of Tx-Est1
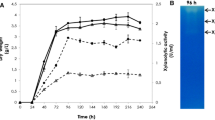
We investigated the biochemical properties of Tx-Est1 by expressing the protein with a six-Histidine-tag at its N-terminus in E. coli JM109 DE3. The transformant successfully expressed the recombinant protein after induction with 1.5 mM IPTG. The protein was present in the soluble fraction without inclusion bodies and represented around 20% of the whole proteins (Fig. 2, lane 1). One liter of a culture of recombinant E. coli gave 12.5 mg of purified protein with a specific activity of 25.41 IU.mg−1 (measured in relation to methyl ferulate 250 μM) (Table 2).

SDS-PAGE electrophoresis of the purified Tx-Est1 revealed by Coomassie blue detection, lane 1 soluble fraction induced with 1.5 mM IPTG, lane 2 protein molecular weight standards, lane 3: purified 6×His-Tx-Est1 eluted with imidazole 100 mM
The molecular mass of the purified protein obtained by SDS-PAGE was around 41 kDa (Fig. 2, lane 3). The measured molecular mass from the MALDI–LC-MS value was 37.4 kDa. This difference could be explained by the 36 amino acids, including the 6-histidine tag, at the N-terminus part of the protein which was removed during the Maldi analysis. These results were confirmed by analyzing the purified protein by Western blotting with an anti-histidine serum. The recombinant protein probed by the anti-His serum migrated at the expected size (data not shown). The MALDI-TOF-MS analysis indicated that the purified recombinant protein corresponded to the theoretical sequence obtained by genome-walking procedures as 21 experimental peptides perfectly matched the theoretical sequence of Tx-Est1. This represents 62% coverage, with a Mascot score of 213, between the experimentally obtained peptides and the theoretical Tx-Est1 protein sequence.
Enzyme characterization
The optimal pH for esterase activity was determined at 50 °C, using methyl ferulate as substrate. Activity was measured over a pH range of 3.0 to 9.0. Tx-Est1 exhibited between 17% and 66% of its maximal activity in the pH range of 5.0 to 8. The highest activity was measured at pH 8.5 (Fig. 3b). The enzyme exhibited 80% of its maximal activity after incubation for 48 h at pH 8.5 and 50 °C and could be kept at 4 °C for several weeks at this pH without denaturation (data not shown).

Effect of pH (a) and temperature (b) on activity of Tx-Est1. Measurements were performed in duplicate, represented values corresponded to mean of two values with standard deviation
The effect of temperature on esterase activity was examined between 20 and 80 °C. Optimal Tx-Est1 activity was obtained at 65 °C. The activities at 50, 55, and 80 °C were 85%, 92% and 80% of the maximal activity, respectively (Fig. 3a). The thermostability of the esterase was examined at 50, 55, 60, 65, 70, and 80 °C. Tx-Est1 displayed high thermal stability at 50 and 55 °C with half-lives >24 and 8 h, respectively. An activity loss of 16% was observed after incubation for 24 h at 50 °C. Stability of the enzyme decreased at 65 °C, with a half-life of approximately 30 min and was less than 10 min at 70 and 80 °C (data not shown).
The calculated kinetic parameters and activities against various substrates are listed in (Table 2). The specific activity of the purified enzyme on methyl ferulate was 25.41 ± 0.41 IU.mg−1. The enzyme exhibited great affinity for MS and MF (K m 103 and 107 μM, respectively). The K m observed with MpC was very high (1,285 μM) and significantly different to that of MF and MS. A very low K m (12.5 μM), close to the minimal substrate concentration tested in the enzyme reaction, was observed with FAX4 indicating that Tx-Est1 showed high affinity for this substrate. This affinity for FAX4 was associated with the high turnover number and catalytic efficiency of the enzyme (k cat 27 s−1 and k cat/K m 2,170 s−1.mM−1) Furthermore, the specific activity against FAX4 was significantly higher than against MF (40.90 IU.mg−1). These results indicate that Tx-Est1 hydrolyzed the feruloylated oligosaccharide more rapidly and more efficiently than the methyl ester substrates. Tx-Est1 exhibited low specific activity and low affinity with pNP-acetate.
The effects of esterase and lipase inhibitors, well described in the literature, on Tx-Est1 activity showed that this activity was strongly inhibited by PMSF, a serine protease inhibitor. Only 3.9% ± 0.04 and 1.69% ± 0.03 of the activity remained with 1 and 5 mM of PMSF, respectively. Diethylpyrocarbonate, a histidine modifier, also led to inhibition. Residual activity in the presence of DEPC 1 mM represented 83% of the total activity. The decrease after contact with DEPC 5 mM was around 55%. These results indicate that serine and histidine residues are important for Tx-Est1 activity.
Characterization of Tx-Est1 mutants
The results obtained with these inhibitors were confirmed by substituting the amino acids predicted to be involved in the triad catalytic site of the enzyme by site-directed mutagenesis. Mutants Ser202Ala, Asp285Ala, Asp287Ala, and His322Leu were constructed, expressed and purified. Table 3 gives the relative activities of the mutant proteins compared to the activity of the wild-type enzyme. The Tx-Est1-Asp285Ala mutant showed similar activity to that of the wild-type esterase indicating that aspartic acid 285 was not involved in the catalytic triad. No activity was observed with the mutants Ser202Ala, Asp287Ala, and His322Leu, which confirmed the importance of all three residues in Tx-Est1 activity.
Alkali-extractable phenolic acid content of lignocellulose substrates
Destarched wheat bran (DWB), destarched maize bran (DMB), and wheat straw (WS) contain different amounts of alkali-extractable phenolic acids. A high content of ferulic acid (FA) was determined in DMB (18.86 ± 1.22 mg.g−1) compared to DWB and WS that contained 4.97 ± 0.03 mg.g−1 and 2.41 ± 0.01 mg.g−1 of dry matter, respectively. In addition to FA, the amounts of p-coumaric acid recovered from DWB, DMB, and WS were 0.13 ± 0.01, 2.24 ± 0.12 mg.g−1, and 3.44 ± 0.10 mg.g−1 of dry matter, respectively. A large quantity of total alkali-extractable diferulic acid was obtained with DMB (8.72 ± 0.35 mg.g−1) compared with 0.77 ± 0.08 mg.g−1 and 0.30 ± 0.002 mg.g−1 from DWB and WS, respectively. The diferulic acids detected in the alkali-extractable phenolic acids from the three lignocelluloses were mainly 5-5′ diferulate and 8-O-4 diferulate (data not shown).
Phenolic acid release from lignocelluloses and synergistic effects between Tx-Xyl11 xylanase and Tx-Est1
Figure 4 shows the amounts of phenolic acid released from DWB, DMB, and WS after 24 h incubation with Tx-Est1, Tx-Xyl11, or both enzymes. Tx-Est1 was able to release FA from the three lignocellulose substrates tested. Tx-Est1 was more active on DWB than on WS or DMB. The amount of FA released from DWB was 500 ± 21 μg per gram of substrate which corresponded to approximately 10% of the ester-linked FA. Tx-Est1 released around 4.7% of the alkali-extractable FA in WS (110 ± 9.80 μg FA per gram of WS). Very little FA was solubilized from DMB, representing 1.7% of the alkali-extractable FA content of DMB (320 ±70 μg.g−1). The amount of FA released by Tx-Est1 from the three substrates tested was significantly higher than the amount of FA released by Tx-Xyl11 alone.

Phenolic acids release from DWB, DMB, and WS obtained with Tx-Est1, Tx-Xyl11, and Tx-Est1 associated with Tx-Xyl11. a Ferulic acid, b p-Coumaric acid, c Diferulic acids. Measurements were performed in duplicate; represented bars corresponded to mean of two values with standard deviation
A synergistic effect was observed when Tx-Est1 was used in combination with Tx-Xyl11 on all three substrates tested. On destarched maize bran, the amount of FA released by the two enzymes was around 2.5% of the DMB alkali-extractable FA (480 ± 60 μg.g−1). This value was 1.5-fold higher than with Tx-Est1 alone (p ≤ 0.03). On wheat straw, the FA released by the two enzymes represented 17% of the alkali-extractable FA of WS (410 ± 10 μg.g−1), which was 3.7-fold higher than with esterase (p ≤ 0.001). The best synergism was obtained with destarched wheat bran, the FA released by the two enzymes representing 78% of the DWB ester-linked FA (3,890 ± 30 μg.g−1) which was 7 times higher than the amount released with Tx-Est1 alone (p ≤ 0.03).
Tx-Est1 released 10 ± 1, 140 ± 30, and 180 ± 5 μg.g−1 of p-coumaric acid (pCA) from DWB, DMB, and WS, respectively. These values represent 7.5%, 6.2%, and 5.2% of the alkali-extractable p-coumaric acid from DWB, DMB, and WS. Although the pCA released by Tx-Est1 was higher than that liberated by Tx-Xyl11, the differences were not statistically significant except for the result obtained for WS. The combination of Tx-Est1 and Tx-Xyl11 enhanced pCA release from DSB and WS. The amount of pCA released by the two enzymes was sevenfold and 2.5-fold higher than with Tx-Est1 alone and represented 54% and 13% of the alkali-extractable pCA in DWB and WS, respectively. No synergism between Tx-Est1 and Tx-Xyl11 was observed on DMB.
Tx-Est1 was able to release diferulic acids from DWB and WS (20 ± 1, 40 ± 2 μg.g−1, respectively). These values accounted for 2.5% and 13% of the alkali-extractable diferulic acids from DWB and WS, respectively. When Tx-Est1 was used in combination with Tx-Xyl11, the amount of diferulate released from DWB and WS was significantly higher (150 ± 10 and 120 ± 10 μg.g−1, respectively) indicating that 19% and 40% of the alkali-extractable diferulic acids was released. In contrast, the amount of diferulic acids released from destarched maize bran, by either Tx-Est1 or both Tx-Est1 and Tx-Xyl11, was very low (<1% of the total diferulic acid from DMB). A significant increase in diferulic acid release was observed with Tx-Xyl11 but synergism remained limited. In all cases, diferulic acids were mainly 8-O-4 diferulate (80% of diferulic released) and 5-5′ diferulate (20% of diferulic released) (data not shown).
Discussion
Although T. xylanilyticus is an efficient producer of different xylanolytic enzymes (Debeche et al. 2000; Samain et al. 1997; Connerton et al. 1999), no esterase from this bacterium had been identified or characterized before this study. We focused on identifying a single domain intracellular esterase gene in the T. xylanilyticus genome. Within the hemicellulolytic enzymes identified in this bacterium, until now, only one family 11 xylanase was secreted in the medium. As for Tx-Est1, a family 10 xylanase (Connerton et al. 1999) and an arabinofuranosidase (Debeche et al. 2000) are intracellular enzymes. In another hemicellulolytic aerobic bacterium (Bacillus stearothermophilus), it is known that a main extracellular xylanase cleaves xylan and generates short oligomers with various substitutions. These oligosides are internalized and are then degraded to monomers by intracellular hemicellulases (Shulami 1999). This strategy allows the utilization of the substrates mainly by the bacterium. So one could suppose that in T. xylanilyticus oligoxylosides produced by the family 11 xylanase could be internalized and cleaved by intracellular hemicellulases such as Tx-Est1.
Tx-Est1 shares homologies with the feruloyl-esterase derived from cellulolytic bacteria and fungi such as C. thermocellum, R. flavefaciens, R. albus, and Orpinomyces sp. (Blum et al. 2000; Fontes et al. 1995; Aurilia et al. 2000; Dodd et al. 2009; Grepinet et al. 1988a, b; Nakamura et al. 2002). According to the functional classification of feruloyl esterases proposed by Crepin et al. (2004) four functional classes (A, B, C, D) have been ascribed, based on their primary amino acid sequence, activities on hydroxycinnamic acid methyl esters, ability to release diferulic acid and their preferential induction medium. According to this classification, the above enzymes were not classified in these four groups due to the absence of biochemical characterization. With Tx-Est1, those enzymes belong to carbohydrate esterase family 1 (CAZY) and are phylogenetically close, so that they could cluster in the same group (Crepin et al. 2004).
Several bacterial esterases have been identified, including an intracellular esterase from the wine-associated lactic acid bacteria Oenococcus oeni, two intestinal feruloyl esterases from Lactobacillus johnsonii and EstE1 from the ruminal hemicellulolytic Butyrivibrio proteoclasticus (Sumby et al. 2009; Lai et al. 2009; Goldstone et al. 2010), but these enzymes do not share significant homology with Tx-Est1 except for some conserved motifs representative of the esterase family. Tx-Est1 is a thermostable feruloyl-esterase showing optimal activity at 65 °C and a thermostability >24 h at 50 °C. This temperature is similar to the optimal temperature of feruloyl-esterase from Clostridium stercorarium and higher than the optimal temperatures of fungal feruloyl-esterases, which were found to be optimally active at mesophilic temperatures between 37 °C to 60 °C (Donaghy et al. 2000; Koseki et al. 2009b). In the same way, the optimal pH for Tx-Est1 is 8.5. Although some fungal feruloyl-esterases are relatively stable under alkaline conditions (Fae1 and Fae-2 from Aspergillus niger, FaeB from Aspergillus nidulans, rAoFaeC from Aspergillus oryzae, FoFaeC from Fusarium oxysporum, StFaeA from Sporotrichum thermophile) their optimal pH is still neutral or acidic except for Fae1 from A. niger for which the optimal pH is 9.0 (Topakas et al. 2004; Hegde and Muralikrishna 2009; Shin and Chen 2007; Koseki et al. 2009b; Moukouli et al. 2008). Currently, few thermostable and alkaline resistant feruloyl-esterases have been characterized (Donaghy et al. 2000; Huang et al. 2010; Abokitse et al. 2010). However, for the development of industrial applications as biorefineries, thermostable and robust enzymes are generally required (Turner et al. 2007). In this issue, the identification and characterization of this new debranching feruloyl thermostable esterase is very interesting in the objective to better fractionate the hemicelluloses part of plant cell walls. Biochemical data showed that the specific activity of Tx-Est1 on methyl ferulate is lower than that of purified esterase from C. stercorarium (25 IU.mg−1 vs 75 IU.mg−1) but higher than that of other previously reviewed bacterial and fungal esterases (Topakas et al. 2007). The substrate specificity of Tx-Est1 was compared with that of previously described fungal feruloyl-esterases. Tx-Est1 exhibits higher affinity for MS and MF than fungal FAEs (Crepin et al. 2003; Moukouli et al. 2008; Shin and Chen 2007; Rumbold et al. 2003; Poidevin et al. 2009; Benoit et al. 2006; Topakas et al. 2003; Topakas et al. 2004; Kroon et al. 2000; Faulds et al. 2005; Koseki et al. 2009b). Different feruloylated oligosaccharides have been used in various studies to characterize feruloyl-esterases (Vardakou et al. 2004; Faulds and Williamson 1995; Kroon et al. 2000; Benoit et al. 2006; Ralet et al. 1994) and showed that the affinity and catalytic efficiency of feruloyl-esterase increased with increasing sugar chain length. In our case, Tx-Est1 presented a very high affinity for feruloylated arabino-xylotetraose and was able to release FA from this oligosaccharide with greater efficiency than from methyl esters of hydroxycinnamic acids. This result could be correlated with the possible physiological role of this intracellular enzyme, in which preferential substrate could be branched oligoxylosides.
In the aim to investigate the interest of Tx-Est1 on hemicelluloses fractionation, we then evaluated the ability of Tx-Est1 to release phenolic acids from a lignocellulose biomass by testing the activity of Tx-Est1 on agricultural by-products such as wheat bran, wheat straw, and maize bran. These substrates were chosen for their very different compositions. Destarched maize bran is very rich in FA and diferulic acids compared to destarched wheat bran. The FA content of wheat straw, compared with wheat bran, is slightly lower, but wheat straw contains higher amount of pCA. These results were in agreement with previous studies of alkali-extractable phenolic acid compositions (Saulnier and Thibault 1999; Beaugrand et al. 2004b; Lequart et al. 1999).
Our data demonstrated that Tx-Est1 alone was able to release phenolic acids from all three lignocellulose biomasses. A synergistic effect was observed, when associated with Tx-Xyl11, whatever the substrate used. Tx-Est1 was able to release FA, pCA, and diferulic acid from destarched wheat bran. When Tx-Est1 was associated with GH11 xylanase from T. xylanilyticus, its efficiency in releasing these products was seven to eightfold higher. The combination of Tx-Est1 and Tx-Xyl11 reported here is among the best synergistic action described up till now (Topakas et al. 2004; Topakas et al. 2003; Faulds et al. 2006; Bartolome et al. 1997; Kroon et al. 2000; Faulds et al. 2005). Very few studies have focused on the release of pCA from destarched wheat bran. Faulds et al. (2004) reported that 50% of pCA was liberated from DWB by a heat-stable multiactive enzyme preparation from H. insolens.
The total FA and pCA released from wheat straw by Tx-Est1 was around 10% of the total alkali-extractable FA and pCA. A threefold increase was obtained in the presence of Tx-Xyl11. These modest yields could be explained by the high content of etherified forms in the case of FA (46%) and the high lignin content of wheat straw (Pan et al. 1998). Other studies have often used pretreated wheat straw due to the high recalcitrance of this lignified biomass. Purified FaeA and FaeB from A. niger were thus able to release 45% and 74% of alkali-extractable FA and pCA, respectively from steam-exploded wheat straw (Benoit et al. 2006). Furthermore, release of diferulic acid from wheat straw was more important even when Tx-Est1 was used alone, and a synergic effect was observed when Tx-Est1 was associated with xylanase. Given the recalcitrance of wheat straw, this high release of diferulic acid, combined with the release of FA and pCA, demonstrates the efficiency of Tx-Est1 and the synergism of the two enzymes on this substrate.
Tx-Est1 had a very limited effect on destarched maize bran, whatever the phenolic acid considered. Hence, no synergism with xylanase was observed as regards pCA release and only a twofold increase in FA and diferulic acid production was obtained by the combined action of Tx-Est1 and Tx-xyl11. This was probably due to the complexity of maize bran hemicelluloses which are highly substituted and contain large amounts of diferulic acid cross-links in the hemicellulose chains (Saulnier and Thibault 1999). In several studies, thermal or enzyme pretreatments were applied to improve the enzyme digestibility of maize bran (Benoit et al. 2006; Saulnier et al. 2001; Faulds and Williamson 1995). The FaeA of A. niger released 40% and 20% of FA and pCA, respectively from autoclaved maize bran, whereas the same enzyme only liberated 4% of FA from non-treated maize bran (Benoit et al. 2006; Poidevin et al. 2009). Autoclaving or a flash explosion pre-treatment of maize bran increased ferulic acid release, with the enzyme preparation Novozym 342, to 80% and 70%, respectively (Saulnier et al. 2001). A milder treatment was used in our study, since the maize bran was destarched by washing with hot water (60 °C). This could explain the weak hydrolysis yield by our enzyme with this substrate. The enzymatic hydrolysis of hemicelluloses implies a use of accessory enzymes such as feruloyl-esterases. Most of the feruloyl-esterases studied are of fungal origin and the majority have been found to be optimally active at, or in the region of, mesophilic temperatures and under acidic conditions.
Our results show that the feruloyl-esterase from T. xylanilyticus is robust, thermostable and alkali tolerant. Tx-Est1 is able to efficiently release ferulic acid as well as p-coumaric and diferulic acids from the plant cell wall either alone or in synergism with xylanase. The de-esterification by Tx-Est1 is more effective on non-pretreated destarched wheat bran and wheat straw, particularly with regard to diferulic and p-coumaric acid release. On more complex biomass such as maize bran, pretreatments will be necessary before application of Tx-Est1.
These characteristics may be advantageous in industrial or biotechnological applications, such as the bioconversion of a lignocellulose biomass, in which thermostability, pH resistance, and biocatalyst efficiency are important factors. Hydrolysis of the hemicellulose part of the plant cell wall requires the cooperation of several enzymes. The solubilization of arabinoxylan is usually increased by combining endoxylanases with esterase or using complex multi-enzyme preparations (Faulds et al. 2006; Topakas et al. 2004; Panagiotou et al. 2007). The interest of using Tx-Est1 for lignocellulose conversion is currently being validated, and synergism experiments with three other hemicellulases from T. xylanilyticus (GH10 and GH11 xylanases and GH51 arabinosidase) are in progress.
References
Abokitse K, Wu M, Bergeron H, Grosse S, Lau PCK (2010) Thermostable feruloyl esterase for the bioproduction of ferulic acid from triticale bran. Appl Microbiol Biotechnol 87:195–203
Aman P, Nordkvist E (1983) Chemical composition and in vitro degradability of botanical fractions of cereal straw. Swe J Agric Res 13:61–67
Aurilia V, Martin JC, McCrae SI, Scott KP, Rincon MT, Flint HJ (2000) Three multidomain esterases from the cellulolytic rumen anaerobe Ruminococcus flavefaciens 17 that carry divergent dockerin sequences. Microbiology 146:1391–1397
Bartolome B, Faulds CB, Kroon PA, Waldron K, Gilbert HJ, Hazlewood G, Williamson G (1997) An Aspergillus niger esterase (ferulic acid esterase III) and a recombinant Pseudomonas fluorescens subsp cellulosa esterase (Xy1D) release a 5-5′ ferulic dehydrodimer (diferulic acid) from barley and wheat cell walls. Appl Environ Microbiol 63:208–212
Beaugrand J, Chambat G, Wong V, Goubet F, Remond-Zilliox C, Paes G, Benamrouche S, Debeire P, O’Donohue M, Chabbert B (2004a) Impact and efficiency of GH10 and GH11 thermostable endoxylanases on wheat bran and alkali-extractable arabinoxylans. Carbohydr Res 339:2529–2540
Beaugrand J, Croner D, Debeire P, Chabbert B (2004b) Arabinoxylan and hydroxycinnamate content of wheat bran in relation to endoxylanase susceptibility. J Cereal Sci 40:223–230
Benamrouche S, Crônier D, Debeire P, Chabbert B (2002) A chemichal and histological study of the effect of 1-4-b-endoxylanase treatment on wheat bran. J Cereal Sci 36:253–260
Benoit I, Navarro D, Marnet N, Rakotomanomana N, Lesage-Meessen L, Sigoillot JC, Asther M (2006) Feruloyl esterases as a tool for the release of phenolic compounds from agro-industrial by-products. Carbohydr Res 341:1820–1827
Blum DL, Kataeva IA, Li XL, Ljungdahl LG (2000) Feruloyl esterase activity of the Clostridium thermocellum cellulosome can be attributed to previously unknown domains of XynY and XynZ. J Bacteriol 182:1346–1351
Connerton I, Cummings N, Harris GW, Debeire P, Breton C (1999) A single domain thermophilic xylanase can bind insoluble xylan: evidence for surface aromatic clusters. Biochim Biophys Acta 1433:110–121
Crepin VF, Faulds CB, Connerton IF (2003) Production and characterization of the Talaromyces stipitatus feruloyl esterase FaeC in Pichia pastoris: identification of the nucleophilic serine. Protein Expr Purif 29:176–184
Crepin VF, Faulds CB, Connerton IF (2004) Functional classification of the microbial feruloyl esterases. Appl Microbiol Biotechnol 63:647–652
Debeche T, Cummings N, Connerton I, Debeire P, O’Donohue MJ (2000) Genetic and biochemical characterization of a highly thermostable α-l-arabinofuranosidase from Thermobacillus xylanilyticus. Appl Environ Microbiol 66:1734–1736
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:465–469
Dodd D, Kocherginskaya SA, Spies MA, Beery KE, Abbas CA, Mackie RI, Iko C (2009) Biochemical analysis of a β-d-xylosidase and a bifunctional xylanase-ferulic acid esterase from a xylanolytic gene cluster in Prevotella ruminicola 23. J Bacteriol 191:3328–3338
Donaghy JA, Bronnenmeier K, Soto-Kelly PF, McKay AM (2000) Purification and characterization of an extracellular feruloyl esterase from the thermophilic anaerobe Clostridium stercorarium. J Appl Microbiol 88:458–466
Faulds CB (2010) What can feruloyl esterases do for us? Phytochem Rev 9:121–132
Faulds CB, Williamson G (1995) Release of ferulic acid from wheat bran by a ferulic acid esterase (Fae-III) from Aspergillus niger. Appl Microbiol Biotechnol 43:1082–1087
Faulds CB, Mandalari G, LoCurto R, Bisignano G, Waldron KW (2004) Arabinoxylan and mono- and dimeric ferulic acid release from brewer’s grain and wheat bran by feruloyl esterases and glycosyl hydrolases from Humicola insolens. Appl Microbiol Biotechnol 64:644–650
Faulds CB, Molina R, Gonzalez R, Husband F, Juge N, Sanz-Aparicio J, Hermoso JA (2005) Probing the determinants of substrate specificity of a feruloyl esterase, AnFaeA, from Aspergillus niger. FEBS J 272:4362–4371
Faulds CB, Mandalari G, Lo Curto RB, Bisignano G, Christakopoulos P, Waldron KW (2006) Synergy between xylanases from glycoside hydrolase family 10 and family 11 and a feruloyl esterase in the release of phenolic acids from cereal arabinoxylan. Appl Microbiol Biotechnol 71:622–629
Fontes CM, Hazlewood GP, Morag E, Hall J, Hirst BH, Gilbert HJ (1995) Evidence for a general role for non-catalytic thermostabilizing domains in xylanases from thermophilic bacteria. Biochem J 307:151–158
Goldstone DC, Villas-Boas SG, Till M, Kelly WJ, Attwood GT, Arcus VL (2010) Structural and functional characterization of a promiscuous feruloyl esterase (EstE) from the rumen bacterium Butyrivibrio proteoclasticus. Proteins 78:1457–1469
Grepinet O, Chebrou MC, Beguin P (1988a) Nucleotide sequence and deletion analysis of the xylanase gene (XynZ) of Clostridium thermocellum. J Bacteriol 170:4582–4588
Grepinet O, Chebrou MC, Beguin P (1988b) Purification of Clostridium thermocellum xylanase Z expressed in Escherichia coli and identification of the corresponding product in the culture medium of C. thermocellum. J Bacteriol 170:4576–4581
Hegde S, Muralikrishna G (2009) Isolation and partial characterization of alkaline feruloyl esterases from Aspergillus niger cfr 1105 grown on wheat bran. World J Microbiol Biotechnol 25:1963–1969
Huang YC, Chen GH, Chen YF, Chen WL, Yang CH (2010) Heterologous expression of thermostable acetylxylan esterase gene from Thermobifida fusca and its synergistic action with xylanase for the production of xylooligosaccharides. Biochem Biophys Res Commun 400:718–723
Ishii T (1997) Structure and functions of feruloylated polysaccharides. Plant Sci 127:111–127
Koseki T, Fushinobu S, Ardiansyah SH, Komai M (2009a) Occurrence, properties, and applications of feruloyl esterases. Appl Microbiol Biotechnol 84:803–810
Koseki T, Hori A, Seki S, Murayama T, Shiono Y (2009b) Characterization of two distinct feruloyl esterases, AoFaeB and AoFaeC, from Aspergillus oryzae. Appl Microbiol Biotechnol 83:689–696
Kroon PA, Williamson G, Fish NM, Archer DB, Belshaw NJ (2000) A modular esterase from Penicillium funiculosum which releases ferulic acid from plant cell walls and binds crystalline cellulose contains a carbohydrate binding module. Eur J Biochem 267:6740–6752
Lai KK, Lorca GL, Gonzalez CF (2009) Biochemical properties of two cinnamoyl esterases purified from a Lactobacillus johnsonii strain isolated from stool samples of diabetes-resistant rats. Appl Environ Microbiol 75:5018–5024
Lequart C, Nuzillard J-M, Kurek B, Debeire P (1999) Hydrolysis of wheat bran and straw by an endoxylanase: production and structural characterization of cinnamoyl-oligosaccharides. Carbohydr Res 319:102–111
Moukouli M, Topakas E, Christakopoulos P (2008) Cloning, characterization and functional expression of an alkalitolerant type C feruloyl esterase from Fusarium oxysporum. Appl Microbiol Biotechnol 79:245–254
Nakamura M, Nagamine T, Takenaka A, Aminov RI, Ogata K, Tajima K, Matsui H, Benno Y, Itabashi H (2002) Molecular cloning, nucleotide sequence and characteristics of a xylanase gene (XynA) from Ruminococcus albus 7. Anim Sci J 73:347–352
Pan GX, Bolton JL, Leary GJ (1998) Determination of ferulic and p-coumaric acids in wheat straw and the amounts released by mild acid and alkaline peroxide treatment. J Agric Food Chem 46:5283–5288
Panagiotou G, Olavarria R, Olsson L (2007) Penicillium brasilianum as an enzyme factory; the essential role of feruloyl esterases for the hydrolysis of the plant cell wall. J Biotechnol 130:219–228
Poidevin L, Levasseur A, Paes G, Navarro D, Heiss-Blanquet S, Asther M, Record E (2009) Heterologous production of the Piromyces equi cinnamoyl esterase in Trichoderma reesei for biotechnological applications. Lett Appl Microbiol 49:673–678
Ralet MC, Faulds CB, Williamson G, Thibault JF (1994) Degradation of feruloylated oligosaccharides from sugar-beet pulp and wheat bran by ferulic acid esterases from Aspergillus niger. Carbohydr Res 263:257–269
Remond C, Boukari I, Chambat G, O’Donohue M (2008) Action of a GH 51 α-l-arabinofuranosidase on wheat-derived arabinoxylans and arabino-xylooligosaccharides. Carbohydr Polym 72:424–430
Rose TM, Schultz ER, Henikoff JG, Pietrokovski S, McCallum CM, Henikoff S (1998) Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res 26:1628–1635
Rumbold K, Biely P, Mastihubova M, Gudelj M, Gübitz G, Robra KH, Prior BA (2003) Purification and properties of a feruloyl esterase involved in lignocellulose degradation by Aureobasidium pullulans. Appl Environ Microbiol 69:5622–5626
Samain E, Debeire P, Touzel JP (1997) High level production of a cellulase-free xylanase in glucose-limited fed batch cultures of a thermophilic Bacillus strain. J Biotechnol 58(2):71–78
Saulnier L, Thibault JF (1999) Ferulic acid and diferulic acids as components of sugar-beet pectins and maize bran heteroxylans. J Sci Food Agric 79:396–402
Saulnier L, Marot C, Elgorriaga M, Bonnin E, Thibault JF (2001) Thermal and enzymatic treatments for the release of free ferulic acid from maize bran. Carbohydr Polym 45:269–275
Shallom D, Shoham Y (2003) Microbial hemicellulases. Curr Opin Microbiol 6:219–228
Shin HD, Chen RRZ (2007) A type B feruloyl esterase from Aspergillus nidulans with broad pH applicability. Appl Microbiol Biotechnol 73:1323–1330
Shulami S, Gat O, Sonenshein AL, Shoham Y (1999) The glucuronic acid utilization gene cluster from Bacillus stearothermophilus T-6. J Bacteriol 181:3695–3704
Sumby KM, Matthews AH, Grbin PR, Jiranek V (2009) Cloning and characterization of an intracellular esterase from the wine-associated lactic acid bacterium Oenococcus oeni. Appl Environ Microbiol 75:6729–6735
Topakas E, Stamatis H, Biely P, Kekos D, Macris BJ, Christakopoulos P (2003) Purification and characterization of a feruloyl esterase from Fusarium oxysporum catalyzing esterification of phenolic acids in ternary water-organic solvent mixtures. J Biotechnol 102:33–44
Topakas E, Stamatis H, Biely P, Christakopoulos P (2004) Purification and characterization of a type B feruloyl esterase (StFae-A) from the thermophilic fungus Sporotrichum thermophile. Appl Microbiol Biotechnol 63:686–690
Topakas E, Vafiadi C, Christakopoulos P (2007) Microbial production, characterization and applications of feruloyl esterases. Process Biochem 42:497–509
Touzel J-P, O’Donohue M, Debeire P, Samain E, Breton C (2000) Thermobacillus xylanilyticus gen. Nov., sp. Nov., a new aerobic thermophilic xylan-degrading bacterium isolated from farm soil. Int J Evol Syst Biol 50:315–320
Turner P, Mamo G, Karlsson EN (2007) Potential and utilization of thermophiles and thermostable enzymes in biorefining. Microb Cell Fact 6:1–23
Vardakou M, Katapodis P, Topakas E, Kekos D, Macris BJ, Christakopoulos P (2004) Synergy between enzymes involved in the degradation of insoluble wheat flour arabinoxylan. Innov Food Sci Emerg Technol 5:107–112
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rakotoarivonina, H., Hermant, B., Chabbert, B. et al. A thermostable feruloyl-esterase from the hemicellulolytic bacterium Thermobacillus xylanilyticus releases phenolic acids from non-pretreated plant cell walls. Appl Microbiol Biotechnol 90, 541–552 (2011). https://doi.org/10.1007/s00253-011-3103-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3103-z