
Abstract
A hypothetical protein AN1772.2 of Aspergillus nidulans was found to have a 56% identity with a known type C ferulic acid esterase (FAE) from Talaromyces stipitatus. In addition, it contained a 13-amino acid conserved region flanking the characteristic G-X-S-X-G motif of a serine esterase, suggesting a FAE function for the protein. The putative FAE was successfully cloned from the genomic DNA and expressed in Saccharomyces cerevisiae. The recombinant protein exhibited high FAE activities. Therefore, its function as an FAE was unequivocally determined. About 86% of the enzyme activity was found in the growth medium, indicating that the native signal peptide was effective in the yeast expression system. The recombinant FAE was purified to its homogeneity, and subsequently characterized. The FAE is stable over an unusually wide range of pH (4.0–9.5), has a pH optimum of 7.0, and a temperature optimum of 45°C. A substrate specificity profiling reveals that the enzyme is a type B FAE, despite its strong sequence homology with type C FAEs, raising an interesting question on the role of the conserved region in substrate specificity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The prospect of broad applications of feruloyl esterase or ferulic acid esterase (FAE) has fueled much of the interest in the enzyme. Phenolic acids released from plant cell wall by the action of FAEs have long been used as food preservatives to inhibit microbial cell growth. Ferulic acid is an effective natural antioxidant with potential applications in pharmaceutical and food industries (Crepin et al. 2003a,b). FAE could be used in pulp and paper process and as animal feed additive to facilitate nutrient assimilation (Mathew and Abraham 2004; Record et al. 2003). In addition, FAE could be used to release ferulic acid from biomass and subsequently be converted to natural vanillin. Further, as the cross-linking through ferulic acid ester bond substantially increases the recalcitrance of biomass and the resistance to enzymatic hydrolysis, FAE is expected to play a vital role in the delignification and depolymerization of polysaccharide chains. Furthermore, besides being exploited as a hydrolase, FAE was recently shown to be a good catalyst in synthesizing sugar–phenolic esters (Topakas et al. 2003a,b) and has the potential to be used to functionalize sugar polymers by adding phenolic derivatives onto the natural biopolymers. These varied applications require FAE to work under a range of pHs and temperatures. The extremely diverse substrates may also require multiple FAEs with different specificities. The subtle specificity differences, often underappreciated, may very well be important in determining the optimal synergy between FAEs and other hemicellulases. Therefore, the discovery of novel FAE continues to be an active research area.
Traditional enzyme screening played an important role in identifying many of the FAEs known today (Crepin et al. 2003a; Faulds and Williamson 1995; Rumbold et al. 2003; Shin and Chen 2006; Topakas et al. 2003a,b, 2004, 2005; Topakas and Christakopoulos 2004; Wang et al. 2004). However, the available genome sequence for an ever-increasing number of microorganisms allows the development of a complementary, yet, perhaps more powerful tool for enzyme discovery. In this paper, we report discovery of the first FAE from Aspergillus nidulans through the use of its published genome sequence. AN1772.2 of FGSC A4, annotated as a hypothetical protein, was found to have a 56% sequence identity with a known FAE. The gene was cloned from the genomic DNA and the protein was successfully expressed in Saccharomyces cerevisiae. The protein was confirmed to be an FAE. Subsequent purification and characterization reveal that it has unique properties that make it valuable in many potential applications.
Materials and methods
Strains and culture media
Escherichia coli JM109 was used for construction and propagation of vectors and Luria–Bertani medium was used for its cultivation. S. cerevisiae INVSc.1 (Invitrogen, Carlsbad, CA, USA) was the host for heterologous expression of the hypothetical protein AN1772.2. The complete synthetic medium without uracil (referred as SC-U medium) for yeast strains contained 0.67%(w/v) of yeast nitrogen base, 0.192% (w/v) of yeast synthetic medium supplements without uracil (Sigma, St. Louis, MO, USA), and 2%(w/v) of a carbon source such as glucose, raffinose, or galactose, as indicated. Genomic DNA was extracted from A. nidulans FGSC A4 and used as a template for subsequent PCR amplification.
Vectors and chemicals
Taq DNA polymerase and Phusion DNA polymerase were purchased from New England Biolabs (Beverly, MA, USA). pGEM-T Easy vector and pYES2.1-TOPO vector were purchased from Promega (Madison, WI, USA) and Invitrogen, respectively. Methyl caffeate (MCA), methyl sinapate (MSA), methyl ferulate (MFA), and methyl-p-coumarate (MpCA) were from Apin Chemicals (Oxon, UK), and ferulic acid, caffeic acid, p-coumaric acid, and sinapic acid were from Sigma.
Vector constructions and transformations
The hypothetical protein AN1772.2 gene was PCR-amplified from genomic DNA using primers designed based upon the available AN1772.2 gene sequence (GenBank NW101269) AN-F (forward) and ANHIS-R (reverse) (Table 1). The DNA amplification was carried out with 35 cycles of denaturation (30 s at 95°C), annealing (30 s at 68°C), and extension (2 min at 72°C), followed by 10 min of further extension at 72°C. The PCR product was cloned into the pGEM-T Easy vector to determine the DNA sequence by a Perkin-Elmer ABI Prism 310 fluorescent DNA analyzer. To splice the intron contained within the gene (between 18,758 and 18,811 bp), a PCR was performed using primers designed for splicing, with SP-F (forward) and SP-R (reverse) flanking the intron (Table 1). A high fidelity Phusion DNA polymerase producing blunt ends was used for the PCR amplification. The primers were phosphorylated at 5′-ends for self-ligation of the PCR product. The PCR was carried out with 35 cycles of denaturation (15 s at 95°C), annealing (30 s at 68°C), and extension (1 min at 72°C), followed by 10 min of further extension at 72°C. The amplified PCR product (4.6 kb) was self-ligated by a T4 DNA ligase, from which the spliced gene was PCR-amplified using Taq DNA polymerase using primers AN-F and ANHIS-R. The amplified PCR product was cloned into pYES2.1/V5-HIS TOPO vector, and then transformed into S. cerevisiae INVSc.1 using S.c. EasyComp™ Transformation kit of Invitrogen.
Screening of recombinant S. cerevisiae transformants
The transformed S. cerevisiae INVSc.1 was plated on a selective SC-U medium and incubated for 2 days at 30°C. Twenty individual clones were cultivated in liquid SC-U supplemented with 2% (w/v) galactose as carbon source and the inducer of GA, and FAE activity was measured after 20 h of induction. One milliliter of liquid culture medium of each clone was centrifuged at 3,000×g for 10 min to remove cells, and then FAE activity were assayed with MFA as substrate as previously described (Shin and Chen 2006).
Production and purification of the recombinant feruloyl esterase
For production of recombinant feruloyl esterase, the recombinant S. cerevisiae harboring AN1772.2 gene was cultivated in SC-U medium supplemented with 2% (w/v) raffinose as the carbon source for 1 day at 30°C, and then inoculated into a production medium, SC-U supplemented with 2% (w/v) galactose, to OD600=0.4. The culture was incubated for 20 h at 30°C in a biological shaker (250 rpm).
To purify the recombinant feruloyl esterase, 500 ml of culture broth was centrifuged and concentrated 100-fold by an ultrafiltration system with a Pellicon 10,000 MWCO membrane (Millipore, Bedford, MA, USA). The culture supernatant was dialyzed overnight at 4°C against a 50-mM sodium phosphate buffer (pH 8.0), applied to a HIS-Select™ Nickel HC Affinity Gel from Sigma. After washing three times with a 50-mM sodium phosphate buffer (pH 8.0) containing 0.3 M NaCl, 10 mM imidazole, and 1% Triton X-100, the bound proteins were eluted twice with 250 μl of elution buffer [50 mM sodium phosphate buffer (pH 8.0) containing 0.3 M NaCl and 250 mM Imidazole]. The eluted protein was quantified using the Bradford method (Bradford 1976) and its homogeneity was analyzed with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Deglycosylation and determination of molecular weight
To determine the exact molecular weight of purified FAE, the eluted protein was treated with Endo-H, N-glycosidase from New England Biolabs, at 37°C for 1 h. The treated and untreated samples were analyzed with SDS-PAGE for molecular weight with and without glycosylation using the Mini-Protean II electrophoresis kit (BioRad, CA, USA) on a 10–20% gradient PAGE Ready gel.
Determination of pH, temperature optima, and pH stability and thermostability
The optimal pH was determined by measuring the FAE activity with pH varied over the range of 4.0–10.0 by carrying out the reaction in the following buffers: 50 mM acetate–phosphate buffer (pH 4.0–7.5), 50 mM Tris–HCl buffer (7.5–9.0), and 50 mM glycine–NaOH buffer (pH9.0–10.0). The pH stability was determined by analyzing the residual enzyme activity after incubating the enzyme in an appropriate buffer at 4°C for 12 h. The optimal temperature was determined by measuring the enzyme activity at various temperatures (30–65°C) in a phosphate buffer (100 mM, pH 7.0). The thermostability was determined by measuring the residual enzyme activity after incubating the enzyme solution at a temperature (30–65°C) for 30 min.
Enzyme assays
FAE activity was assayed by analyzing the ferulic acid released from MFA as previously described (Shin and Chen 2006). The assay was routinely carried out at 40°C for 30 min in a 50-mM Na–phosphate buffer (pH 7.0) containing 1 mM MFA, and the liberated ferulic acid was analyzed by high-performance liquid chromatography (HPLC). One unit of FAE activity was defined as the amount of enzyme that liberates 1 μmol ferulic acid/min.
Analytical methods
Cell growth was determined by the measurement of absorbance of culture broth at 600 nm. Protein concentration was determined by the Bradford method (Bradford 1976) using a commercial kit (BioRad) in which bovine serum albumin was used as the standard.
Quantitative analysis of ferulic acid was performed by HPLC on a LiChrospher 100 RP-18 column (Agilent Technologies, CA, USA). Detection was carried out at 280 nm based on calibration curves prepared using standard ferulic acid (Sigma) in water. A gradient elution was used with 0.01% acetic acid (A) and methanol (B) at a flow rate of 1.0 ml/min. A linear gradient was run as follows: t=0 min, 80:20 (A:B); t=4 min, 80:20; t=24 min, 60:40; t=27 min, 0:100; t=29 min, 0:100; t=30 min, 80:20; and t=35 min, 80:20.
Results
Identification of AN1772.2 as a putative type C feruloyl esterase
FAEs were classified into four types (A–D) based on their substrate specificity and amino acid sequence (Crepin et al. 2004). Type C FAEs with broad substrate specificity were found in Aspergillus niger (FAE-B) (de Vries et al. 2002), Talaromyces stipitatus (FAEC) (Crepin et al. 2003a,b), and Sporotrichum thermophile (StFaeC) (Topakas et al. 2005). A Basic Local Alignment Search Tool sequence analysis indicated that AN1772.2 from A. nidulans FGSC A4 had a 56% identity with FAEC from T.stipitatus. The corresponding protein was annotated as a hypothetical protein with unknown functions. The full sequence of 527 amino acids includes a secretion signal peptide of 19 amino acids (MALLRHLLPVLTVGSAVQS) based upon the prediction using SignalP, which is a web-based program (http://www.cbs.dtu.dk/services/SignalP). The predicted mass and isoelectric point (pI) of the mature protein were 56,107.32 Da and pH 4.66, respectively, by calculations using the Compute pI/Mw tool of ExPASY (http://ca.expasy.org/tools/pi_tool.html). As shown in Table 2, this protein has one motif that is characteristic of the serine esterase family (G-X-S-X-G) [15], suggesting that it is an esterase. In particular, the motif (G-C-S-T-G) matches exactly with that of the other two known type C FAEs, whereas G-H-S-L-G is typical of type A FAEs. For type B and type D FAEs, however, the motif was less conserved than type A and C FAEs. Moreover, there appears a conserved region of 13 amino acids (SYYLGCSTGGRQG) encompassing the motif among the type C FAEs. Similarly, there is a conserved region of 16 amino acids (ALTVTGHSLGASLAAL) for three type A FAEs (with a one amino acid exception with A. niger FAE), suggesting that the region surrounding the esterase motif may be important in determining the substrate specificity. This analysis strongly suggests that the hypothetical protein AN1772.2 is a putative type C FAE.
Cloning of the AN1772.2 gene
Figure 1 shows the vector constructions for cloning and expression of AN1772.2 gene in S. cerevisiae. The cloning strategy was designed such that the native signal sequence of the gene, required for translocation of the expressed protein from endoplasmic reticulum to extracellular locale, is retained and a six-histidine tag added at the C-terminal for facile purification. The AN1772.2 gene was first amplified from A. nidulans FGSC A4 genomic DNA using primers (AN-F and ANHIS-R, Table 1) designed based upon the AN1772.2 gene sequence (GenBank accession no. NW101269). The PCR product was cloned into the pGEM-T Easy vector for DNA sequence confirmation. A strategy was developed to splice the intron contained before cloning into the yeast expression vector. A pair of primers flanking the intron (SP-F and SP-R, Table 1) was designed to amplify the entire vector pGEM-T-AN1772.2 (4.6 kb) except the intron. A high fidelity polymerase was used for the PCR amplification due to the large size of the DNA. Sequence analysis confirmed the success of splicing. The amplified vector pGEM-T-AN1772.2 containing no intron (pGEM-T-SPAN1772.2) was self-ligated. The spliced gene, amplified from pGEM-T-SPAN1772.2 using AN-F and ANHIS-R primers (Table 1), was then cloned into pYES2.1/V5-HIS-TOPO vector and subsequently transformed into the S. cerevisiae INVSc.1. Twenty transformants were selected for their ability to grow on a minimal medium without uracil and subsequently screened for FAE activities as detailed in the “Materials and method.” The best clone showing the highest FAE activity was selected for further study.

Construction of vectors for expression of hypothetical protein AN1772.2 gene in S. cerevisiae
Production and purification of the recombinant FAE
Figure 2 shows a typical time profile of FAE production in the heterologous host. Cells harboring the expression vector were grown with a minimum medium supplemented with galactose. FAE activity could be first detected in the medium 8 h after inoculation into the galactose-containing medium, and peaked at 18 h with a titer of 4.5 U/l. The FAE production was highly growth-associated and a rapid decrease of FAE activity was observed after the cessation of cell growth. A majority of FAE activity (86%) was found in the culture medium, indicating an efficient secretion of the expression system with the native signal peptide. Only a small portion of the recombinant FAE was present intracellularly, yet a slow but steady increase over time was observed even after the sharp decrease of the extracellular FAE activity. The sharp decrease of extracellular FAE activity might be due to the proteolytic degradation occurring at the end of the cell growth.

Feruloyl esterase production in the recombinant S. cerevisiae harboring AN1772.2 gene. The expression of AN1772.2 gene in the recombinant S. cerevisiae was induced by cultivation on the SC-U minimal medium containing 2% (w/v) galactose at 30°C and 250 rpm. Open squares: extracellular FAE activity, open circles: intracellular FAE activity, filled square: total FAE activity, and filled triangles: biomass
The recombinant FAE was purified from the concentrated culture broth by immobilized metal affinity chromatography (IMAC) (Table 3). This affinity chromatography allows a one-step purification to its homogeneity. Forty-seven micrograms of the purified protein was obtained from 500 ml of culture broth, corresponding to a yield of 36% based on total activity and a purification factor of 3 based on specific activity (Table 3).
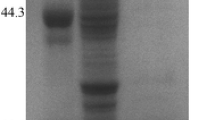
The homogeneity of the purified recombinant AN1772.2 FAE was examined on a SDS-PAGE, which appeared as a single band (Fig. 3). The molecular weight was estimated to be 130 kDa, which more than doubled its predicted molecular weight (56,107.32 Da). This discrepancy suggests that this recombinant protein might be heavily glycosylated as glycosylation of recombinant proteins is common in a yeast expression system (Benitez et al. 1989). This is probable as the mature protein has six potential N-glycosylation sites (56, 86, 139, 277, 312, and 356 amino acid residues) when analyzed using NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc). To determine the molecular weight of the naked AN1772.2 FAE, the purified FAE was treated with an N-glycosidase, Endo-H, and subsequently analyzed with SDS-PAGE (Fig. 3). Deglycosylated protein appeared as a single band of 56 kDa, which is in good agreement with the theoretical value.

SDS-PAGE of the purified AN1772.2 FAE and its deglycosylated form after Endo-H treatment. M: standards of molecular weights, A: purified AN1772.2 FAE, B: deglycosylated AN1772.2 FAE by Endo-H, and C: Endo-H
Characterization of the recombinant FAE
To examine the effect of pH on the activities of the purified recombinant FAE, enzyme activities were measured at different pHs, and their relative activities are displayed in Fig. 4. The enzyme exhibited pH optima at pH 7. To examine the pH stability, the enzyme was incubated at 4°C for 12 h in a pH buffer (pH ranging from 4 to 10). The residual enzyme activity was then analyzed after adjusting the pH to 7. The enzyme was stable over a very wide range of pH and over 80% of its activity was retained at pH 4.0 and 9.5. Thus, the enzyme is more stable in acidic or alkaline conditions than other fungal FAEs discovered so far (Table 4). Taking pH optima and stability into consideration, the enzyme could be used in broad pH conditions, ranging from 5.0 to 8.5 with at least 50% of its optimal activity (pH 7) (Fig. 4). Optimal temperature for FAE activity was 45°C. The enzyme is stable at a temperature up to 45°C (Fig. 4).

Effects of pH and temperature on activity (filled symbols) and stability (open symbols) of the purified FAE. To investigate the optimal pH and stability of FAE activity, the three different kinds of buffers were used as follows: 50 mM acetate–phosphate buffer (pH 4.0–7.5, filled circles and open circles), 50 mM Tris–HCl buffer (7.5–9.0, filled squares and open squares), and 50 mM glycine–NaOH buffer (pH 9.0–10.0, filled triangles and open triangles)
To examine the substrate specificities of the purified AN1772.2 FAE, enzyme activities were measured with different synthetic substrates, including MFA, MpCA, MCA, and MSA, and kinetic constants K m and V max were determined. As shown in Table 5, the purified FAE is capable of catalyzing the hydrolysis of three methyl hydroxycinnamates, with the activity for MpCA being the highest, followed by those for MCA and MFA. There was no detectable activity toward MSA. The catalytic efficiency (V max/K m) revealed a value of threefold greater for MpCA than for MFA, whereas similar values were measured for MCA and MpCA (Table 5). These results suggest that this FAE should be functionally classified as a type B FAE according to the classification criteria (Crepin et al. 2004), despite its high sequence identity with type C FAEs in the vicinity of the active site.
Discussion
This work has unequivocally proved that the hypothetical protein AN 1772.2 is a FAE esterase. The protein was successfully cloned and expressed in S. cerevisiae as a hyperglycosylated enzyme. The native signal peptide was functional in the heterologous host, allowing a majority of recombinant enzymes (about 86%) secreted into the medium.
The recombinant FAE was purified to its homogeneity and fully characterized. It exhibits temperature and pH optima at 45°C and pH 7, respectively. While its thermal stability is typical for a mesophilic fungal enzyme, it appears to be stable over a wider range of pH than most of the fungal FAE. The high stability in acidic or alkaline conditions of AN1772.2 FAE could be advantageous in many biotechnological applications that require acidic or alkaline conditions (such as alkaline pulping) (Record et al. 2003; Sigoillot et al. 2005). The high stability allows the enzyme to recover its optimal activity from temporary pH upsets often encountered in large scale operations with nonideal pH controls and overshooting of acid or base. Although the conserved 13 amino acids in the vicinity of the active site suggest a type C FAE, its substrate specificity profile depicts a typical type B FAE, raising an interesting question about the role of the conserved region.
As shown in this work, a bioinformatics-assisted enzyme discovery is highly effective and can identify useful enzymes in an expeditious manner. Similar strategies can be used to identify other enzymes for biotechnological applications.
References
Benitez J, Silvat A, Vazquez R, Noa MD, Hollenbergs CP (1989) Secretion and glycosylation of Clostridium thermocellum endoglucanase A encoded by the CelA gene in Saccharomyces cerevisiae. Yeast 5:299–306
Blum DL, Kataeva IA, Li XL, Ljungdahl LG (2000) Feruloyl esterase activity of the Clostridium thermocellum cellulosome can be attributed to previously unknown domains of XynY and XynZ. J Bacteriol 182:1346–1351
Borneman WS, Ljungdahl LG, Hartley RD, Akin DE (1992) Purification and partial characterization of two feruloyl esterases from the anaerobic fungus Neocallimastix strain MC-2. Appl Environ Microbiol 58:3762–3766
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Crepin VF, Faulds CB, Connerton IF (2003a) A non-modular type B feruloyl esterase from Neurospora crassa exhibits concentration-dependent substrate inhibition. Biochem J 370:417–427
Crepin VF, Faulds CB, Connerton IF (2003b) Production and characterization of the Talaromyces stipitatus feruloyl esterase FAEC in Pichia pastoris: identification of the nucleophilic serine. Protein Expr Purif 29:176–184
Crepin VF, Faulds CB, Connerton IF (2004) Functional classification of the microbial feruloyl esterases. Appl Microbiol Biotechnol 63:647–652
de Vries RP, vanKuyk PA, Kester HC, Visser J (2002) The Aspergillus niger faeB gene encodes a second feruloyl esterase involved in pectin and xylan degradation and is specifically induced in the presence of aromatic compounds. Biochem J 363:377–386
Donaghy JA, Bronnenmeier K, Soto-Kelly PF, McKay AM (2000) Purification and characterization of an extracellular feruloyl esterase from the thermophilic anaerobe Clostridium stercorarium. J Appl Microbiol 88:458–466
Faulds CB, Williamson G (1995) Release of ferulic acid from wheat bran by a ferulic acid esterase (FAE-III) from Aspergillus niger. Appl Microbiol Biotechnol 43:1082–1087
Ferreia LMA, Wood TM, Williamson G, Faulds C, Hazewood GP, Black GW, Gilbert HJ (1993) A modular esterase from Pseudomonas fluorescence subsp. cellulosa contains a non-catalytic cellulose-binding domain. Biochem J 294:349–355
Fillingham IJ, Kroon PA, Williamson G, Gilbert HJ, Hazlewood GP (1999) A modular cinnamoyl ester hydrolase from the anaerobic fungus Piromyces equi acts synergistically with xylanase and is part of a multiprotein cellulose-binding cellulase-hemicellulase complex. Biochem J 343:215–224
Mathew S, Abraham TE (2004) Ferulic acid: an antioxidant found naturally in plant cell walls and feruloyl esterases involved in its release and their applications. Crit Rev Biotechnol 24:59–83
Record E, Asther M, Sigoillot C, Pages S, Punt PJ, Delattre M, Haon M, van den Hondel CA, Sigoillot JC, Lesage-Meessen L, Asther M (2003) Overproduction of the Aspergillus niger feruloyl esterase for pulp bleaching application. Appl Microbiol Biotechnol 62:349–355
Rumbold K, Biely P, Mastihubova M, Gudelj M, Gubitz G, Robra KH, Prior BA (2003) Purification and properties of a feruloyl esterase involved in lignocellulose degradation by Aureobasidium pullulans. Appl Environ Microbiol 69:5622–5626
Shin HD, Chen RR (2006) Production and characterization of a type B feruloyl esterase from Fusarium proliferatum NRRL 26517. Enzyme Microb Technol 38:478–485
Sigoillot C, Camarero S, Vidal T, Record E, Asther M, Perez-Boada M, Martinez MJ, Sigoillot JC, Asther M, Colom JF, Martinez AT (2005) Comparison of different fungal enzymes for bleaching high-quality paper pulps. J Biotechnol 115:333–343
Topakas E, Christakopoulos P (2004) Production and partial characterization of alkaline feruloyl esterase by Fusarium oxysporum during submerged batch cultivation. World J Microbiol Biotechnol 20:245–250
Topakas E, Stamatis H, Biely P, Kekos D, Macris BJ, Christakopoulos P (2003a) Purification and characterization of a feruloyl esterase from Fusarium oxysporum catalyzing esterification of phenolic acids in ternary water-organic solvent mixtures. J Biotechnol 102:33–44
Topakas E, Stamatis H, Mastihubova M, Biely P, Kekos D, Macris BJ, Christakopoulos P (2003b) Purification and characterization of a Fusarium oxysporum feruloyl esterase (FoFAE-I) catalyzing transesterification of phenolic acid esters. Enzyme Microb Technol 33:729–737
Topakas E, Stamatis H, Biely P, Christakopoulos P (2004) Purification and characterization of a type B feruloyl esterase (StFAE-A) from the thermophilic fungus Sporotrichum thermophile. Appl Microbiol Biotechnol 63:686–690
Topakas E, Vafiadi C, Stamatis H, Christakopoulos P (2005) Sporotrichum thermophile type C feruloyl esterase (StFaeC): purification, characterization, and its use for phenolic acid (sugar) ester synthesis. Enzyme Microb Technol 36:729–736
Wang X, Geng X, Egashira Y, Sanada H (2004) Purification and characterization of a feruloyl esterase from the intestinal bacterium Lactobacillus acidophilus. Appl Environ Microbiol 70:2367–2372
Acknowledgement
This research is funded by a grant from USDA, CSREES 2004-35503-1520.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shin, HD., Chen, R.R. A type B feruloyl esterase from Aspergillus nidulans with broad pH applicability. Appl Microbiol Biotechnol 73, 1323–1330 (2007). https://doi.org/10.1007/s00253-006-0612-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0612-2