
Abstract
Laccases are copper-containing phenol oxidases that are commonly found in many types of plant, insect, fungi and bacteria. Whilst phenol oxidases have been well characterized in fungal species, laccase-type enzymes originating from bacteria have been much less well defined. Bacteria belonging to the family Azotobacteraceae share many morphological characteristics with strains already known to exhibit polyphenol and phenol oxidase activity; and hence the aim of this work was to identify and characterize a novel laccase from the isolated strain Azotobacter chroococcum SBUG 1484 in an attempt to provide further understanding of the roles such enzymes play in physiological development. Laccase activity was clearly observed through oxidation of 2,6-dimethoxyphenol, other typical substrates including: methoxy-monophenols, ortho- and para-diphenols, 4-hydroxyindole, and the non-phenolic compound para-phenylenediamine. A. chroococcum SBUG 1484 showed production of a cell-associated phenol oxidase when grown under nitrogen-fixing conditions, and was also observed when cells enter the melanogenic and encystment stages of growth. Catechol which is structurally related to melanin compounds was also released from Azotobacter cells into the surrounding culture medium during nitrogen-fixing growth. From our results we propose that a membrane-bound laccase plays an important role in the formation of melanin, which was monitored to correlate with progression of A. chroococcum SBUG 1484 cells into the encystment stage of growth.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Collectively termed within phenol oxidases (POs), and belonging to the group of multi-copper protein family, the enzymatic class of laccases (benzenediol:oxygen oxidoreductases, EC 1.10.3.2.) encompasses a wealth of functional diversity. A commonly described attribute of this group of enzymes is a wide spectrum of substrate acceptance which includes: substituted ortho- and para-dihydroxylated monoaromatics, polyaromatic hydrocarbons, aromatic (di)amines, as well as inorganic compounds (Alcalde 2007). Polyphenol oxidases (PPOs) differ from laccase not only on basis of enzyme substrates, but also in sensitivity towards selected inhibitors, and the amount and type of copper atoms in active site. The assembly of PPOs includes monophenol oxidases (tyrosinases, EC 1.14.18.1) and catechol oxidases (o-diphenol:oxygen oxidoreductases, EC 1.10.3.1). Tyrosinases are known to exhibit ortho-hydroxylation of monophenols, which includes substrates such as l-tyrosine, dopamine and p-cresol (cresolase activity), with subsequent oxidation of generated dihydroxylated compounds into the corresponding quinoid products (Pomerantez 1966). In contrast to tyrosinases, catechol oxidases show an inability to hydroxylate monoaromatic compounds; however, these enzymes do share functional similarity in PO activity towards ortho-dihydroxylated compounds such as catechol to form ortho-benzoquinone. PPO and PO enzymes share the characteristic of oxygen requirement, whereby molecular oxygen as an electron acceptor is reduced during reaction cycle to water. Despite the aforementioned differences between laccases, tyrosinases and catechol oxidases, many authors collectively refer to these enzymes under the subject of PPOs (Messerschmidt 1997). However, strictly speaking, laccases should be termed within the PO enzymatic class on the basis of their substrate scope and general aforementioned characteristics. Enzymatic oxidation of substrate molecules by these enzymes leads to appearance of reactive forms of the original aromatic structure. This mechanism can be observed in degradation processes of complex natural macromolecules, e.g. lignin, and can also lead to subsequent non-enzymatic oxidative coupling reactions (Mikolasch and Schauer 2009). The interest in exploitation of these abilities in industrial applications has stimulated the implementation of PO-mediated reactions during processes of pulp delignification (Palonen and Viikari 2004), textile dye bleaching (Abadulla et al. 2000), and biopolymer synthesis (Mikolasch and Schauer 2009). Significant interest in the application of POs has also been generated in scientific fields concerning the detoxification and degradation of environmental pollutants (Brenna and Bianchi 1994), and also concerning the production of fine chemicals (Mikolasch et al. 2008a) and antibiotics (Anyanwutaku et al. 1994; Mikolasch et al. 2008b). Most POs have been described in filamentous fungi, insects and several plants. The conclusions of distribution, occurrence, structural organization and localization of prokaryotic POs seemed to be restricted to special species, or to those at distinctive morphological or physiological stages of cell differentiation (Claus et al. 2002; Sharma et al. 2007). Estimating the physiological functions from eukaryotic and prokaryotic POs, some parallels including pigmentation processes and morphological alterations can be drawn (Sanchez-Amat et al. 2001; Takami et al. 2002).
In Cryptococcus neoformans, production of a polysaccharide capsule with concurrent formation of a melanin-like pigment in the presence of phenolic substrates was evoked by the expression of laccase and highlighted as a virulence factor (Ikeda et al. 1993; Williamson 1994; Zhu et al. 2001). Endospore generation of melanogenic Bacillus species reveals PO and PPO activity exclusively localized in spore coats (Hullo et al. 2001; Martins et al. 2002). Apart from unequal mechanisms of development, progression into dormant stages by means of Azotobacter cyst bodies is comparable to endospore generation of melanogenic Bacillus species. In this regard, some species of the genus Azotobacter undergo morphological alterations in the process of encystment, which was inducible with a variety of environmental conditions such as: desiccation, heat, UV light, organic acids, nutritional deficits or availability of metal ions (Layne and Johnson 1964; Wyss et al. 1969; Stevenson and Socolofsky 1972). With many reports describing the appearance of POs and PPOs in bacterial groups known for development of melanogenic cells or dormant stages (Claus and Filip 1997), the presence of a PO in melanogenic members of the family Azotobacteraceae is highly presumed. The aim of this work was the discovery of a novel bacterial PO, investigated by means of directed isolation of bacteria belonging to the genus Azotobacter which were competent for encystment and production of melanin. The strain Azotobacter sp. SBUG 1484 isolated from soil was monitored for extracellular and intracellular PO production with regard to a variety of culture conditions. We demonstrated that occurrence of a PO could be induced with nutritional deficits, especially exogenous nitrogen sources, and that it is characterized by culture pigmentation and incidence of dormant cell stages.
Materials and methods
General
All substrates for characterization of the strain Azotobacter sp. SBUG 1484 and the bacterial PO were purchased from Sigma-Aldrich (Taufkirchen, Germany) in the highest purity available. SIM agar, Kovacs indole reagent, Griess–Ilosvay reagent and crystal violet solution were obtained from Merck KGaA (Darmstadt, Germany). Primers for 16S rRNA sequencing were purchased from biomers.net GmbH (Ulm, Germany).
Isolation and growth conditions
Azotobacter sp. SBUG 1484 was isolated from composted earth (Greifswald, Germany) using the classical silica-gel plate method, as previously described for selective isolation of nitrogen-fixing bacteria of the genus Azotobacter (Winogradski 1926), and is publicly available from the SBUG (Strain Collection, Department of Biology, University of Greifswald, Germany). The strain was initially cultivated on solid medium plates of Winogradski’s nitrogen-free mineral salt medium supplemented with 0.1 g/l of CaCO3 (w/v) and 1% glucose (v/v) and incubated for 5 days at 37 °C before storing at room temperature for several days. The liquid pre-culture was prepared with 100 m Winogradski’s medium supplemented with 5 ml of a stock solution containing (per litre): 50 g KH2PO4, 25 g MgSO4∙7H2O, 25 g NaCl, 1 g FeSO4∙7H2O, 1 g Na2MoO4∙2H2O and 1 g MnSO4∙4H2O, and also supplemented with 1.5 ml of trace element solution containing (per litre): 70 mg ZnCl2, 6 mg H3BO3, 190 mg CoCl2∙6H2O, 2 mg CuCl2∙7H2O, 24 mg NiCl2∙6H2O) and 0.1 g/l CaCO3. The pH was adjusted to 7.2 using 25% NaOH. Sterile medium supplemented with 1% glucose and 0.4% yeast extract (v/v) as an additional organic nitrogen source, was inoculated with one inoculating loop of cell material in 500-ml shaking flasks (modified according to Winogradski 1935). Pre-cultures were incubated on a rotary shaker (Infors AG, Bottmingen, Switzerland) at 30 °C and 180 rpm for 12 h. For further characterization studies and enzyme assays, pre-cultured biomass was harvested by centrifugation (10 °C, 20 min, 7,000×g) (Sorvall® RC-53 Refrigerated Superspeed Centrifuge, DuPont Instruments, Bad Homburg, Germany) and the cells washed twice with nitrogen-free medium to eliminate residual compounds. Short term strain cultivation was replaced by activation and incubation of fresh stock culture derived from −80 °C storage.
Determination of growth-promoting substrates and further taxonomic tests
For growth tests in liquid medium, strain SBUG 1484 was pre-cultivated on solid medium plates of Winogradski’s nitrogen-free mineral medium supplemented with 0.1 g/l of CaCO3 (w/v) and 1% glucose (v/v) and incubated for 5 days at 37 °C. After growth on agar, the cell material was carefully removed with nitrogen-free mineral medium under sterile conditions. The utilization of diverse carbon sources was investigated in 100-ml Erlenmeyer flasks containing 40 ml nitrogen-free mineral medium at an optical density (OD500 nm) of 0.2. As a source of carbon, filter-sterilized sugar solutions (20%) were added at a concentration of 1% (v/v). In the case of determining growth with aliphatic hydrocarbons (concentrations of 0.001% and 0.01%) as sole source of carbon and energy, a 1% methanol stock solution of these substrates was prepared and added into sterile 100-ml Erlenmeyer flasks 24 h before the experiment was started. The cultures were incubated on a rotary shaker at 30 °C and 180 rpm and change in OD500 nm monitored at regular intervals. All growth experiments were carried out in duplicate using Winogradski’s nitrogen-free mineral medium. A negative control of cells without substrate, and a positive control of cells with 1% glucose were used. In addition to growth experiments with different sugars as carbon sources in liquid medium, their utilization by Azotobacter sp. SBUG 1484 was also monitored in test tubes containing 8 ml of a bromothymol blue dyed nutrient broth and 1% (v/v) of the corresponding sugar inoculated with cell material. To determine potential anaerobic sugar utilization, Durham tubes were used. Every experiment was performed in duplicate and cultures were incubated at 37 °C over 72 h. With a view to identification of the species and determining physiological properties of Azotobacter sp. SBUG 1484, several methods for identification have been investigated as described by Thompson and Skerman (1979) and Bergey’s Manual of Systematic Bacteriology (Kennedy et al. 2005). Motility and formation of indole have been analyzed using SIM-agar inoculated with a stitch needle. Indole formation from tryptophan was monitored with Kovacs indole reagent. The degradation of sulphurous substances was checked using a sodium thiosulphate (0.1%, v/v) supplemented nutrient broth, which was inoculated with one inoculating loop of cells and a lead acetate filter attached 1 cm above the surface of the medium. In the case of a positive reaction the filter discoloured black caused by the liberation of hydrogen sulphide. Nitrate respiration was studied using a method described by Neyra and van Berkum (1977). Nitrate ammonification was assayed using Griess-Ilosvays reagent. Denitrification was monitored by gas accumulation in a Durham tube. Residual nitrate was detected using diphenylamine reagent resulting in a blue colouration of the medium. For testing urease activity a Christensen urea agar was used (Christensen 1946). Hydrolysis of starch was monitored using the method described by Wheater (1955). All tests were performed in duplicate and incubated at 37 °C for 48 h.
Microscopy examination
Examinations of cell morphology and differentiation processes were carried out using a ZEISS model Axiolab light microscope possessing bright-field and phase-contrast optics equipped with a ProgRes C5 digital camera (Jenoptik Germany, Jena, Germany). Gram characteristics were determined using crystal violet solution. Staining methods as described by Wirtz (1908) were used for examinations in distinguishing between cysts and endospores. Poly-β-hydroxybutyrate (PHB) granula became observable after introduction of cell material into a drop of filtered 70% ethanolic sudan black B solution placed on a microscope slide. Stained preparations were monitored using bright-field optics, whereby microscopy of unstained samples was conducted under phase-contrast adjustment.
16S rRNA sequencing and strain identification
DNA from the isolated strain was extracted using the TRI Reagent® protocol (Sigma-Aldrich) and the concentration quantified using a NanoDrop® ND-1000 spectrophotometer (PEQLAB Biotechnologie GmbH, Germany) prior to PCR amplification of the 16S rRNA using standard 16S rRNA primers 27f and 1492r (Peace et al. 1994). Using an NZY-A PCR cloning kit (nzytech, Portugal), the PCR product was cloned into the pNZY28 vector and blue-white selection performed. Colony PCR using the 27f and 1492r primers confirmed successful cloning of the 16S rRNA gene. DNA from overnight cultures was extracted using a GeneJet™ Plasmid Miniprep kit (Fermentas, St. Leon-Rot, Germany) and the sequence determined (GATC, Konstanz, Germany) prior to performing BLAST and sequence alignment (EMBL-EBI) to provide strain identification based upon homology to known 16S rRNA gene sequences in the NCBI database.
Studies of enzyme production
For studies of PO production by Azotobacter sp. SBUG 1484, a defined volume of the pre-culture was added to 500-ml shake flasks containing 100 ml Winogradski’s nitrogen-free medium supplemented with 1% filter-sterilized glucose to reach an initial OD500 nm of approximately 0.2. In the case of incubation with nitrogen rich mineral medium, 0.4% of organic or 0.3% of inorganic nitrogen source was added to the cultures followed by incubation under the same conditions described above. After incubation for different time periods (6–120 h) the cultures were harvested (4 °C, 20 min, 7,000×g) (Sorvall® RC-53 Refrigerated Superspeed Centrifuge, DuPont Instruments, Bad Homburg, Germany) and the supernatant used for studies of extracellular PO production. The cells where washed twice with phosphate buffered saline (pH 7) to remove divalent cations that may have bound to the cells. The cell pellet was resuspended in 100 mM sodium acetate buffer (NAc buffer) pH 5, and subsequently disrupted by passing the cells through a pre-cooled French press cell (SLM AMINCO®, Mini-Cell FA-003, Rochester, NY, USA) four times. The turbid crude extracts obtained after centrifugation (4 °C, 5 min, 4,500×g; Eppendorf AG, Centrifuge 5417R, Hamburg, Germany) of the cell lysate were used for further studies to determine the activities of the herein described bacterial PO. Preparation of cellular fractions, including the cytosolic fraction as well as parts of cytoplasmic and outer membrane fractions, were accomplished according to Kumar et al. (2001). The washed cell pellet (4 °C, 20 min, 7,000×g) was resuspended in NAc buffer (100 mM, pH 5) containing 10 mM EDTA and disrupted using a French press as previously described. Particulate cell debris was removed by centrifugation at 6,000×g for 5 min and the remaining supernatant subjected to ultracentrifugation (Sorvall®, Ultra Centrifuge CombiPlus, DuPont Company, Newton, UK) at 100,000×g for 1 h. After ultracentrifugation the clear supernatant was removed as the cytosolic fraction. The membrane fraction was redissolved in NAc buffer and incubated in the presence of 1% sodium lauryl sarcosinate (Sarkosyl) (v/v) at 4 °C for 1 h with occasional vortexing. After solubilisation, samples were subjected to ultracentrifugation as described previously. The supernatant obtained was taken as the cytoplasmic membrane fraction, whereby the remaining Sarkosyl insoluble pellet was taken as the outer membrane fraction. Subcellular fractions (cytosolic fraction, cytoplasmic and outer membranes) were prepared at different times of incubation (24, 48, 56 and 72 h) and monitored for PO production, whereas a washed pre-culture (0 h sample) was prepared under the same conditions and taken as a negative control. In addition to determination of 2,6-dimethoxyphenol (2,6-DMP) oxidizing activity in several fractions, PO activity was also tested in whole cells. The protein content of each sample was normalised to 0.5 mg/ml using 100 mM NAc buffer (pH 5).
Determination of protein concentration
For quantification of the produced cell protein which could be used as an estimation of growth, biomass was resuspended in 8 mM Tris/HCl buffer (pH 7.8) and protein extraction accomplished with 0.1 M NaOH for 1 h at 80 °C. Protein content was monitored spectrophotometrically (Thermo Fisher UV1 spectrophotometer, Schwerte, Germany) at a wavelength of 595 nm according to the Bradford assay (Bradford 1976).
Estimation of melanin and catechol
Separation and preparation of melanin from cell pellets was accomplished according to the method described by Whittaker (1963). Characterization of the isolated pigment was determined as published by White (1958) and Shivprasad and Page (1989). Dopa-melanin was synthesized from 3,4-dihydroxyphenylalanine as described by Arnow (1938), and catechol-melanin prepared from 1,2-benzenediol using identical conditions. The absorption spectra of the purified melanin from Azotobacter SBUG 1484 and synthetic melanins were recorded by scanning with a Thermo Fisher UV1 spectrophotometer (Schwerte, Germany), and was facilitated by dissolving melanin in warm 0.8 M KOH. Accumulated melanin in culture supernatants was determined after removal of the cells by centrifugation and measuring the absorbance at a wavelength of 400 nm. Released catechol was extracted three times from acidified cell-free culture medium (pH 1.8) using ethyl acetate. Extracts were concentrated and dried by rotary evaporation at 35 °C and re-dissolved in 2 ml methanol. Analysis of extracts was performed by gas chromatography on a BPX5 capillary column (length 25 m, ID 0.25 mm), coupled to a mass spectrometer (GCMS-QP2010S, Shimadzu Corporation, Germany). Quantification of catechol released into the culture medium was carried out according to the procedure described by Barnum (1977). For validation of results, all measurements were conducted with tenfold repetition.
Enzyme assays
PO activity was determined spectrophotometrically (Thermo Fisher UV1 spectrophotometer, Schwerte, Germany) at 468 nm using the substrate 2,6-DMP \( \left( {{\varepsilon_{{\left( {\rm{468nm}} \right)}}} = {3}.{5} \times {1}{0^{{4}}}{{\hbox{M}}^{{ - {1}}}}{\hbox{c}}{{\hbox{m}}^{{ - {1}}}}} \right) \). Standard assays were carried out in 700 μl of 100 mM NAc buffer (pH 5) supplemented with 100 μl aqueous 2,6-DMP (5 mM) and the reaction initiated by adding 200 μl of the diluted enzyme preparation. Laccase substrates including substituted o-, m- and p-phenols and aromatic amines at a concentration of 5 mM (2 mM in the case of syringaldazine) were added to start the reaction, and the rate of enzymatic oxidation determined spectrophotometrically over a defined time range, whereas 1 unit of activity is defined as the amount of enzyme that catalyzes the conversion of 1 μmol ml−1 min−1 substrate at 25 °C. All assays were performed four times.
SDS- and native polyacrylamide gel electrophoresis
Analytical SDS-polyacrylamide gel electrophoresis (PAGE) was performed as described by Laemmli (1970), with an acrylamide concentration of 10% in the resolving gel and 4% in the stacking gel. Using a common SDS procedure, samples were mixed in a 2:1 ratio (v/v) with 2-mercaptoethanol-containing sample buffer (0.18 M Tris–HCl, pH 6.8, 15% glycerol, 0.075% bromophenol blue, 3% SDS), heated for 10 min at 95 °C and electrophoresis performed at 200 mA for 1 h. Protein bands were stained with Coomassie brilliant blue R-250 followed by de-staining in a aqueous acetic acid/ethanol solution. 2,6-DMP activity staining of proteins on semi-denaturated SDS-PAGE was accomplished according to the method described by Solano et al. (2001). 2-Mercaptoethanol was omitted from the sample buffer and electrophoresis was run initially at 15 mA for 20 min, followed by a further 1 h at 25 mA. Activity staining of PO was conducted in 100 mM NAc buffer (pH 5) with 5 mM 2,6-DMP at 37 °C until active proteins became visible. Native PAGE was conducted under conditions already described with exclusion of SDS and 2-mercaptoethanol from the sample buffer, and by using a running buffer consisting of 0.05 M Tris–HCL and 0.38 M glycine (pH 8.3). Native gels were run at 20 mA for 2 h, prestained for 5 min with Coomassie brilliant blue R-250 followed by staining for activity as described previously. The molecular masses of proteins were compared using a prestained low-range SDS-PAGE standard marker (PageRuler Plus Prestained Protein Ladder, Fermentas, St. Leon-Rot, Germany).
Results
Isolation, physiological characterization and identification
Strain SBUG 1484 was isolated from composted earth using a silica-gel plate method for selective enrichment of nitrogen-fixing Azotobacter species. The isolated brown-pigmented strain SBUG 1484 was tested for its growth dependency and cell shape on various nitrogen-sources and incubation times, and on its ability to produce copper-containing enzymes, especially laccase (PO), under different conditions of cultivation. Due to the great variability of cell morphology and the pronounced differentiation cycle of Azotobacter cells it was necessary to guarantee specific cultivation conditions, so that enzyme activity could be related to pronounced physiological characteristics.
Taxonomic identification of strain SBUG 1484 was accomplished using the physiological characterization procedures described in the Material and methods section. Results were compared to other members belonging to the genus Azotobacter, whereby utilization and physiological abilities were compared with selected characteristics published in the latest version of Bergey’s Manual of Systematic Bacteriology. A partial sequence with a fragment length of 715 base pairs of the cloned 16S rRNA gene from isolated Azotobacter sp. SBUG 1484 revealed 100% sequence similarity to the 16S rRNA gene of Azotobacter chroococcum AM12A, which was isolated from rhizosphere soil of Miscanthus sinensis in studies to find a potent nitrogen fixer. Comparison of 16S rRNA sequences also revealed the type strain A. chroococcum ATCC 9043 to be of close phylogenetic relation (see Table S1). According to results gained from physiological and morphological tests in addition to sequencing the 16S rRNA gene, Azotobacter SBUG 1484 is herein identified as A. chroococcum SBUG 1484. The partial 16S rRNA sequence for this strain has been deposited with the EMBL-EBI/GenBank accession number FR731999. A full table showing results of physiologic studies on A. chroococcum SBUG 1484 in comparison to type strains of the genus Azotobacter can be found in the supplementary material (Fig. S1).
Microscopic studies of cell growth
Cell shape was strongly influenced by the growth phase as well as the availability and character of nitrogen sources (Fig. 1). Under nitrogen-fixing conditions, germinating cysts (Fig. 1a) formed rod shaped filaments comprising between four and six fragments. After linear growth, separation and shortening of filaments occurred during exponential phase where diplococcoid states were found to dominate; however, tetrameric structures could also be monitored (Fig. 1b). With 72 h of incubation, the presence of completely encysted cells with an intensive black-brown colouration was observed with formation of cellular aggregates (Fig. 1c). Glucose cultivated cells were determined as Gram-negative by cell wall staining with crystal violet and Gram-staining, whereas Gram-testing with 3% KOH of cultures growing on solid medium showed Gram-variability depending on incubation time and supplementation. Staining according to Wirtz (1908) revealed no green colouration of cyst bodies as described for spores of Bacillaceae and related spore forming Gram-positive bacteria, although similarities in morphological characteristics have been discussed (Bisset 1955).

a Microphotographs of swollen cysts showing germination by out-growing of cell bodies with rupture of cyst coat and reserve substances in vegetative cell bodies of a 4-h culture, cultivated on nitrogen-free mineral glucose agar. Bar = 5 μm. b Phase contrast micrograph from cells of A. chroococcum SBUG 1484 showing the so-called diplococcus stage and also tetrameric structures, when cultivated in glucose-starved medium supplemented with yeast extract after 120 h incubation. Bar = 10 μm. c Microphotographs of aggregates of crystal violet stained matured cysts, showing the poly-β-hydroxybutyrate central granules of A. chroococcum SBUG 1484 grown in Winogradski’s nitrogen-free liquid medium supplemented with glucose as the sole source of carbon and energy after 72 h incubation. Bar = 10 μm. d Phase contrast micrograph of a peptone supplemented culture with pleomorphic cells representing atypical morphology for the genus Azotobacter. Bar = 5 μm. e Bright field micrograph of a crystal violet-stained nitrogen-fixing culture after 6 h of incubation showing enlarged cell bodies in the process of division, with uniform distribution of cell material. Bar = 5 μm. f Bright field micrograph of initiating encystment by contraction and concentration of cell material, with partial remains of cell envelopes in a nitrogen-fixing culture stained with crystal violet after 48 h incubation. Bar = 5 μm
In contrast to the assimilation of atmospheric nitrogen, additional nitrogen sources gave vigorous pleomorphic growing cells that did not show signs of encystment or pigmentation during 8 days of study. Independent from available inorganic or organic nitrogen compounds, cells react in the presence of additional carbohydrates with retarded diminishment of cell size. Only when glucose was omitted did cells commence to a diplococcoid stage (after 120 h of incubation); however, no further development to encystment was determined. The most interesting morphology was viewed in cultures growing with peptone as the nitrogen source (Fig. 1d). Compared to other cultivation procedures involving exogenous nitrogen, cells of A. chroococcum SBUG 1484 showed a spindle shape with slightly pointed ends. Otherwise, cells in rod-shaped pairs or even single cells showed rounded ends and equal distribution of storage substances and nucleotides during contraction (Fig. 1e and f). Storage substances were revealed in phase contrast microscopy as strongly refracting and bright cell embedments, and were identified as an energy reservoir of Poly-β-hydroxybutyrate (PHB) via staining with sudan black.
Growth dependency on various nitrogen sources
When cultivating A. chroococcum SBUG 1484 with inorganic nitrogen compounds such as nitrate derivatives (Ca(NO3)2, NaNO3 and KNO3) or ammonium salts ((NH4)2SO4, NH4Cl, NH4NO3, CH3COONH4) in the presence of glucose, the duration of lag phase prior to growth differed depending on the available nitrogenous mineral (Fig. 2). In the case of nitrate derivatives, cells generally need 10 h for adaption, whereas the presence of ammonium salts results in a reduced lag period of 5 h. Nevertheless, if nitrate compounds are provided as the accessible source of nitrogen, cells reach higher optical densities during stationary phase than cultures grown in the presence of ammonium salts (with the exception of ammonium acetate, Fig. 2). Utilising CH3COONH4 as the single source of carbon for growth and energy, A. chroococcum SBUG 1484 reached the same biomass as observed with other ammonium sources despite an extended adaption phase. In the case of organic nitrogen sources (yeast extract, peptone, urea) growth was determined in cultures individually supplemented with glucose or with 0.4% nitrogenous components. According to pre-culture conditions, cells immediately showed exponential growth in the presence of organic nitrogen with lack of clear adaption phase. Pre-cultures in each instance, were cultivated under the same conditions to provide comparable results. Growth of pre-cultures in the absence of additional nitrogen was also conducted in parallel and yielded similar results. When yeast extract, peptone and urea were provided as N source in glucose-containing media, exponential growth occurred during the first 20 h of cultivation with comparable profiles plotting optical density measurements (Fig. 2). Only when peptone was provided as the source of carbon and energy was a comparable slight decrease in biomass observed (data not shown). In the absence of another carbon source, urea was found not to be suitable as a growth substrate. However, cells utilizing organic nitrogen sources showed strong correlation in biomass production to cultures in which nitrates or ammonium acetate were present. In contrast, nitrogen-fixing cells generally showed more extended lag periods followed by exponential growth until 45 h, but still resulted in comparable optical density measurements to cultivations in several nitrogen-rich media (Fig. 2).

Growth curves for A. chroococcum SBUG 1484 cultivated in Winogradski’s medium in the presence of 1% (v/v) glucose, with supplementation of nitrogen sources: KNO3 (open triangle), CH3COONH4 (open circle), (NH4)2SO4 (open square) and yeast extract (open diamond). Cells growing in nitrogen-free medium (closed square) showed a generally distinctive lag-period compared to glucose-grown cultures with different inorganic or organic nitrogen compounds. Incubation was carried out at 180 rpm and 30 °C over a period of 120 h
Analysis of cell pigment and culture supernatants
Under nitrogen-fixing conditions, cells of A. chroococcum SBUG 1484 after 48 h cultivation became more visibly pigmented with increasing incubation time, whereby cell-free culture supernatants were also found to exhibit a brownish to black colouration. Examination of the extracted cell pigment based on physicochemical characteristics was made in comparison to synthesised melanin compounds, and gave evidence that the cell-associated pigment belongs to the class of melanins. The yellow-white powder was insoluble in hot and cold water as well as cold 0.5 M NaOH and organic solvents (chloroform, ethanol, acetone), but was soluble in aqueous 0.5 M NaOH (heated to 80 °C) and 1 M Na2CO3. Precipitation in conjunction with appearance of an ochre colouration was monitored by adding 0.1 M FeCl3 to the dried melanin substance. Comparison of the absorption spectra of extracted melanin (λ max = 289 nm) with synthesised dopa-melanin and catechol-melanin showed comparable absorption characteristics. Measuring OD400 nm of cell-free supernatants which is applied for spectrophotometric determination of melanins revealed increased absorption that strongly relates to cellular protein content, and remained static after 56 h of incubation (Fig. 3).

Nitrogen-fixing growth of A. chroococcum SBUG 1484 (filled square) and change in melanin (OD400 nm) (filled circle), and catechol concentration (filled triangle), dependent on incubation time. The strain was incubated in Winogradski’s nitrogen-free medium containing 1% glucose for 72 h at 30 °C with shaking at 180 rpm. The cellular protein content was determined as a function of growth. Error bars refer to standard deviations by means of ten replicates
Mass spectrometric analysis of brownish coloured extracts (methanolic, underivatized) gained after ethyl acetate extraction of acidified culture supernatants (56 and 72 h), showed a dominant peak at a retention value of 13.9 min and mass data as follows: m/z 110 (base peak), 92 (13%), 81 (15%), 64 (38%), 53 (9%), 39 (6%). In alignment with mass spectra and retention times of a catechol standard, this substance could be identified as 1,2-benzenediol with reference to Shivprasad and Page (1989). Estimation of catechol concentration in cell-free supernatants showed an initial increase at an incubation time of 30 h and subsequent accumulation up to 0.34 mM after 72 h (Fig. 3). The increase in catechol strongly correlates with the observed formation of intensively coloured dark-brown/black melanized cell extracts at stationary phase.
Production of a PO
PO activity was not detected in cultures grown on Winogradski’s medium supplemented with organic or inorganic nitrogen sources; however, positive results were obtained when A. chroococcum SBUG 1484 was cultivated without a nitrogen source but with 1% glucose (v/v) as the sole source of carbon and energy. Under nitrogen-fixing conditions, cultures respond with the production of melanin after 48 h of incubation, whereby cultures in the presence of additional nitrogen did not become melanogenic or encysted. Generally, no enzyme activity could be monitored in the culture medium; however, it was found in the cell lysates after disruption of cells and cysts. To obtain evidence of when PO production occurs and under which culture conditions, samples from nitrogen-fixing cells growing in lag phase, early exponential phase, as well as those in the transition from exponential to stationary phase, and in stationary phase were analyzed for the presence of PO. Estimation of 2,6-DMP oxidizing activity in whole cells was also performed. In nitrogen-fixing whole cells 2,6-DMP oxidizing activity could be determined after 24 h of incubation, whereby cells after 48 h cultivation showed the highest substrate conversion. Subsequent prolonged incubation revealed decreased levels of activity, which could be explained with the concurrent change in cell morphology, in which encapsulated cells lose their original cell wall and membrane constitutions to form multilayer membranes (Fig. 4). As a consequence of this process, cellular localization of PO is substantially affected. After preparative centrifugation (6,000×g) of the lysate to separate particulate fractions, enzyme activity was determined in the turbid supernatant of 24-h samples, but was, however, found even more intensely in the fraction of cell debris. Activity testing of cytoplasmic membrane and cytosolic fractions also resulted in PO determination only in preparations gained from 24-h cultures and not in samples at later incubation stages. Furthermore, greater PO activity was found in crude lysate samples from prolonged incubation periods, with a general increase in PO activity in outer membrane fractions at later incubation times (correlating with the formation of the outer cyst coat) (Fig. 4). Referring to these observations, we postulate that the PO of A. chroococcum SBUG 1484 is associated with the particulate fraction of cells (particularly cell walls, cyst envelopes and outer membrane components) and could not be detected in the supernatants after ultracentrifugation.

PO activity monitored by oxidation of 2,6-DMP of whole cells and prepared cell fractions. Error bars refer to standard deviations by means of three replicates
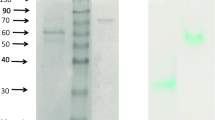
In addition to activity measurements, PAGE confirms the existence of PO activity in late growth phases (Fig. 5). The appearance of cultures was initially milky white (lag phase), and with an increase in incubation time cellular colouration turned from tan to nearly fuliginous. Accordingly, PO production could intensively be monitored in cells progressing from exponential growth to stationary phase and later stationary phase for A. chroococcum SBUG 1484. Activity staining of SDS-PAGE analysis according to the method described by Solano et al. (2001) revealed active bands in a sample taken at 24 h and also after subsequent incubation times, and suggests an active monomeric form of the enzyme exists at a apparent molecular weight of 40 kDa (Fig. 5, Gel 2). A broad active band in the range of 130 kDa from native-PAGE analysis (Fig. 5, Gel 3) suggests the formation of an active homo-trimer. Referring to these results, we assumed that the change in cellular colouration bears direct relation to PO production, and whereby encystment is initialized. With passing from exponential to stationary phase, the process of encystment is typified by cells building a protective coat and consequently turning brownish in colour.

SDS- and native PAGE analysis of A. chroococcum SBUG 1484 cell extracts. Gel 1: Coomassie Brilliant Blue staining of protein from samples prepared after different incubation times including lag-phase (6 h), early exponentially growth phase (24 h) as well as during transition from exponential to stationary phase (48 h) and stationary phase (72 and 96 h). Gel 2: Identical sample from 48 h cultivation prepared according to the method described by Solano et al. (2001) and stained for activity highlighting a 2,6-DMP oxidizing protein. Gel 3: Native PAGE with sample (48 h) prepared under non-denaturating conditions and stained for activity with 2,6-DMP. Arrows reference phenol oxidase protein and activities
Substrate specificity of PO
A. chroococcum SBUG 1484 PO within crude extracts of a 56-h cultivation was able to oxidize typical substrates, including methoxy-monophenols as well as ortho- and para-diphenols, and the non-phenolic compounds para-phenylenediamine and 4-hydroxyindole (Table 1). Tyrosinase or cresolase activity could not be determined due to a lack of conversion of 1 mM l-tyrosine (λ = 280 nm) and 5 mM p-cresole (λ = 300 nm) at the conditions tested. Oxidation of 1,3-dihydroxylated resorcinol (λ = 276 nm) and its derivatives orcinol (λ = 278 nm) and olivetol (λ = 281 nm) could not be detected despite incubation of reaction mixtures for over 24 h. Of all the tested phenolic compounds, the dimethyl ether 2,6-DMP was discovered to yield the highest enzymatic preference and was followed by transformation of mono-methoxylated guaiacol. Interestingly the methoxy-activated substance syringaldazine which is often described as an applicable substrate in determining PO activity, especially those of bacterial laccases, was not oxidized at the conditions tested. The same results were found with ferulic acid, whereas the structural analogue hydrocaffeic acid showed only 11% relative activity.
Through the oxidation of para-hydroquinone, the enzyme exhibits the commonly described specific feature of laccases in the preferred oxidation of para-diphenols in the presence of oxygen (Alcalde 2007; Xu 1996). Alkylsubstituted hydroquinones like methylhydroquinone and tert-butylhydroquinone were also transformed with comparable activities. Nevertheless the bacterial PO also showed activity in the case of 1,2-benzenediol and methyl substituted catechols. Generally, A. chroococcum SBUG 1484 exhibits laccase characteristics on the basis of substrate level, whereby existence of tyrosinase, cresolase and also the occurrence of an exclusive catecholase can be excluded based upon these observations.
Discussion
The aim of the present study was to characterize a novel bacterial PO from the isolated strain A. chroococcum SBUG 1484 produced when cell were grown under nitrogen-fixing conditions, and when entering the melanogenesis and encystment stage. Growth in the presence of additional organic and inorganic nitrogen sources resulted in an absence of PO activity, and correlates with a lack in pigmentation and absence of cyst formation. PO was produced under nutrient limited conditions and when cells reached the encystment stage, and therefore we postulate that under conditions of normal metabolism without nitrogen fixation, the PO of A. chroococcum SBUG 1484 is physiologically irrelevant.
The cell-associated PO was produced in nitrogen-fixing cultures during the transition from exponential growth to stationary phase, and was accompanied by morphological changes leading to encystment, and simultaneous production of a brown-black pigment. This development of specialised resting cells has been studied for many decades and is described as a cyclic process of cellular differentiation, with the appearance of modified vegetative cells comprising a PHB granulated central body surrounded by a triple-layered wall and triple-layered membrane. Encystment has also been described as a consequence of growth in nitrogen-free media with simple sources of carbon (Winogradski 1939). When reviewing the occurrence of prokaryotic laccases and laccase-like proteins in other strains (Sharma et al. 2007), comparisons can be made to the herein described enzyme of A. chroococcum SBUG 1484. As PO production in A. chroococcum SBUG 1484 was determined in cultures adjudged in cell differentiation, parallels can be drawn to the occurrence of PPO in Streptomyces griseus (Endo et al. 2002). This strain exhibits an extracytoplasmic laccase-like enzyme in succession of mycelium-producing aerial hyphae, which cumulate into spore chains via septum formation, and is caused by nutritional limitations and various environmental signals (Endo et al. 2002). In contrast to EpoA from S. griseus which is secreted in liquid culture, the PO of A. chroococcum SBUG 1484 was always found to be associated with insoluble membrane containing fractions, and prompts our suggestion that this PO is involved in protective mechanisms during cellular differentiation to form cysts. When cells are growing aerobically under nitrogen-fixing conditions, establishment of adaptive features in the closely related A. chroococcum 184 (Shivprasad and Page 1989) revealed low levels of PPO activity (catecholase activity) concurrent with pigmentation and morphologic alteration. However, intensive studies in characterization of this PPO activity with a view to determining exact enzyme classification were not pursued. For the prevention of cellular damage caused by the presence of highly reactive hydroxyl radicals arising from the Fenton reaction, Azotobacteraceae and other related free-living nitrogen-fixing bacteria have established catalases, peroxidases and superoxide dismutases (Moore et al. 1984).
Melanin and catecholates assumed to be involved in PO induction are thought to be responsible for the distinctive colouration of A. chroococcum SBUG 1484 cultures. Exhibition of PO activity was also found in melanogenic cell cultures of the genus Bacillus, especially B. subtilis (Hullo et al. 2001), B. licheniformis (Koschorreck et al. 2008) and Bacillus HR03, whereby in melanised cells of Bacillus HR03 all types of copper-containing enzymes including tyrosinase, catecholase, and laccase occurred in culture supernatants (Dalfard et al. 2006). Interestingly catechol release from cells of A. chroococcum SBUG 1484 into the culture supernatants could also be determined. Catechol, an accepted substrate of POs, is closely related in structure to the compounds involved in synthesis of melanins. The release of catechol in this instance is presumed to be linked to the formation of melanins which yield the observed characteristic change in cellular colouration. Attempts to uncover the cellular localization and constitution of A. chroococcum SBUG 1484 yielded evidence for PO occurrence in the particulate cell fractions. With increasing incubation time, PO activity could predominantly be determined in components of the outer membrane and residues of cell and cyst walls. After prolonged cultivation times and with appearance of cyst formation, no PO activity could be found in water soluble cell fractions.
PO activity for A. chroococcum SBUG 1484 was clearly determined through 2,6-DMP oxidation which revealed the highest activities of all tested phenolic substrates, comparable to the substrate preference of Streptomyces coelicolor laccase SLAC (Machczynski et al. 2004).
Most bacterial POs and fungal laccases (e.g. laccase of P. oryzae) show activity in the oxidation of syringaldazine (Fauré et al. 1994). Enzyme preparations from A. chroococcum SBUG 1484 were not active towards syringaldazine at the tested conditions and showed no tendency towards tyrosinase and cresolase activity when using l-tyrosine and p-cresole as substrates. However, catechol oxidizing activity, which is also an accepted feature of laccases, was exhibited in the oxidation of 1,2-dihydroxylated catechol and its methylated derivatives. Oxidation of the model laccase substrate syringaldazine could not be monitored although its functional groups are structurally similar to the highly preferred substrate 2,6-DMP. However, the reactive C4 position of 2,6-DMP is bonded to the azine substituent in the syringaldazine structure, and may account for this difference. The native laccase-like PPO EpoA of the saprophytic soil bacterium S. griseus was also unable to oxidize methoxy-activated syringaladazine and a range of other characteristic laccase substrates including: 2,6-DMP, guaiacol, para-hydroquinone and catechol (Endo et al. 2003). Solano et al. (2001) defined 2,6-DMP as a useful indicator in differentiating laccase activity into three groups according to the rate of substrate turnover, or upon dependency of copper supplementation (pseudo-laccase). According to their classification of microbial blue multi-copper proteins, the PO detected in A. chroococcum SBUG 1484 belongs to the first group expressing real laccase activity, signalised by a rapid 2,6-DMP oxidation without copper supplementation. As laccases and laccase-like multi-copper oxidases have been determined in genomic studies to be widespread in bacteria (Alexandre and Zhulin 2000), they were previously reviewed in their functions as somehow being comparable to those of extracellular fungal laccases (Sharma et al. 2007). Related characteristics were discussed in physiological features such as pigmentation and sporulation/morphogenesis (Claus and Filip 1997). The oxidation of phenolic compounds, destruction of toxic oxygen species (Bains et al. 2003) and effects of metals (Roberts et al. 2002; Cha and Cooksey 1991) also provide a context for comparative analysis of these enzymes. These factors have enabled parallels to be drawn between the novel herein described PO of the isolated strain A. chroococcum SBUG 1484 and previously described enzymes of this class from other microbial origins. Whilst prior knowledge of laccases facilitated an in-depth characterization of the enzyme from A. chroococcum SBUG 1484 in these studies, it has also served to highlight some remarkably contrasting features of this enzyme such as its strict pattern of localisation to cell-membrane containing fractions, and a distinct substrate scope when compared to previously described bacterial POs and laccase-like enzymes. The proposal of specific physiological and biochemical roles for the expression of this enzyme in A. chroococcum SBUG 1484 awaits further investigation.
References
Abadulla E, Tzanko T, Costa S, Robra KH, Cavaco-Paulo A, Gübitz GM (2000) Decolorization and detoxification of textile dyes with a laccase from Trametes hirsuta. Appl Environ Microbiol 66:3357–3362
Alcalde M (2007) Laccases: biological functions, molecular structure and industrial applications. In: Polaina J, MacCabe AP (eds) Industrial enzymes, 1st edn. Springer, New York, pp 461–476
Alexandre G, Zhulin IB (2000) Laccases are widespread in bacteria. Trends Biotechnol 18:41–42
Anyanwutaku I, Petroski R, Rosazza J (1994) Oxidative coupling of mithramycin and hydroquinone catalyzed by copper oxidases and benzoquinone. Implications for the mechanism of action of aureolic acid antibiotics. Bioorg Med Chem 2:543–551
Arnow LE (1938) The preparation of dopa-melanin. Science 82:308
Bains J, Capalash N, Sharma P (2003) Laccase from a nonmelanogenic, alkalotolerant γ-proteobacterium JB isolated from industrial wastewater drained soil. Biotechnol Lett 25:1155–1159
Barnum DW (1977) Spectrophotometric determination of catechol, epinephrine, dopa, dopamine and other aromatic vic-diols. Anal Chim Acta 89:157–166
Bisset KA (1955) Evidence from the cytology of Azotobacter chroococcum of a relationship with Rhizobium and the Bacillaceae. J Gen Microbiol 13(3):442–445
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Brenna O, Bianchi E (1994) Immobilized laccase for phenolic removal in must and wine. Biotechnol Lett 24:35–40
Cha J, Cooksey DA (1991) Copper resistance in Pseudomonas syringae by periplasmic and outer membrane proteins. Proc Natl Acad Sci USA 88:8915–1919
Christensen WB (1946) Urea decomposition as a means of differentiating Proteus and paracolon cultures from each other and from Salmonella and Shigella types. J Bacteriol 52:461–466
Claus H, Filip Z (1997) The evidence of a laccase-like activity in a Bacillus sphaericus strain. Microbiol Res 152:209–215
Claus H, Faber G, König H (2002) Redox-mediated decolorization of synthetic dyes by fungal laccases. Appl Microbiol Biotechnol 59:672–678
Dalfard AB, Khajeh K, Soudi MR, Naderi-Manesh H, Ranjbar B, Sajedi RH (2006) Isolation and biochemical characterization of laccase and tyrosinase activities in a novel melanogenic soil bacterium. Enzyme Microb Technol 39:1409–1416
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:465–469
Endo K, Hosono K, Beppu T, Ueda K (2002) A novel extracytoplasmatic phenol oxidase of Streptomyces: its possible involvement in the onset of morphogenesis. Microbiology 148:1767–1776
Endo K, Hayashi Y, Hibi T, Hosono K, Beppu T, Ueda K (2003) Enzymological characterization of EpoA, a laccase-like phenol oxidase produced by Streptomyces griseus. J Biochem 133:671–677
Fauré D, Boullant ML, Bally R (1994) Isolation of Azospirillium lipoferum 4T Tn5 mutants affected in melanization and laccase activity. Appl Environ Microbiol 60:3412–3415
Hullo MF, Moszer I, Danchin A, Martin-Verstraete I (2001) CotA of Bacillus subtilis is a copper-dependent laccase. J Bacteriol 183:5426–5430
Ikeda R, Shinoda T, Morita T, Jacobson ES (1993) Characterization of a phenol oxidase from Cryptococcus neoformans var. neoformans. Microbiol Immunol 37:759–764
Kennedy C, Rudnick P, MacDonald ML, Melton T (2005) Genus III. Azotobacter Beijerinck 1901, 567AL. In: Brenner DJ, Krieg NR, Staley JT (eds) Bergey’s manual of systematic bacteriology, vol 2, 2nd edn. Springer, New York, pp 384–402
Koschorreck K, Richter S, Ene A, Roduner E, Schmid R, Urlacher V (2008) Cloning and characterization of a new laccase from Bacillus licheniformis catalyzing dimerization of phenolic acids. Appl Microbiol Biotechnol 79:217–224
Kumar SS, Sankaran S, Haigh R, Williams PH, Balakrishnan A (2001) Cytopathic effects of outer-membrane preparations of enteropathogenic Escherichia coli and co-expression of maltoporin with secretory virulence factor EspB. J Med Microbiol 50:602–612
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Layne JS, Johnson EJ (1964) Natural factors involved in the induction of cyst formation in Azotobacter. J Bacteriol 87:684–689
Machczynski MC, Vijgenboom E, Samyn B, Canters GW (2004) Characterization of SLAC: a small laccase from Streptomyces coelicolor with unprecedented activity. Protein Sci 13:2388–2397
Martins LO, Soares CM, Pereira M, Teixeira M, Costa T, Jones GH, Jones AO (2002) Molecular and biochemical characterization of a high stable bacterial laccase that occurs as a structural component of the Bacillus subtilis endospore coat. J Biol Chem 277:389–397
Messerschmidt A (1997) Spatial structures of ascorbate oxidase, laccase and related proteins: implications for the catalytic mechanism. In: Messerschmidt A (ed) Multi-copper oxidases, 1st edn. World Scientific, Singapore, pp 23–79
Mikolasch A, Schauer F (2009) Fungal laccases as tools for the synthesis of new hybrid molecules and biomaterials. Appl Microbiol Biotechnol 82:605–624
Mikolasch A, Matthies A, Lalk M, Schauer F (2008a) Laccase-induced C–N coupling of substituted p-hydroquinones with p-aminobenzoic acid in comparison with known chemical routes. Appl Microbiol Biotechnol 80:389–397
Mikolasch A, Wurster M, Lalk M, Witt S, Seefeldt S, Hammer E, Schauer F, Jülich WD, Lindequist U (2008b) Novel β-lactam antibiotics synthesized by amination of catechols using fungal laccase. Chem Pharm Bull 56:902–907
Moore ERB, Pinina Norrod E, Jurtshuk P (1984) Superoxide dismutases of Azotobacter vinelandii and other aerobic, free-living nitrogen-fixing bacteria. FEMS Microbiol Lett 24:261–265
Neyra CA, van Berkum P (1977) Nitrate reduction and nitrogenase activity in Spirillum lipoferum. Can J Microbiol 23:306–310
Palonen H, Viikari L (2004) Role of oxidative enzymatic treatments on enzymatic hydrolysis of softwood. Biotechnol Bioeng 68(5):550–557
Peace TA, Brock KV, Stills HF (1994) Comparative analysis of the 16S rRNA gene sequence of the putative agent of proliferative ileitis of hamsters. Int J Syst Bacteriol 44:832–835
Pomerantez SH (1966) The tyrosine hydroxylase activity of mammalian tyrosinase. J Biol Chem 241:161–168
Roberts SA, Weichsel A, Grass G, Thakali K, Hazzard JT, Tollin G, Rensing C, Montfort WR (2002) Crystal structure and electron transfer kinetics of CueO, a multicopper oxidase required for copper homeostasis in Escherichia coli. Proc Natl Acad Sci USA 99(5):2766–2771
Sanchez-Amat A, Lucas-Elio P, Ferandez E, Garcia-Borron JC, Solano F (2001) Molecular cloning and functional characterization of a unique multipotent polyphenol oxidase from Marinomonas mediterranea. Biochim Biophys Acta 1547:104–116
Sharma R, Goel R, Capalash N (2007) Bacterial laccases. World J Microbiol Biotechnol 23:823–832
Shivprasad S, Page WJ (1989) Catechol formation and melanization by Na+-dependent Azotobacter chroococcum: a protective mechanism for aeroadaption? Appl Environ Microbiol 55(7):1811–1817
Solano F, Lucas-Elio P, Lopez-Serrano D, Fernandez E, Sanchez-Amat A (2001) Dimethoxyphenol oxidase activity of different microbial blue multicopper proteins. FEMS Microbiol Lett 204:175–181
Stevenson LH, Socolofsky MD (1972) Encystment of Azotobacter vinelandii in liquid culture. Antonie Leeuwenhoek 38:605–616
Takami H, Takaki Y, Chee G (2002) Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res 30:3927–3935
Thompson JP, Skerman VBD (1979) Azotobacteraceae: the taxonomy and ecology of the aerobic nitrogen fixing bacteria. Academic, New York
Wheater DM (1955) The characteristics of Lactobacillus acidophilus and Lactobacillus bulgaricus. J Gen Microbiol 12:123–132
White LP (1958) Melanin, a naturally occurring cation exchange material. Nat Lond U K 182:1427–1428
Whittaker JR (1963) Changes in the melanogenesis during the dedifferentiation of chick retinal pigment cells in cell culture. Dev Biol 8:99–127
Williamson PR (1994) Biochemical and molecular characterization of the diphenol oxidase of Cryptococcus neoformans: identification as a laccase. J Bacteriol 176(3):656–664
Wirtz R (1908) Eine einfache Art der Sporenfärbung. Zbl Bakt Hyg I Abt Orig 46:727–728
Wyss O, Smith DD, Pope LM, Olson KE (1969) Endogenous encystment of Azotobacter vinelandii. J Bacteriol 100(1):475–479
Winogradski S (1926) Études sur la Microbiologie du sol. Ann Inst Pasteur 40:455–520
Winogradski S (1935) The method in soil microbiology as illustrated by studies on Azotobacter and the nitrifying organisms. Soil Sci 40:59–76
Winogradski S (1939) Sur la synthése biogéne de l’ammoniac dans le sol et les eaux. 3rd Commission Intern Soc Soil Sci Trans B:37–39
Xu F (1996) Oxidation of phenols, anilines, and benzenethiols by fungal laccases: correlation between activity and redox potentials as well as halide inhibition. Biochemistry 35:7608–7614
Zhu X, Gibbons J, Garcia-Rivera J, Casadevall A, Williamson PR (2001) Laccase of Cryptococcus neoformans is a cell wall-associated virulence factor. Infect Immun 69:5589–5596
Acknowledgements
The government of Mecklenburg-Vorpommern, Germany, in the framework of Landesgraduiertenförderung, is gratefully acknowledged for financial support.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Herter, S., Schmidt, M., Thompson, M.L. et al. A new phenol oxidase produced during melanogenesis and encystment stage in the nitrogen-fixing soil bacterium Azotobacter chroococcum . Appl Microbiol Biotechnol 90, 1037–1049 (2011). https://doi.org/10.1007/s00253-011-3093-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3093-x