
Abstract
Fungal laccases (benzenediol:oxygen oxidoreductase, EC 1.10.3.2) from Pycnoporus cinnabarinus and Myceliophthora thermophila were used as biocatalysts for enzymatic reaction of halogen-, alkyl-, alkoxy-, and carbonyl-substituted p-hydroquinones (laccase substrates) with p-aminobenzoic acid (no laccase substrate). During this reaction, the laccase substrate was oxidized to the corresponding quinones, which react with p-aminobenzoic acid by amination of the laccase substrate. The different substitutions at the hydroquinone substrates were used to prove whether the substituents influence the position of amination and product yields. The cross-coupling of methoxy-p-hydroquinone (alkoxylated) and 2,5-dihydroxybenzaldehyd (carbonyl-substituted) with p-aminobenzoic acid resulted in the formation of one monoaminated product (yield alkoxylated 52%). If monohalogen- or monoalkyl-substituted p-hydroquinones were used as laccase substrates, two monoaminated products (constitution isomers) were formed. The simultaneous formation of two different monoaminated products from the same hydroquinone substrate is the first report for laccase-mediated synthesis of aminated constitution isomers. Depending from the type of substituent of the hydroquinone, the positions of the two monoaminations are different. While the amination at the monoalkylated hydroquinone occurs at the 5- and 6-positions (yield 38%), the amination at monohalogenated hydroquinones was detectable at the 3- and 5-positions (yield 53%). The same product pattern could be achieved if instead of the biocatalyst laccase the chemical catalyst sodium iodate was used as the oxidant. However, the yields were partially much lower (0–45% of the yields with laccase).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
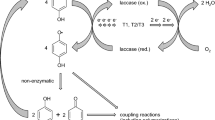
Fungal laccases (EC 1.10.3.2) are multicopper oxidases (Thurston 1994; Yaropolov et al. 1994), which are able to transfer electrons from phenolic hydroxyl groups to molecular oxygen, resulting in unstable phenoxy radicals and water (Bollag 1992; Tatsumi et al. 1994; Heinzkill et al. 1998). Because of their high stability and wide substrate spectrum, laccases are important enzymes increasingly applied in white biotechnology. Laccases have so far been applied mainly in the fields of waste detoxification, textile dye transformation, biosensors, and applications in the food industry and in pulp bleaching (Mayer and Staples 2002; Milligan and Ghindilis 2002; Bollag 1992; Minussi et al. 2002; Rochefort et al. 2004; Mickel et al. 2003; Murugesan et al. 2006; Nagai et al. 2002). On the basis of using aqueous solutions and the mild reaction conditions, the interest for applying laccases in green chemistry grew constantly during recent years (Agematu et al. 1993; Anyanwutaku et al. 1994; Bhalerao et al. 1994; Ikeda et al. 1996, 2001; Osiadacz et al. 1999; Majcherczyk et al. 1999; Schäfer et al. 2001; Uchida et al. 2001; Hosny and Rosazza 2002; Mikolasch and Schauer 2003; Burton 2003; Riva 2006). Most of the laccase-catalyzed syntheses of aromatic amines comprise heterologous coupling reactions performed in aqueous systems resulting in the formation of a monoaminated and/or a diaminated quinone (Niedermeyer et al. 2005; Manda et al. 2005; Mikolasch et al. 2006, 2007). The cross-couplings of 2,5-dihydroxylated arenes with amino compounds are interesting methods for C–N coupling reactions relevant for the organic chemistry and resulted in the formation of one monoaminated quinonoid product per reaction or an unclear product pattern.
To characterize the potential use of laccase-catalyzed reactions in fine chemical synthesis and to gain more basic information about the product pattern, different halogen-, alkyl-, alkoxy-, and carbonyl-substituted p-hydroquinones were brought to reaction with p-aminobenzoic acid by using fungal laccases from Pycnoporus cinnabarinus and Myceliophthora thermophila. The aim of following studies was not only the formation of new products but especially to study the influence of different substituents of laccase substrates on the position of amination in cross-coupling reactions. We show for the first time that laccase-catalyzed amination results in the simultaneous formation of two different monoaminated products (constitution isomers) if monohalogen- or monoalkyl-substituted p-hydroquinones were aminated with p-aminobenzoic acid. Furthermore, we compared the reaction course and the resulting products of laccase-catalyzed oxidative reaction with the synthesis of aminoquinones using the chemical oxidant sodium iodate (Schäfer and Aguado 1971; Pardo et al. 1979; Torres et al. 1985). Advantages of laccase-catalyzed amination are discussed.
Materials and methods
Chemicals
All p-hydroquinones were purchased from Sigma-Aldrich Fine Chemicals (Taufkirchen, Germany). The p-aminobenzoic acid was obtained from Serva Feinbiochemica (Heidelberg, Germany). All chemicals were of p.A. quality.
Enzymes
Fungal strain
P. cinnabarinus SBUG-M 1044 was isolated from an oak tree in northern Germany. The white rot fungus is deposited at the strain collection of the Department of Biology of the University Greifswald (SBUG).
Cultivation of P. cinnabarinus SBUG-M 1044
P. cinnabarinus was initially cultivated on malt agar plates that were incubated for 7 days at 30°C and then kept at 4°C. The liquid culture was prepared by inoculating a nitrogen-rich medium containing 5 g glucose, 1 g KH2PO4, 0.52 g l-asparagine, 0.5 g yeast extract, 0.5 g KCl, 0.5 g MgSO4·7H2O, 50 mL mineral salt solution, and 50 mL FeSO4 solution (0.2 g·L−1) with three 1-cm3 agar culture fragments. The mineral salt solution contained 1 g Ca(NO3)2·4H2O, 0.06 g CuSO4·5H2O, and 0.04 g ZnSO4·7H2O per liter (modified according to Braun-Lüllemann et al. 1997). Incubation was performed without shaking at 30°C for 7 days. A uniform inoculum was obtained by homogenization of this culture with an Ultra-Turrax homogenizer T25 (IKA Labortechnik, Staufen, Germany) at 8,000 rpm. For the production of the ligninolytic enzyme laccase, 40 mL medium inoculated with 2 mL of the homogenized preculture was incubated in 100-mL Erlenmeyer flasks for 7 days with 3,4-dimethoxybenzyl alcohol (10 mM), a known inducer of laccase. Cultures were shaken in a water bath (GFL model 1092, Burgwedel, Germany) at 30°C and 158 rpm.
Preparation of laccase from P. cinnabarinus SBUG-M 1044
P. cinnabarinus was cultivated as described above. Under these conditions, P. cinnabarinus produced laccase as a single extracellular enzyme with activity at a level of 500 nmol·mL−1·min−1 (substrate: 2,2′-amino-bis-3-ethylbenzthiazoline-6-sulfonic acid [ABTS]). The culture medium was filtered through a glass fiber filter in a Büchner funnel to separate the medium from whole cells. The cell-free culture medium was stirred with diethylaminoethyl (DEAE)-Sephacel (Sigma, Steinheim, Germany) for 1 h, and the adsorbed enzymes were eluted from the DEAE-Sephacel with 20 mM sodium acetate buffer (pH 5). This enzyme extract was desalted using a Sephadex G-25 Superfine column (Pharmacia, Freiburg, Germany). This enzyme preparation contains only isoenzymes of laccase and had an activity of 2 U mg−1 and was used always in sodium acetate buffer pH 5.0 because of the pH optimum around pH 5.0 (Feng et al. 1996; Eggert et al. 1996; Jonas 1998).
Laccase from M. thermophila
Laccase from M. thermophila (expressed in genetically modified Aspergillus sp.) was bought from Novozymes (Bagsvaerd, Denmark). It was used as received (activity 1,000 U g−1; substrate: syringaldazine) always in citrate–phosphate buffer pH 7.0 because of the pH optimum around pH 7.0 (Feng et al. 1996; Berka et al. 1997).
Measurement of laccase activity
The activity of laccases was determined spectrophotometrically at 420 nm with ABTS as substrate (Bourbonnais and Paice 1990) using the method described by Jonas et al. (1998). One unit is defined as 1 μmol·mL−1·min−1.
Laccase-catalyzed amination of different substituted p-hydroquinones and quinones with p-aminobenzoic acid
For analytical scale experiments, the compounds were incubated at equimolar concentrations of 1 mM with 0.5, 1, 2, 4, or 10 U/ml of laccases (substrate: ABTS) in 1 ml of sodium acetate buffer (20 mM, pH 5.0) and citrate–phosphate buffer (18 mM citrate, 165 mM phosphate, pH 7.0) for laccases from P. cinnabarinus and M. thermophila, respectively. The reaction mixture was incubated at room temperature for 24 h.
For preparative scale, laccase of P. cinnabarinus (final activity 1.0 U in reaction mixture) was added to 50 mL of a solution of the compounds dissolved in sodium acetate buffer, pH 5.0. For the synthesis of monoaminated quinones, hydroquinone excess was used (2:1 mM). Products of alkyl-substituted p-hydroquinones were isolated 24 h after addition of the enzyme, and products of halogen-, alkoxy-, and carbonyl-substituted p-hydroquinones and of p-hydroquinone were isolated 60 min after starting the reaction.
Chemical product synthesis
Final concentrations in each reaction of p-hydroquinones and p-aminobenzoic acid 2a were 1 mM. Final concentrations for sodium iodate were 6, 24, 50, 100, and 320 mM in sodium acetate buffer, pH 5.0, or in citrate–phosphate buffer, pH 7.0, resulting comparable oxidative reactivity as 0.5, 1, 2, 4, and 10 U activity of laccase. The reaction mixtures were incubated at room temperature for 24 h.
Isolation of products
All products were isolated from reaction mixture of preparative scale as described above.
Product 3a
Product 3a (yield 32.0%) was isolated from the reaction mixture of p-hydroquinone 1a and p-aminobenzoic acid 2a. All isolation steps were performed by solid-phase extraction with an RP18 silica gel column (StrataC18-E, 55 μm, 70 Å, 10 g/60 ml Phenomenex, Aschaffenburg, Germany). After activation with methanol and equilibration with water, the column was loaded with 50 mL of the reaction mixture. Column was washed with 25 mL of water and 25 mL of methanol/water (10:90 v/v) to remove laccase and polar impurities from the column. 3a was eluted with methanol/water 30:70 v/v. The red eluate was collected in fractions, which were analyzed by high-performance liquid chromatography (HPLC) for pure products. After solid-phase extraction, the product containing eluates were combined and dried under vacuum at 35°C.
Products 3b1, 3b2
Products 3b 1 and 3b 2 together (yield together 38.9%) were isolated from the reaction mixture of 2-methyl-p-hydroquinone 1b and p-aminobenzoic acid 2a by the same isolation steps as described for 3a.
Product 3c
Product 3c (yield 6.7%) was isolated from the reaction mixture of 2-tert-butyl-p-hydroquinone 1c and p-aminobenzoic acid 2a by the same isolation steps as described for 3a with one exception. 3c was eluted with methanol/water 50:50 v/v.
Product 3d
Product 3d (yield 51,9%) precipitated from the reaction mixture of 2-methoxy-p-hydroquinone 1d with p-aminobenzoic acid 2a as a red solid. The red precipitate was washed three times with 50 mL of water and dried on the atmosphere.
Products 3g1, 3g2
Product 3g 1 (yield 38.17%) and 3g 2 (yield 15.1%) were isolated from the reaction mixture of 2-chloro-p-hydroquinone 1g and p-aminobenzoic acid 2a by the same isolation steps as described for 3a until the elution of the product from the RP18 silica gel column. 3g 1 and 3g 2 together were eluated from the column with 100% methanol. This eluate was evaporated to a smaller volume of 10 ml. 3g 1 and 3g 2 were separated by preparative HPLC (Merk-HITATCHI, column: LiChroCART® 125-4 RP 18e; 5 μm [Merck, Darmstadt, Germany]). One RP18 silica gel column was loaded with the fractions of separated product 3g 1, and an other RP18 silica gel column was loaded with the fractions of separated product 3g 2 to remove the products from the water phase. The products were eluated with 100% methanol. The methanol eluates with pure products were dried under vacuum at 35°C.
Product 3h
Product 3h (yield 38.45%) was isolated from the reaction mixture of 2,6-dichloro-p-hydroquinone 1h and p-aminobenzoic acid 2a by the same isolation steps as described for 3a with one exception. 3h was eluted with 100% methanol.
For all products, yields have not been optimized.
Characterization of products
Analytical high-performance liquid chromatography
For routine analysis, samples of the incubation mixture were analyzed using an HPLC system LC-10ATvP (Shimadzu, Germany) consisting of an FcV-10ATvP pump, a SPD-M10AvP photodiode array detector (200–595 nm), and a SCL-10AvP control unit controlled by VPClass 5.0. The separation of the substances was achieved on an end-capped, 5-μm, LiChroCart 125-4 RP 18 column (Merck). A solvent system consisting of phosphoric acid, 0.1% pH 2 (eluent A), and methanol (eluent B), starting from an initial ratio of 90% A and 10% B and reaching 100% B within 14 min, was used at a flow rate of 1 mL/min.
Mass spectrometry
The products were characterized by mass spectrometry (MS) and liquid chromatography/MS (LC/MS) using a Bruker-Daltoniks micrOTOF instrument (Bremen, Germany; ionization method: electrospray ionization [ESI], dry and nebulizer gas: nitrogen, software: HyStar).
Nuclear magnetic resonance
The nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 600 instrument (Rheinstetten, Germany) at 600 MHz.
3a (2-[4′-(Carboxyphenyl)amino]-1,4-benzoquinone)
Synthesis and isolation are as described above. The characteristics are as follows: Red precipitate. Yield 32.0%. R f (HPLC) 9.56 min, UV-Vis (MeOH) λ max 201, 285, 497 nm.1H NMR (methanol-d4) δ 8.07(d, 3J = 8.6 Hz, 2H, H3′, H5′), 7.41 (d, 3J = 8.5 Hz, 2H, H2′, H6′), 6.80 (d, 3J = 10.1 Hz, 1H, H6), 6.73 (dd, 3J = 10.1 Hz, 4J = 2.5 Hz, 1H, H5), 6.22 (d, 4J = 2.5 Hz, 1H, H3). 13C NMR δ 187.0 (C4), 182.5 (C1), 170.2 (C7′), 143.5 (C2), 142.0 (C1′), 138.5 (C5), 131.3 (C3′, C5′), 128.2 (C4′), 121.5 (C2′, C6′). Heteronuclear multiple bond correlations (HMBC): data shown in supporting material. Atmospheric pressure ESI (AP-ESI): neg. mode [M − H]− 242.046063 m/z (calculated 242.04585 m/z).
3b1 (5-[4′-(Carboxyphenyl)amino]-2-methyl-1,4-benzoquinone)
Synthesis and isolation are as described above. The characteristics are as follows: Red precipitate. Yield together with 3b 2 38.9%. R f (HPLC) 10.86 min, UV-Vis (MeOH) λ max 201, 283, 488 nm. 1H NMR (acetonitrile-d3) δ 8.00 (d, 3J = 8.6 Hz, 2H, H3′, H5′), 7.38 (d, 3J = 8.6 Hz, 2H, H2′, H6′), 6.22 (s, 1H, H6), 6.18 (d(s), 4J = 2.8 Hz, 1H, H3), 2.06 (d, 4J = 2.8 Hz, 1H, H7), 2.04 (s, 2H, H7). 13C NMR δ 185.0 (C1), 184.3 (C4), 167.1 (C7′), 143.9 (C1′), 143.0 (C5), 135.0 (C2) 132.0 (C3′, C5′), 124.6 (C2′, C6′) 103.0 (C3), 103.2 (C6), 15.2 (C7). HMBC: data shown in supporting material. AP-ESI: neg. mode [M − H]− 256.057324 m/z (calculated 256.06155 m/z). AP-ESI: pos. mode [M + H+] 258.07495 m/z (calculated 258.07605 m/z).
3b2 (6-[4′-(Carboxyphenyl)amino]-2-methyl-1,4-benzoquinone)
Synthesis and isolation are as described above. The characteristics are as follows: Red precipitate. Yield together with 3b 1 38.9%. R f (HPLC) 11.11 min, UV-Vis (MeOH) λ max 201, 283, 496 nm. 1H NMR (acetonitrile-d3) δ 8.00 (d, 3J = 8.6 Hz, 2H, H3′, H5′), 7.38 (d, 3J = 8.6 Hz, 2H, H2′, H6′), 6.62 (m, J = 1.9 Hz, J = 2.8 Hz, 1H, H3), 6.52 (m, J = 1.9 Hz, 1H, H5), 2.05 (d, J = 2.8 Hz, 1H, H7), 2.02 (d(s), J = 1.9 Hz, 2H, H7). 13C NMR δ 187.7 (C1), 167.1 (C7′), 149.7 (C6), 143.9 (C1′), 132.0 (C3′, C5′), 130.7 (C2), 124.6 (C2′, C6′), 100.1 (C3), 100.4 (C5), 16.1 (C7). HMBC: data shown in supporting material. AP-ESI: neg. mode [M − H]− 256.057324 m/z (calculated 256.06155 m/z). AP-ESI: pos. mode [M + H+] 258.07495 m/z (calculated 258.07605 m/z).
3c (6-[4′-(Carboxyphenyl)amino]-2-tert-butyl-1,4-benzoquinone)
Synthesis and isolation are as described above. The characteristics are as follows: Black precipitate. Yield 6.7%. R f (HPLC) 11.96 min, UV-Vis (MeOH) λ max 201, 282, 495 nm. 1H NMR (methanol-d4) δ 8.04 (d, 3J = 8.4 Hz, 2H, H3′, H5′), 7.37 (d, 3J = 8.4 Hz, 2H, H2′, H6′), 6.51 (d(s), 4J = 1.8 Hz, 1H, H3), 6.13 (d(s), 4J = 1.8 Hz, 1H, H5), 1.33 (s, 9H, H8). 13C NMR δ 182.8 (C1), 168.9 (C7′), 152.4 (C2), 142.2 (C1′), 133.0 (C3), 130.6 (C3′, C5′), 128.3 (C4′), 121.4 (C2′, C6′), 99.7 (C5), 34.9 (C7), 27.9 (C8). HMBC: data shown in supporting material. AP-ESI: neg. mode [M − H]− 298.105948 m/z (calculated 298.10845 m/z).
3d (5-[4′-(Carboxyphenyl)amino]-2-methoxy-1,4-benzoquinone)
Synthesis and isolation are as described above. The characteristics are as follows: Red precipitate. Yield 51.9%. R f (HPLC) 9.58 min, UV-Vis (MeOH) λ max 201, 279, 499 nm. 1H NMR (DMSO-d6) δ 9.26 (s, 1H, NH), 7.94 (d, 3J = 8.6 Hz, 2H, H5′, H3′), 7.46 (d, 3J = 8.6 Hz, 2H, H6′, H2′), 6.08 (s, 1H, H3), 5.9 (s, 1H, H6), 3.81 (s, 3H, H7). 13C NMR δ 183.3 (C4), 180.3 (C1), 167.1 (C7′), 161.2 (C2), 144.1 (C5), 142.5 (C1′), 131.3 (C3′, C5′), 127.2 (C4′), 122.7 (C2′, C6′), 104.8 (C3), 100.2 (C6). HMBC: data shown in supporting material. AP-ESI: neg. mode [M − H]− 272.054910 m/z (calculated 272.056398 m/z).
3f1 (3-[4′-(Carboxyphenyl)amino]-2-bromo-1,4-benzoquinone)
The characteristics are as follows: R f (HPLC) 9.44 min, UV-Vis (MeOH) λ max 201, 221, 288, 515 nm. LC/MS m/z AP-ESI: pos. mode; [M + H]+ 322.0 and 324.0 m/z (calculated 322.12 m/z).
3f2 (5-[4′-(Carboxyphenyl)amino]-2-bromo-1,4-benzoquinone)
The characteristics are as follows: R f (HPLC) 10.91 min, UV-Vis (MeOH) λ max 201, 289, 510 nm. LC/MS m/z AP-ESI: pos. mode [M + H]+ 322.0 and 324.0 m/z (calculated 322.12 m/z), [M + Na]+ 344.0 and 346.0 m/z, [M + H–COOH]+ 277.0 and 279.0 m/z, [2M + H]+ 644.9 m/z, [2M + Na]+ 666.9 m/z.
3g1 (3-[4′-(Carboxyphenyl)amino]-2-chloro-1,4-benzoquinone)
Synthesis and isolation are as described above. The characteristics are as follows: Black precipitate. Yield 38.17%. R f (HPLC) 9.58 min, UV-Vis (MeOH) λ max 201, 223, 287, 513 nm. 1H NMR (methanol-d4) δ 7.96 (d, 3J = 8.4 Hz, 2H, H3′, H5′), 7.09 (d, 3J = 8.4 Hz, 2H, H2′, H6′), 6.95 (d, 3J = 10.1 Hz, 1H, H6), 6.85 (d, 3J = 10.1 Hz, 1H, H5). 13C NMR δ 181.9 (C4), 179.8 (C1), 172.1 (C7′), 142.6 (C1′), 140.9 (C3), 137.5 (C6), 133.3 (C5), 129.5 (C3′, C5′), 127.2 (C4′), 122.3 (C2′, C6′), 113.8 (C2). HMBC: data shown in supporting material. AP-ESI: neg. mode [M − H]− 276.015621 m/z (theoretical value 276.006910 m/z).
3g2 (5-[4′-(Carboxyphenyl)amino]-2-chloro-1,4-benzoquinone)
Synthesis and isolation are as described above. The characteristics are as follows: Black precipitate. Yield 15.1%. R f (HPLC) 10.86 min, UV-Vis (MeOH) λ max 201, 285, 513 nm. 1H NMR (methanol-d4) δ 8.14 (d, 3J = 8.4 Hz, 2H, H3′, H5′), 7.42 (d, 3J = 8.4 Hz, 2H, H2′, H6′), 7.09 (s, 1H, H3), 6.29 (s, 1H, H6). 13C NMR δ 181.3 (C4), 178.5 (C1), 169.5 (C7′), 146.1 (C2), 145.2 (C5), 141.5 (C1′) 131.2 (C3′, C5′), 130.8 (C3), 128.3 (C4′), 122.5 (C2′, C-6′), 100.1 (C6). HMBC: data shown in supporting material. AP-ESI: neg. mode [M − H]− 276.010965 m/z (theoretical value 276.006910 m/z).
3h (3-[4′-(Carboxyphenyl)amino]-2,6-dichloro-1,4-benzoquinone)
Synthesis and isolation are as described above. The characteristics are as follows: Black–violet precipitate. Yield 38.45%. R f (HPLC) 11.34 min, UV-Vis (MeOH) λ max 201, 225, 284, 525 nm. 1H NMR (acetonitrile-d3) δ 7.98 (s, 1H, NH), 7.95 (d, 3J = 8.6 Hz, 2H, H3′, H5′), 7.12 (d, 3J = 8.6 Hz, 2H, H2′, H6′), 7.09 (s, 1H, H5). 13C NMR δ 180.9, 177.2, 173.6, 146.4, 143.2, 141.8, 131.8, 130.8, 127.1, 124.0, 114.5. AP-ESI: neg. mode [M − H]− 309.965546 m/z (calculated 309.9685 m/z). AP-ESI: pos. mode [M + H+] 311.982417 m/z (calculated 311.98245 m/z).
3i (3-[4′-(Carboxyphenyl)amino]-2,5-dichloro-1,4-benzoquinone)
The characteristics are as follows: R f (HPLC) 10.97 min, UV-Vis (MeOH) λ max 201, 287, 525 nm. LC/MS m/z AP-ESI: pos. mode [M + H]+ 312.0 and 314.0 m/z (calculated 312.12 m/z), [M + Na]+ 334.0 m/z, [M + H–Cl]+ 278.0 m/z, [M + H–COOH]+ 267.0 m/z, [M + H–Cl–COOH]+ 233.0 m/z.
3j (3-[4′-(Carboxyphenyl)amino]-2-acetyl-1,4-benzoquinone)
The characteristics are as follows: R f (HPLC) 9.64 min, UV-vis (MeOH) λ max 201, 263, 483 nm. LC/MS m/z AP-ESI: pos. mode [M + H]+ 286.1 m/z (calculated 285.26 m/z), [M + Na]+ 308.1 m/z, [M + H–H2O]+ 268.1 m/z, [M + H–COOH]+ 241.0 m/z.
Results
A number of different substituted p-hydroquinones 1a to 1j (Table 1) were subjected to laccase-catalyzed transformation with p-aminobenzoic acid 2a. Resulting from these reactions, 12 N–C coupling dimers consisting of p-quinone and aminobenzoic acid moieties could easily be detected by HPLC by using a diode array detector. With exception of 1e, all substrates were rapidly transformed by laccases independent of the laccase source (substrate utilization 100% within 1h 1a–1d and 1f–1h, 100% within 3h 1i–1j, determined by HPLC).
Using p-hydroquinone (1a) or monoalkylated derivates (1b, 1c) in equimolar reactions, the substrates were oxidized to the corresponding quinones as described for various p-hydroquinones (Niedermeyer et al. 2008; Leontievsky et al. 2001; Liu et al. 1981). The corresponding quinones (the first transformation products of the laccase mediated reaction) of 1a, 1b, and 1c were detectable for a long time by HPLC/UV-Vis, and the yields of monoaminated products (3a, 3b 1,3b 2, 3c) in solution were comparatively low (Fig. 1). Aminations of monohalogenated p-hydroquinones with p-aminobenzoic acid were faster than the reactions of p-hydroquinone or monoalkylated derivates. Furthermore, the yields of monoaminated products (3g 1, 3g 2) were higher if monohalogenated p-hydroquinones were used as laccase substrates. When dihalogenated p-hydroquinones were used, a large amount of products were formed. Methoxy-p-hydroquinone reacted very rapidly to one monoaminated product in quantitative yield, whereas 2,6-dimethoxy-p-hydroquinone was not transformed.

Reaction course of product formation (analyzed with HPLC) for 3a, 3b 1,3b 2, 3c, 3d, 3g 1, 3g 2, and 3h in equimolar reactions (1 mM), using laccase from P. cinnabarinus
Structural characterization of monoaminated main products
Mass spectral and NMR analyses of the products 3a, 3c, 3d, 3h, 3i, and 3j yielded structural data attributed to the coupling of one p-hydroquinone (1a, 1c, 1d, 1h, 1i, 1j) with one molecule p-aminobenzoic acid accompanied by loss of four hydrogen atoms. The structural data of 3a, 3c, 3d, 3h, 3i, and 3j are described in “Materials and methods” and led to the identification of the products as carboxyphenylamino-1,4-benzoquinones.
Laccase-catalyzed amination of methyl-p-hydroquinone and chloro-p-hydroquinone with p-aminobenzoic acid resulted in the formation of 3b 1, 3b 2 and 3g 1, 3g 2 (Fig. 2), respectively. Whereas 3b 1 (t R = 10.86 min) and 3b 2 (t R = 11.11 min) showed nearly the same retention times in the HPLC and could not be separated from each other, 3g 1 (t R = 9.58 min) and 3g 2 (t R = 10.86 min) were isolated as pure substances with distinct chromatographic behavior. After separation of the products 3g 1 and 3g 2 as pure precipitates, mass spectral and NMR spectral data showed that 3g 1 is aminated at the C3 position of the quinone ring and 3g 2 is aminated at the C5 position of the quinone ring.

Principal product patterns of monoaminated products (3b 1, 3b 2 and 3g 1, 3g 2) in equimolar laccase-catalyzed reactions (1 mM) of p-hydroquinones (1b, 1g) with p-aminobenzoic acid (2a)
Although the products 3b 1 and 3b 2 could only be obtained as a mixture, mass spectral and NMR analyses led to the identification of 3b 1 (minor product) as a product which is aminated at the C5 position of the quinone ring and of 3b 2 (major product) as a product which is aminated at the C6 position as described for chemical aminations of methyl-p-quinone with primary aromatic amines (Yogo et al. 1991; Kallmayer and Tappe 1986).
Comparison of laccase-catalyzed reaction with different laccase sources and with established synthesis
Using laccase from P. cinnabarinus, the monoaminated products were the main products sometime accompanied by one or more diaminated products (consisting of para-quinone and two aminobenzoic acid moieties) in low rates (data not shown). The yields of monoaminated products were nearly independent of the activity of laccase from P. cinnabarinus (Fig. 3a). In contrast to the laccase of P. cinnabarinus, higher activity of laccase from M. thermophila results in higher amount of formed products (Fig. 3b). So, the kind of laccase did influence the quantity of monoaminated products but not the product pattern.

Comparison of product formation (analyzed with HPLC) for 3a (triangles), 3b 1 and 3b 2 together (circles), 3g 1 and 3g 2 together (squares) in equimolar reactions (1 mM) using laccase from P. cinnabarinus pH 5.0 (a), laccase from M. thermophila pH 7.0 (b), sodium iodate pH 5.0 (c), and pH 7.0 (d). Final concentrations for sodium iodate were 6, 24, 50, 100, and 320 mM, resulting comparable oxidative reactivity as 0.5, 1, 2, 4, and 10 U activity of laccase
Compared with the laccase-catalyzed reaction (100% relative yield), after 1 h, the accumulated amounts of 3a, 3g 1, and 3g 2 from the reactions with sodium iodate as the oxidant is low at a low concentration of sodium iodate (0–45% relative yield, Fig. 3). With increasing concentration of laccases and sodium iodate, the relative yields of the synthetic method with sodium iodate increased, but a higher concentration of sodium iodate as 320 mM is impossible, because 320 mM means saturated solution. In contrast to sodium iodate, the activity of laccase can be much higher without any problem. The accumulated amounts of 3b 1 and 3b 2 were very low with every method tested, but they were somewhat higher with laccases than with sodium iodate.
Discussion
Most of the p-hydroquinonoid substrates were readily utilized by laccase of P. cinnabarinus and of M. thermophila independent of source of laccases and were oxidized to the corresponding quinones as described for various p-hydroquinones (Leontievsky et al. 2001; Liu et al. 1981; Nakamura 1960; Brown 1967). Interestingly, 1e was not transformed into the quinone under the reaction conditions used due to the fact that 1e was not a substrate of laccases and of sodium iodate, whereas 1d reacted very rapidly to the corresponding quinone. The fact that 1e is not a substrate of laccases and of sodium iodate and did not react to the corresponding quinone explains the lack of amination products because di- and trimethoxylated p-quinones could be chemically aminated (Schäfer et al. 1971).
Structural characterization of halogen-, alkyl-, alkoxy-, and carbonyl-substituted reaction products showed that in the course of laccase-catalyzed reactions, one monoaminated oxidative coupling product or two constitution isomers were formed by amination of p-hydroquinones. The position of the amination was dependent on the character, position, and number of the substituents on the substrate. Using monomethyl-p-hydroquinone and monohalogenated p-hydroquinones, the amination resulted in the formation of two constitution isomers in every case. Whereas in previous experiments, the products of the laccase-catalyzed reaction of monomethyl-p-hydroquinone could not be isolated (Niedermeyer et al. 2005), now we could show that two products were produced. 3b 1 is aminated at the C5 position of the quinone ring, and 3b 2 is aminated at the C6 position. Using monohalogenated p-hydroquinones, the amination took place at C3 and C5. Consequently, the position of amination depends on electronic and steric effects of the substituents. In summary, we could show that for laccase-catalyzed reactions of alkyl-substituted p-hydroquinones with p-aminobenzoic acid, the order of reactivity is as follows: C6 > C5 >> C3 (position C3 did not show any reactivity in enzymatic reaction), according to the order of Brunmark and Cadenas (1989) for chemical reactions.
Laccase-catalyzed amination at the aromatic ring system of carbonyl-substituted p-hydroquinones (2,5-dihydroxyacetophenon, 2,5-dihydroxybenzoic acid derivatives) led to one monoaminated product with C–N coupling at the ortho-position (C3) to the carbonylic groups as described for aminations with primary aromatic amines (Manda et al. 2005; Niedermeyer et al. 2005), with amino acids (Manda et al. 2006), or with amino-β-lactams (Mikolasch et al. 2006, 2007). In summary, we could show that for the laccase-catalyzed reaction of carbonyl-substituted p-hydroquinones with p-aminobenzoic acid, the order of reactivity is as follows: C3 >> C6 > C5 (position C5 and C6 did not show any reactivity in enzymatic reaction), according to the order of Brunmark and Cadenas (1989) for chemical reactions.
To further determine the potential use of laccase-catalyzed reactions in fine chemical synthesis, we compared reactions catalyzed by laccase of P. cinnabarinus and of M. thermophila with an established synthetic method using sodium iodate as the oxidant (Schäfer and Aguado 1971; Pardo et al. 1979; Torres et al. 1985).
If we compare the accumulated amounts of monoaminated products for one reaction, catalyses with laccases cause better yields than with sodium iodate that means that laccase is the better oxidant for the reaction of halogen-, alkyl-, alkoxy-, and carbonyl-substituted p-hydroquinones with p-aminobenzoic acid. Laccase from P. cinnabarinus (pH 5.0) should be applied for the aminations of halogen-, alkoxy-, and carbonyl-substituted p-hydroquinones, whereas for alkyl-substituted p-hydroquinones, the laccase from M. thermophila seems to be more effective. Derived from all results, laccase-induced C–N coupling has some advances over reactions using sodium iodate as the oxidant.
References
Agematu H, Tsuchida T, Kominato K, Shibamoto N, Yoshioka T, Nishida H, Okamoto R, Shin T, Murao S (1993) Enzymatic dimerization of penicillin-X. J Antibiot 46:141–148
Anyanwutaku IO, Petroski RJ, Rosazza JPN (1994) Oxidative coupling of mithramycin and hydroquinone catalysed by copper oxidases and benzoquinone. Implications for the mechanism of action of aureolic acid antibiotics. J Bioorg Med Chem 2:543–551
Berka RM, Schneider P, Golightly EJ, Brown SH, Madden M, Brown KM, Halkier T, Mondorf K, Xu F (1997) Characterization of the gene encoding an extracellular laccase of Myceliophthora thermophila and analysis of the recombinant enzyme expressed in Aspergillus oryzae. Appl Environ Microbiol 63:3151–3157
Bhalerao UT, Muralikrishna C, Rani BR (1994) Laccase enzyme-catalyzed efficient synthesis of 3-substituted-1,2,4-triazolo(4,3-b)(4,1,2)benzothiadiazine-8-ones. Tetrahedron 50:4019–4024
Bollag JM (1992) Enzymes catalyzing oxidative coupling reactions of pollutants. In: Sigel H, Sigel A (eds) Metal ions in biological systems. vol. 28. Marcel Dekker, New York, pp 205–217
Bourbonnais R, Paice MG (1990) Oxidation of non-phenolic substrates—an expanded role for laccase in lignin biodegradation. FEBS Lett 267:99–102
Braun-Lüllemann A, Majcherczyk A, Hüttermann A (1997) Degradation of styrene by white-rot fungi. Appl Microbiol Biotechnol 47:150–155
Brown BR (1967) Biochemical aspects of oxidative coupling of phenols. In: Taylor WJ, Battersby AR (eds) Oxidative coupling of phenols. Marcel Dekker, New York, pp 167–201
Brunmark A, Cadenas E (1989) Redox and addition chemistry of quinoid compounds and its biological implications. Free Radical Bio Med 7:435–477
Burton SG (2003) Laccases and phenol oxidases in organic synthesis—a review. Curr Org Chem 7:1317–1331
Eggert C, Temp U, Eriksson KEL (1996) The Ligninolytic System of the White Rot Fungus Pycnoporus cinnabarinus: Purification and Characterization of the Laccase. Appl Environ Microbiol 62:1151–1158
Feng X, Woonsup S, Brown SH, Wahleithner JA, Sundaram UM, Solomon EI (1996) A study of a series of recombinant fungal laccases and bilirubin oxidase that exhibit significant differences in redox potential, substrate specificity, and stability. Biochim Biophys Acta 1292:303–311
Heinzkill M, Bech L, Halkier T, Schneider P, Anke T (1998) Characterization of laccases and peroxidases from wood-rotting fungi (family Coprinaceae). Appl Environ Microbiol 64:1601–1606
Hosny M, Rosazza JPN (2002) Novel oxidations of (+)-catechin by horseradish peroxidase and laccase. J Agr Food Chem 50:5539–5545
Ikeda R, Uyama H, Kobayashi S (1996) Novel synthetic pathway to a poly(phenylene oxide). Laccase-catalyzed oxidative polymerization of syringic acid. Macromolecules 29:3053–3054
Ikeda R, Tanaka H, Uyama H, Kobayashi S (2001) A new crosslinking method of vinyl polymers having a phenol moiety via oxidative coupling. Polym J 33:959–961
Jonas U (1998) Biotransformation von Biarylverbindungen durch Weißfäulepilze unter besonderer Berücksichtigung des ligninolytischen Enzymsystems von Pycnoporus cinnabarinus und Trametes versicolor. Ph.D. thesis, Greifswald, Germany
Jonas U, Hammer E, Schauer F, Bollag J-M (1998) Transformation of 2-hydroxy dibenzofuran by laccases of the white rot fungi Trametes versicolor and Pycnoporus cinnabarinus and characterization of oligomerisation products. Biodegradation 8:321–328
Kallmayer HJ, Tappe C (1986) Quinone-amine reactions. 18. 2-Methyl-1,4-benzoquinone derivatives from psychopharmacological agents with secondary amine structure. Pharmazie 41:29–33
Leontievsky AA, Myasoedova NM, Baskunov BP, Golovleva LA, Bucke C, Evans CS (2001) Transformation of 2,4,6-trichlorophenol by free and immobilized fungal laccase. Appl Microbiol Biotechnol 57:85–91
Liu SY, Minard RD, Bollag JM (1981) Coupling reactions of 2,4-dichlorophenol with various anilines. J Agric Food Chem 29:253–257
Majcherczyk A, Johannes C, Hüttermann A (1999) Oxidation of aromatic alcohols by laccase from Trametes versicolor mediated by the 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulphonic acid) cation radical and dication. Appl Microbiol Biotechnol 51:267–276
Manda K, Hammer E, Mikolasch A, Niedermeyer THJ, Dec J, Jones AD, Benesi AJ, Schauer F, Bollag JM (2005) Laccase-induced cross-coupling of 4-aminobenzoic acid with para-dihydroxylated compounds 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide and 2,5-dihydroxybenzoic acid methyl ester. J Mol Catal B Enzym 35:86–92
Manda K, Hammer E, Mikolasch A, Gördes D, Thurow K, Schauer F (2006) Laccase-induced derivatization of unprotected amino acid L-tryptophan by coupling with p-hydroquinone 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide. Amino Acids 31:409–419
Mayer A, Staples R (2002) Laccase: new functions for an old enzyme. Phytochemistry 60:551–565
Mickel M, Hee-Cheol K, Hampp N (2003) Origin of the mediator losses in electrochemical delignification processes: primary and secondary reactions of violuric acid and N,N-dimethylvioluric acid radicals with lignin model compounds. Green Chem 5:8–14
Mikolasch A, Schauer F (2003) Biotransformation of N-(2-alkylamino-4-phenylimidazol-1-yl)-acetamides and kinetic studies by using cells and laccase from Trametes versicolor. J Basic Microb 43:508–521
Mikolasch A, Niedermeyer THJ, Lalk M, Witt S, Seefeldt S, Hammer E, Schauer F, Gesell M, Hessel S, Julich WD, Lindequist U (2006) Novel penicillins synthesized by biotransformation using laccase from Trametes spec. Chem Pharm Bull 54:632–638
Mikolasch A, Niedermeyer THJ, Lalk M, Witt S, Seefeldt S, Hammer E, Schauer F, Gesell M, Hessel S, Julich WD, Lindequist U (2007) Novel cephalosporins synthesized by amination of 2,5-dihydroxybenzoic acid derivatives using fungal laccases. Chem Pharm Bull 55:412–416
Milligan C, Ghindilis A (2002) Laccase based sandwich scheme immunosensor employing mediatorless electrocatalysis. Electroanalysis 14:415–419
Minussi R, Pastore G, Duran N (2002) Potential applications of laccase in the food industry. Trends Food Sci Technol 13:205–216
Murugesan K, Arulmani M, In-Hyun N, Young-Mo K, Yoon-Seok C, Kalaichelvan PT (2006) Purification and characterization of laccase produced by a white rot fungus Pleurotus sajorcaju under submerged culture condition and its potential in decolorization of azo dyes. Appl Microbiol Biotechnol 72:939–946
Nagai M, Sato T, Watanabe H, Saito K, Kawata M, Enei H (2002) Purification and characterization of an extracellular laccase from the edible mushroom Lentinula edodes, and decolorization of chemically different dyes. Appl Microbiol Biotechnol 60:327–335
Nakamura T (1960) On the process of enzymatic oxidation of hydroquinone. Biochem Biophys Res Commun 2:111–113
Niedermeyer THJ, Mikolasch A, Lalk M (2005) Nuclear amination catalyzed by fungal laccases: reaction products of p-hydroquinones and primary aromatic amines. J Org Chem 70:2002–2008
Osiadacz J, Al-Adhami AJH, Bajraszewska D, Fischer P, Peczynska-Czoch W (1999) On the use of Trametes versicolor laccase for the conversion of 4-methyl-3-hydroxyanthranilic acid to actinocin chromophore. J Biotechnol 72:141–149
Pardo M, Joos K, Schäfer W (1979) Über die oxidative Aminierung von 1′,4′-dihydroxy-2′-acetonaphthon. Liebigs Ann Chem 1979:503–521
Riva S (2006) Laccases: blue enzymes for green chemistry. Trends Biotechnol 24:219–226
Rochefort D, Leech D, Bourbonnais R (2004) Electron transfer mediator systems for bleaching of paper pulp. Green Chem 6:14–24
Schäfer W, Aguado A (1971) Chemistry of substituted benzoquinones. 8. Oxidative amination of hydroquinones. Angew Chem Int Ed 10:405–406
Schäfer W, Aguado A, Sezer U (1971) Chemistry of Substituted Benzoquinones. 9. New method of preparing heterocyclic quinones. Angew Chem Int Ed 10:406–407
Schäfer A, Specht M, Hetzheim A, Francke W, Schauer F (2001) Synthesis of substituted imidazoles and dimerization products using cells and laccase from Trametes versicolor. Tetrahedron 57:7693–7699
Tatsumi K, Freyer A, Minard RD, Bollag JM (1994b) Enzyme-mediated coupling of 3,4-dichloroanilin and ferulic acid: a model for pollutant binding to humic materials. Environ Sci Technol 28:210–215
Thurston CF (1994) The structure and function of fungal laccases. Microbiology 140:19–26
Torres T, Eswaran SV, Schäfer W (1985) Quinone chemistry. Synthesis of 3-methoxy[2,1]benzisoxazole- and 3-methoxynaphth[2,3-c]isoxazolequinones. J Heterocycl Chem 22:697–699
Uchida H, Fukuda T, Miyamoto H, Kawabata T, Suzuki M, Uwajima T (2001) Polymerization of bisphenol A by purified laccase from Trametes villosa. Biochem Biophys Res Commun 287:355–358
Yaropolov AI, Skorobogatko OV, Vartanov SS, Varfolomeyev SD (1994) Laccase—properties, catalytic mechanism, and applicability. Appl Biochem Biotechnol 49:257–280
Yogo M, Ito C, Furukawa H (1991) Synthesis of some carbazolequinone alkaloids and their analogs—facile palladium-assisted intramolecular ring-closure of arylamino-1,4-benzoquinones to carbazole-1,4-quinones. Chem Pharm Bull 39:328–334
Acknowledgment
We thank K. Weisz (Institute of Biochemistry, University of Greifswald) for providing NMR data and R. Jack (Institute of Immunology, University of Greifswald) for help in preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Laccase-induced C–N coupling of substituted p-hydroquinones with p-aminobenzoic acid in comparison with known chemical routes (DOC 779 KB)
Rights and permissions
About this article
Cite this article
Mikolasch, A., Matthies, A., Lalk, M. et al. Laccase-induced C–N coupling of substituted p-hydroquinones with p-aminobenzoic acid in comparison with known chemical routes. Appl Microbiol Biotechnol 80, 389–397 (2008). https://doi.org/10.1007/s00253-008-1595-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-008-1595-y