
Abstract
Lipase secretion, extracellular lipolysis, and fatty acid uptake were quantified in the yeast Yarrowia lipolytica grown in the presence of olive oil and/or glucose. Specific lipase assays, Western blot analysis, and ELISA indicated that most of the lipase activity measured in Y. lipolytica cultures resulted from the YLLIP2 lipase. Lipase production was triggered by olive oil and, during the first hours of culture, most of the lipase activity and YLLIP2 immunodetection remained associated with the yeast cells. YLLIP2 was then released in the culture medium before it was totally degraded by proteases. Olive oil triglycerides were largely degraded when the lipase was still attached to the cell wall. The fate of lipolysis products in the culture medium and inside the yeast cell, as well as lipid storage, was investigated simultaneously by quantitative TLC–FID and GC analysis. The intracellular levels of free fatty acids (FFA) and triglycerides increased transiently and were dependent on the carbon sources. A maximum fat storage of 37.8% w/w of yeast dry mass was observed with olive oil alone. A transient accumulation of saturated FFA was observed whereas intracellular triglycerides became enriched in unsaturated fatty acids. So far, yeasts have been mainly used for studying the intracellular synthesis, storage, and mobilization of neutral lipids. The present study shows that yeasts are also interesting models for studying extracellular lipolysis and fat uptake by the cell. The quantitative data obtained here allow for the first time to establish interesting analogies with gastrointestinal and vascular lipolysis in humans.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lipases or triacylglycerol hydrolases (E.C.3.1.1.3) are widespread in nature and are particularly common in the microbial world. From the screening of more than 1,200 bacteria, yeasts, and fungi for lipase activity, about 25% of these microorganisms were found to be lipase-positive using soybean oil as substrate (Hou and Johnston 1992; Hou 1997). Although the carbon metabolism of microorganisms is considered to be primarily based on carbohydrates, the presence of lipases enables many microorganisms to utilize “non-conventional” carbon sources such as triglycerides and other lipids with ester bonds. Triglycerides cannot passively cross cell membranes and they have to be degraded into partial glycerides and free fatty acids (FFAs) outside the cell before FFAs are absorbed by the cell. This process requires that microbial lipases are excreted into the medium where lipids are hydrolyzed. Many lipase-secreting microorganisms have thus been isolated from lipid-rich environments such as dairy products and wastes from oil and fat industry.
Some yeasts produce a mixture of extracellular lipases coded by multiple genes (Huge-Jensen et al. 1988; Bertolini et al. 1994; Ferrer et al. 2001; Fickers et al. 2005a), and excreted lipases often show a heterogeneity due to post-translational modifications (Ferrer et al. 2001), including glycosylation (Pignede et al. 2000; Aloulou et al. 2007a; Jolivet et al. 2007). In various lipolytic yeasts, such as Candida rugosa, Yarrowia lipolytica, and Geotrichium candidum, lipases are produced after induction by the fatty acids derived from fat breakdown. Among these yeasts, Y. lipolytica has the ability to produce in large amounts the LIP2 lipase (YLLIP2) in the presence of oleic acid, which is a key factor for the lip2 gene induction and thus for lipase production (Fickers et al. 2004; Beopoulos et al. 2009). Lipase production is also induced in the presence of olive oil from which oleic acid can be released. Other fatty acids, like elaedic and linoleic acids, were also reported as inductors of lipase secretion when used as a carbon source (Hadeball 1991). In yeasts, the oleic acid induction of gene expression was first attributed to activation sequences such as the oleate response element which exists in the upstream regions of genes encoding enzymes involved in β-oxidation, fatty acid metabolism, and proteins involved in peroxisomal biogenesis (Gurvitz and Rottensteiner 2006). Recently, Desfougeres et al. identified the SOA genes (specific for oleic acid), which encode proteins controlling the lip2 lipase gene expression in Y. lipolytica in response to extracellular triacylglycerol (Desfougeres et al. 2009). Yet the overall induction mechanism is not elucidated. Nitrogen source also modulates lipase production and bioactive peptides enhancing lipase production in Y. lipolytica were isolated from peptone and tryptone casein hydrolysates (Turki et al. 2009). In addition to its carbon and nitrogen sources dependency, YLLIP2 production also depends on several physicochemical factors such as pH, oxygen partial pressure (pO2), temperature, and water activity (Hadeball 1991).
Y. lipolytica is frequently isolated from different food media (cheeses, sausages…), from soils, sewage, and natural environments such as oil fields (Barth and Gaillardin 1997). It is a “generally recognized as safe” (GRAS) microorganism with no adverse effect on humans, and various biotechnological applications of this yeast were investigated. Y. lipolytica is used in organic acids production (Finogenova et al. 2005), flavoring lactones production (Wache et al. 2003), single-cell oil production (Papanikolaou et al. 2002), single-cell or heterologous protein production (Senez 1979; Madzak et al. 2004), biosensors (Alkasrawi et al. 1999), and olive mill (Scioli and Vollaro 1997; Lanciotti et al. 2005) and palm oil mill (Oswal et al. 2002) waste water treatments. Many of these applications involve the yeast lipase activity. The purified or partly purified YLLIP2 lipase is under investigation for pharmaceutical (enzyme replacement therapy for pancreatic exocrine insufficiency (Aloulou et al. 2007a)) and chemical (transesterification of 2-bromo-arylacetic acid esters (Guieysse et al. 2004)) applications. These applications of Y. lipolytica lipase are still limited today but optimized and commercially cost-effective processes might soon be accessible. Lipase production was improved by chemical mutagenesis of the wild-type Y. lipolytica strain CBS6303, and a mutant strain (LGx64.81) producing 40-fold more lipase activity in the presence of oleic acid than the wild-type strain was obtained (Fickers et al. 2003). Further enhancement of lipase productivity was obtained by the amplification of the lip2 gene of the LGx64.81 Y. lipolytica strain and the selection of mutant strains (Fickers et al. 2005b). Lipase production was also improved by autocloning and amplification of lip2 gene and the resulting multi-copy strains could produce 0.5 g/L lipase in the culture supernatant, i.e., 40 times more than the single-copy strain (Pignede et al. 2000).
Although Y. lipolytica is a GRAS microorganism, its genetic modification might not be accepted however in many processes and applications particularly when they are related to environment and waste treatment. Therefore, there is still an interest in studying the lipolytic system of the wild-type Y. lipolytica and the natural lipase secretion occurring in the presence of lipids. Apart from biotechnological applications, Y. lipolytica provides an interesting model for investigating fat metabolism, particularly the extracellular lipolysis occurring in connection with lipase secretion. However, many data obtained so far on lipase secretion and lipolysis remain either non-specific or only qualitative. In the present study, the wild-type strain CBS 7504 (W29) was used to perform a full quantitation of lipase secretion and lipolysis in the presence of olive oil in the culture medium. The location and contribution to lipolysis of the YLLIP2 lipase were better characterized using optimized lipase assays (Aloulou et al. 2007a) and specific antibodies (Aloulou et al. 2007b). The fate of lipolysis products in the culture medium and inside the yeast cell, as well as lipid storage, was investigated simultaneously from the complete analysis of lipolysis products derived from olive oil and intracellular neutral lipids.
Materials and methods
Culture conditions
The Y. lipolytica CBS 7504 strain was purchased from the CBS yeasts database CABRI (Common Access to Biological Resources and Information). Batch cultures of the yeast were performed in a 25-mL Erlenmeyer flask containing 5 mL of culture medium. Three different culture media were used: yeast extract—peptone—D-glucose (YPD), yeast extract—peptone—olive oil (YPO), and yeast extract—peptone—D-glucose—olive oil (YPDO). They all contained 1% w/v bacto yeast extract, 2% w/v bacto peptone, and 0.1 M sodium phosphate and the initial pH was adjusted to 6.8. YPD and YPDO media contained 4% w/v (220 mM) dextrose. YPO and YPDO contained 1% w/v (12 mM) olive oil. The bacto yeast extract and bacto peptone solutions were sterilized by autoclaving at 120 °C for 20 min. The sodium phosphate buffer, the D-glucose solution, and the olive oil (extra virgin from Puget, France) were sterilized by filtration through a 0.22-μm membrane (Millipore).
Cultures were inoculated with a 24-h preculture in YPD to get an initial optical density of 1 U at 600 nm, and they were then incubated at 29 °C in an INFORS orbital shaker set at 200 rpm. Individual cultures were stopped at various times (5, 10, 15, 28, and 46 h) and the full contents of the Erlenmeyer flasks were used for analysis.
Cell growth
Biomass growth kinetics were determined by measuring both the dry mass (g of cells/L of culture) and the optical density at 600 nm at different times. For dry mass determination, a 1-mL sample of the culture was transferred into a pre-weighted 1.5-mL microcentrifuge tube. Cells were then separated from the culture supernatant by centrifugation at 10,000 rpm for 10 min at room temperature, washed twice with 1 mL of 0.85% w/v NaCl solution, and dried overnight at 90 °C. After the tube was cooled down to room temperature, it was weighed again for cell dry mass estimation. For optical density measurements, 1 mL of culture was centrifuged and cells were washed twice with 1 mL of a 0.85% w/v NaCl solution. The cell pellet was resuspended in 1 mL of 0.85% w/v NaCl solution before optical density measurements at 600 nm were performed. A correlation between cell dry mass and optical density at 600 nm was then established.
Lipid extraction and analysis
To overcome the heterogeneity of olive oil dispersion in yeast cultures, the whole 5-mL cultures were used for lipid extraction. The full contents of the Erlenmeyer flasks were mixed with 0.2 mL 0.5 N HCl, 1 g of 0.5 mm glass beads, and 5 mL chloroform/methanol (2/1 v/v) containing 1 mg/mL O-cholesteryl ethyleneglycol (CholE1) as an internal standard (IS). Recovery of CholE1 from aqueous biological samples was found to be high and similar to that of triglycerides and lipolysis products (diglycerides (DG), and monoglycerides (MG), FFA) and it was found to be well separated from these products using thin layer chromatography coupled to flame ionization detection (TLC–FID), thus being a suitable IS (Cavalier et al. 2009). This mixture was vortexed in a 25-mL glass tube for 1 min in order to ensure cell disruption and efficient lipid extraction. The tube was then centrifuged at 5,000 rpm (25 °C) for phase separation and the lower organic phase was collected using a Pasteur pipette and transferred to a new 15-mL glass tube. The Erlenmeyer culture flask was washed with a second volume of 5 mL chloroform–methanol (without IS) in order to recover the lipids remaining on the glass surface and this mixture was used for a second lipid extraction of the culture medium. The resulting organic phase was pooled with the previous one. It was checked that two successive extractions were sufficient to extract all lipids from the cultures. The organic phase was dried over anhydrous magnesium sulfate (MgSO4) and MgSO4 was separated by centrifugation at 3,000 rpm for 2 min. The dried organic phase was transferred into a 2-mL glass vial with a Teflon screw cap and the vial was kept at −20 °C until lipid analysis was performed.
Lipids were also extracted from the culture supernatant and the cell fraction separately. In this case, cells were first separated by centrifugation at 5,000 rpm (15 °C) and then washed with 5 mL of a 0.85% w/v NaCl solution. Addition of detergents (Triton X-100, NaTDC) in the cell washing solution was tested and did not result in the higher recovery of lipids (data not shown). After cell separation, the NaCl washing solution and the cell-free culture medium were pooled (approx. 10 mL total volume) and extracted twice with 5 mL chloroform–methanol (2/1) containing 0.5 mg/mL CholE1 and 0.2 mL 0.5 N HCl. Prior to the extraction, the 5-mL chloroform–methanol fractions were used to wash the culture flask as described above. After extraction, the organic phases were pooled, dried on MgSO4 and stored at −20 °C. Lipid extraction from cells was performed by resuspending the cells in 5 mL of water and by adding 0.2 mL 0.5 N HCl, 1 g of glass beads, and 5 mL chloroform–methanol (2/1) containing 0.5 mg/mL CholE1. This procedure was repeated twice and the organic phases were then handled as described previously for the whole culture extraction.
Analysis of lipolysis product was performed by TLC–FID using a Iatroscan MK6 (Iatroscan Laboratories, Japan) as described previously (Cavalier et al. 2009). Heptane–dietyl ether–formic acid (55:45:1, v/v/v) was the mobile phase used for the chromatographic separation of TG, DG, MG, FFA, and CholE1 (IS). The TLC–FID mass detection was calibrated using various amounts of triolein, diolein, monoolein, oleic acid, and O-cholesteryl ethyleneglycol. Lipid mass concentrations in the aqueous samples were estimated from TLC–FID data and the known amounts of IS used for the extraction. The molar masses of triolein (885.43 g/mol), diolein (586.97 g/mol), monoolein (356.54 g/mol), and oleic acid (282.46 g/mol) were used for the conversion of mass values into mole equivalents.
The amounts (mmol) of FFA generated upon lipolysis at a given time t (FFAgt) were estimated from the following equation:
where TG0 is the initial amount of TG in the culture medium and TG t , DG t , and MGt are the residual TG, the DG, and the MG measured at time t, respectively. The amounts of absorbed FFA at the same time t (FFAat) were estimated from the following equation:
where FFA t is the effective amount of FFA measured at time t.
Gas chromatography analysis of fatty acid methyl esters
TGs and FFAs were separated from total lipid extracts using solid phase extraction disposable cartridges. A 1-mL sample of each lipid extract was evaporated to dryness under nitrogen, dissolved into 1 mL chloroform, and then loaded on a silica column (SiOH Chromabond, Macherey-Nagel) previously equilibrated with chloroform. Esterified neutral lipids were first eluted with chloroform (2 × 3 mL) and FFAs were then flushed by 3 mL methanol. These fractions were evaporated to dryness under nitrogen for further derivatization.
The neutral lipid fraction was redissolved in hexane and loaded on a bonded phase aminopropyl-silica column (NH2 Chromabond, Macherey-Nagel) equilibrated with hexane. TGs were eluted with 3 mL hexane/diethyether/dichloromethane (89:1:10, by volume) and dried again under a nitrogen flow before derivatization into fatty acid methyl esters (FAMEs). Two milliliters of 0.5 M sodium methoxide in methanol (1:9, v/v) were added to the TG samples and the reaction mixture was heated at 105 °C for 30 min. After cooling, the FAMEs were extracted in heptane (2 mL), and the upper heptane phase was stored at −20 °C before gas chromatography (GC) analysis.
FFAs were methylated using 2 mL BF3 in methanol (14% w/v, Sigma) and FAMEs were extracted with heptane.
Before GC analysis, each FAME sample in heptane was concentrated under a nitrogen stream until a volume of approx. 50 μL was reached. Two microliters of the sample was then injected into a Thermo Scientific TRACE GC Ultra™ gas chromatograph equipped with a cyanopropyl polysilphenylene-siloxane (10 m × 0.10 mm ID × 0.20 μm film) TR-FAME column. The chromatograph included a split injector at 250 °C and a flame ionization detector (FID) at 280 °C. Hydrogen was used as carrier gas at a flow rate of 0.8 mL/min and a split ratio of 14:1. The oven temperature was programmed as follows: 45 °C for 3 min, from 45 °C to 120 °C at a rate of 30 °C/min, from 120 °C to 165 °C at a rate of 3 °C/min, from 165 °C to 180 °C at a rate of 10 °C/min, from 180 °C to a final temperature of 220 °C at a rate of 30 °C/min, and then the temperature was held at 220 °C for 1 min. The fatty acids were identified by comparison with the separation of a 37-standard-FAME-mixture (Supelco, Sigma).
Lipase activity measurements
Lipase activity in fresh cultures was measured potentiometrically using the pH-stat technique on tributyrin and olive oil in the conditions described by Aloulou et al. (2007a). Activity on tributyrin (0.5 mL) was measured in 14.5 mL of 0.3 mM Tris, 150 mM NaCl, 5 mM CaCl2, and 4 mM sodium taurodeoxycholate (NaTDC) at pH 6.0 and 37 °C. Free butyric acid (pKa = 4.6) released from tributyrin was fully ionized at pH 6 and was measured by direct titration at this pH value with 0.1 N NaOH.
Activity measurements with olive oil were performed at pH 7 and 37 °C in 0.3 mM Tris, 150 mM NaCl, and 5 mM CaCl2 and without bile salts. Although the YLLIP2 lipase was previously found to act optimally on olive oil at pH 6 (Aloulou et al. 2007a), this pH value does not allow a direct titration of long-chain free fatty acids which are poorly ionized at pH 6. YLLIP2 still shows a high specific activity at pH 7.0 and 55% of long-chain free fatty acids are ionized at this pH value. FFAs were directly titrated with 0.1 N NaOH and a correction factor of 1.8 was applied to take into account the partial ionization of fatty acids at pH 7.0. This correction factor was established by back-titration experiments at pH 9 after incubating the purified YLLIP2 lipase and olive oil at pH 7.0. Prior to the assay, an olive oil–gum Arabic (GA) emulsion was prepared by mixing 5 mL of olive oil (extra virgin from Puget, France) with 45 mL of a 10% GA solution in water. Five milliliters of this emulsion were then mixed with 10 mL of a solution containing 0.45 mM Tris, 225 mM NaCl, and 7.5 mM CaCl2 in the pH-stat vessel before the pH was adjusted to 7.0 and the lipase sample was added.
Lipase activities were measured using 10- to 25-μL samples from the culture media and expressed in international units per milliliter. One unit (U) corresponds to 1 μmol of fatty acid released per minute. The specific activities of YLLIP2 under the experimental conditions mentioned above were estimated to be 10,760 ± 115 and 9,180 ± 2,576 U/mg on tributyrin and olive oil, respectively (Aloulou et al. 2007a).
Analysis of proteins from yeast cultures
After centrifugation for removing the cells, the culture medium samples were treated with methanol/chloroform for removing lipids as described by Wessel and Flugge (1984). The protein pellets were resolubilized in a denaturing buffer containing 5% SDS and then subjected to electrophoresis on a 10% polyacrylamide–2% SDS gel. After SDS-PAGE, proteins were detected by acidic silver staining (Blum et al. 1987).
Western blotting analysis on nitrocellulose membrane (Burnette 1981) was performed in order to detect the YLLIP2 lipase in culture media as well as in the washing solutions obtained when the lipase was released from cells (see below). YLLIP2 was detected on membrane by incubating it subsequently with polyclonal anti-YLLIP2 rabbit antibodies (Aloulou et al. 2007b) and then anti-rabbit IgG—alkaline phosphatase conjugate (A0418 from Sigma, diluted 1:10,000). Staining was performed by adding the alkaline phosphatase substrate solution (B6404 BCIP/NBT solution from Sigma) and then stopped by adding 0.5 M H2SO4. Rabbit polyclonal antibodies raised against a specific YLLIP2 peptide (residues 9–17, HIDQESYNF) were also used for Western blot analysis (Aloulou et al. 2007b).
N-terminal amino acid sequencing was performed on an Applied Biosystems Procise 494 automated sequencer after the proteins were blotted on a polyvinylidene difluoride (PVDF) membrane and the protein band excised (Klose 1990). Nitrocellulose and PVDF membranes were purchased from Bio-rad.
Detergents were used to release the lipase bound to cell walls. One-milliliter samples of yeast cultures were centrifuged at 10,000 rpm for 10 min at 20 °C and the resulting cell pellets were first washed with 1 mL 0.85% NaCl, centrifuged again at 10,000 rpm at 20 °C, and then incubated with 1 mL of 13 mM KCl, 5.5 mM K2HPO4/KH2PO4 buffer, pH 6.2, containing either Triton X-100 (Sigma), Tween 80 (Sigma), NaTDC (Sigma), or no detergent. Two detergent concentrations (1.5 and 4 mM) were tested and incubations were performed at room temperature for various times ranging from 5 min to 1 h and 30 min. The lipase activities remaining on cells and released in the washing solution were assayed after separation by centrifugation at 10,000 rpm and 20 °C. The supernatants were also analyzed by Western blot using anti-YLLIP2 antibodies after they were treated with methanol/chloroform for removing lipids (Wessel and Flugge 1984).
A double-sandwich ELISA was performed to measure the amounts of YLLIP2 in samples of Y. lipolytica culture medium (YPO) kept frozen at −80 °C after collection. Rabbit anti-YLLIP2 polyclonal antibodies were used as capture antibodies, and the detector antibody was the rabbit anti-YLLIP2 peptide antibody. The experimental conditions for the ELISA were reported previously (Aloulou et al. 2007b). This assay was validated using known amounts of purified YLLIP2 spiked into samples of Y. lipolytica cultures grown in YPD medium and containing no significant amounts of endogenous lipase (data not shown).
Results
Lipase activity measurements and distribution of lipase activity in cells and culture medium
Lipase activities were first measured in samples collected at various times from Y. lipolytica cultures grown in YPO medium, using both tributyrin and olive oil as substrates (Fig. 1). Since it was already known that a fraction of lipase activity could be bound to yeast cells (Pereira-Meirelles et al. 2000; Fickers et al. 2004), three measurements were performed for each time point using (1) a homogeneous sample of the culture medium containing cells (total lipase activity), (2) the culture supernatant obtained after centrifugation of 1-mL culture sample (unbound lipase activity), and (3) the cell pellet recovered in 1-mL phosphate buffer, pH 6.8 (cell-bound activity).

Distribution of lipase activity in cultures of Y. lipolytica grown in YPO culture medium. a Lipase activity measured using tributyrin as substrate, b lipase activity measured using olive oil as substrate. Each data point (mean ± SD) is the result of triplicate experiments
The time-course variations in lipase activity measured with tributyrin (Fig. 1a) and olive oil (Fig. 1b) showed similar patterns. During the first 15 h of culture, the total lipase activity increased continuously and a large proportion of this activity (60% to 80%) was found to be bound to yeast cells (Fig. 1). After 15 h, the total lipase activity still increased whereas the cell-bound activity had reached a maximum and was gradually released in the culture medium where the optimum unbound activity (75% to 85% of total activity) was obtained at around 30 h of culture. The maximum lipase activities on tributyrin (77 ± 4 U/mL) and olive oil (70 ± 15 U/mL) were recorded during the 15–30 h interval. After that period, the overall lipase activity decreased until no more unbound activity was detected in the culture medium at 70–80 h. A residual lipase activity could still be measured at that time but was entirely bound to yeast cells.
The olive-oil-to-tributyrin-activity ratio was found to vary in a narrow range (1.12 ± 0.37) and was equal to 0.84 ± 0.28 at t = 30 h when most of the activity was released in the culture medium.
In order to improve the quantification of lipase production by Y. lipolytica, it was tested whether all lipase activity could be released in the culture medium before the lipase assay was performed. Yeast cultures were harvested after 15 h of culture when most of the lipase activity was cell-bound and the cells were separated from the culture medium by centrifugation at 10,000 rpm (20 °C). The cells were first washed with a 0.85% NaCl solution and were then incubated for 5 to 90 min with KCL/phosphate buffer alone or in the presence of various detergents. After centrifugation, the lipase release in the supernatant was estimated by both lipase activity measurement with tributyrin as substrate and Western blot using anti-YLLIP2 antibodies (Fig. 2). Some lipase activity was recovered in the NaCl solution that was probably residual unbound lipase since a further incubation of cells with KCL/phosphate buffer alone only allowed to release traces of lipase activity after 90 min of incubation (Fig. 2a). Among the detergents used, Triton-X100 at a concentration of 1.5 mM was the most potent detergent for releasing cell-bound lipase activity with almost all the activity recovered in the supernatant after only 5 min of incubation. Tween 80 and NaTDC at 1.5 mM were less efficient, even after 90 min of incubation (Fig. 2a). Interestingly, the total activity recovered in the supernatant and the cell pellet in the presence of detergents was higher (110% to140%) than the initial cell-bound activity, suggesting that some cell-bound lipase could not express its activity or that the cell-bound lipase had a lower specific activity than the unbound lipase or that some intracellular lipase could also be released by detergents. This latter hypothesis was ruled out by testing cell integrity with the trypan blue dye assay. No coloration of cells was observed whatever the detergent tested was.

Lipase release from cell walls by non-ionic (Trion X-100 and Tween 80) and anionic (NaTDC) detergents. The data shown here were obtained for an incubation time of 90 min but similar results were obtained after only 5 min of incubation of the cells with the detergents at concentrations of 1.5 and 4.0 mM. a Lipase activity measurements. Each data point (mean ± SD) is the result of triplicate experiments. b Detection by Western blot of YLLIP2 release in cell washing buffers, with or without detergents at 1.5 mM
When detergents were tested at a higher concentration (4 mM), the results obtained with Triton X100 were similar to those obtained with 1.5 mM detergent and the effects of Tween 80 were clearly improved with almost all activity recovered in the supernatant (Fig. 2a). With 4 mM NaTDC, most of the activity was recovered in the supernatant but a loss of lipase activity was observed after 90 min of incubation (Fig. 2a). The possibility that the presence of detergents in samples could impact on lipase activity measurements was ruled out. Although these detergents were initially present in the sample at concentrations (1.5 or 4 mM) which have been found to affect lipase activity (Aloulou et al. 2007b), only 100–200 μl of these samples were introduced in the pH-stat vessel containing 15 mL of assay solution and substrate, and their final concentration was always much lower than their critical micellar concentration. Using the purified YLLIP2, it was checked that the addition of detergents at these concentrations in the course of lipase activity measurements had no effect.
Western blot analysis of the various supernatants obtained in the presence of detergents was performed using rabbit polyclonal antibodies produced against purified YLLIP2. They clearly showed that the YLLIP2 lipase was initially cell-bound and was released upon treatment with these detergents (Fig. 2b). A band corresponding to a protein of 38 kDa was detected with anti-YLLIP2 antibodies, the highest intensity being observed after the cells were incubated for 90 min with 1.5 mM Triton X100 and Tween 80, respectively. A similar band was revealed with the yeast culture medium, as well as with a sample of purified YLLIP2 (Fig. 2b). Since YLLIP2 shares some sequence homology with other lipases produced by Y. lipolytica (33–35%) amino acid identities with Lip7 and Lip8 (Fickers et al. 2005a), rabbit polyclonal antibodies produced against a specific YLLIP2 peptide (residues 9–17, HIDQESYNF), poorly conserved in Lip7 and Lip8 (LVSQGFYDA), were also used in Western blot analysis of Yarrowia culture medium and solutions obtained from cell washing with Triton X100. Similar results were obtained with both antibodies, indicating that YLLIP2 was the major lipase detected by Western blot.
Lipase production by Y. lipolytica in different culture conditions
Following the previous experiments which allowed the optimization of the lipase extraction and assay conditions, the lipase production by Y. lipolytica was studied in culture media containing various carbon sources. The yeast was grown in YPO and YPD media containing olive oil and glucose as the sole carbon source, respectively, as well as in YPDO medium containing both olive oil and glucose (Fig. 3). The total lipase activity was measured in homogeneous yeast culture samples, without separating the culture medium from cells but after adding 1.5 mM Triton X100 in each sample. Lipase production was also monitored by Western blot using the anti-YLLIP2 polyclonal antibodies.

Time-course variations in the lipase activity (U/mL), cell growth (dry mass, g/L), and pH in cultures of Y. lipolytica grown in YPO medium (a, a′), YPD medium (b, b′), and YPDO medium (c, c′). Lipase activity was measured using tributyrin as substrate and expressed in units per milliliter of culture medium. Cell growth was estimated from dry mass and expressed in grams per liter of culture medium. Each data point (mean ± SD) is the result of triplicate experiments
When olive oil was used as the sole source of carbon, the lipase activity drastically increased during the early stage of culture and reached a maximum level of 214 ± 72 U/mL at 28 h when the dry mass of yeast cells was 9.7 ± 0.2 g/L (Fig. 3a). As previously observed (Fig. 1), the lipase activity then decreased rapidly and was no longer detected after 70 h whereas the biomass still increased up to 24.2 ± 0.4 g/L at 140 h (Fig. 3a). SDS-PAGE (Fig. 4a) and Western blot (Fig. 4b) analysis of YLLIP2 production showed a very good correlation with lipase activity measurements. The intensity of the 38-kDa band revealed by anti-YLLIP2 antibodies increased with time up to 28–46 h and then totally disappeared at 66 h. The loss of lipase activity at that time was therefore due to the degradation of the lipase. A protein band corresponding to a mass of 31 kDa was also detected after 10 h of culture and its intensity rose up to 66 h (Fig. 4a). After transfer on PVDF membrane, this band was excised and subjected to N-terminal sequence analysis. The 31-kDa protein was identified as the alkaline extracellular protease (AEP) produced by Y. lipolytica (Davidow et al. 1987). Since the pH of the culture medium became alkaline (Fig. 3a′) at the same time that AEP levels increased and lipase activity decreased, it is likely that the YLLIP2 degradation was due to AEP.

SDS-PAGE analysis of the proteins secreted in Y. lipolytica culture medium (YPO) at various culture times (a) and fate of YLLIP2 lipase followed by Western blot analysis (b)
The time-course production of YLLIP2 in YPO culture medium was also measured by using a double-sandwich ELISA with polyclonal anti-YLLIP2 and anti-YLLIP2 peptide antibodies. A very good correlation was obtained between the mass concentrations of YLLIP2 measured by ELISA and the mass concentrations of active YLLIP2 deduced from activity measurements (Fig. 5). A maximum mass concentration of 19.9 ± 6.7 μg/mL YLLIP2 was measured in the YPO culture medium at 28 h.

Comparison between ELISA and lipase activity measurements for determining the mass concentration of YLLIP2 in Y. lipolytica culture medium. Lipase activity measurements (U/mL) using tributyrin as substrate were converted into mass concentration (μg/mL) of active YLLIP2 based on the known specific activity of YLLIP2 on tributyrin (10.760 U/mg). The samples tested were those collected during the experiments with YPO culture medium (see Fig. 3a)
When glucose was used as the sole source of carbon, the lipase activity always remained at very low levels and did not exceed 8 U/mL (Fig. 3b). The yeast growth was similar to that observed in the presence of olive oil with dry masses of 10.7 ± 0.2 g/L at 28 h and 25.0 ± 0.2 g/L at 140 h (Fig. 3c).
When the yeast was cultivated in YPDO medium containing both glucose and olive oil (Fig. 3c), the yeast growth was similar to those observed in YPO and YPD media during the early stage of culture, with a dry mass of 10.4 ± 2.5 g/L at 28 h, but the cell growth was found to be higher in the late stage of culture with a dry mass of 35.0 ± 0.2 g/L at 140 h. Lipase activity remained at low levels (2–19 U/mL) during the early stage of culture, and the maximum lipase activity (33 ± 4 U/mL) was measured at 70 h. The lipase activity levels in YPDO medium were therefore much lower than those observed in YPO medium. In addition, the lipase peak was delayed when compared to lipase secretion in YPO medium (Fig. 3a), indicating a sequential consumption of glucose first and then olive oil. Lipase activity was still detected at 90 h (18 U/mL) in YPDO medium but then fell down to undetectable levels. The fact that lipase activity could be preserved for a longer period of time in YPDO medium could be due to pH variations. The pH value was only 6.6 ± 0.3 at 90 h in YPDO medium (Fig. 3c′) whereas it had already increased up to 8.1 ± 0.3 at 90 h in YPO medium (Fig. 3a′).
Whatever the culture medium in which the yeast cells were grown, the lipase activity was always lost in the late stage of culture when the pH reached alkaline values, suggesting a proteolytic degradation by AEP. It is worth noticing that all cultures were performed in culture media buffered with 0.1 M sodium phosphate in order to limit pH variations.
Lipolysis of olive oil and fat absorption by Y. lipolytica in YPO and YPDO culture media
Y. lipolytica cultures in YPO and YPDO media were collected at various times for total lipid extraction and quantitative analysis of residual triglycerides from olive oil and lipolysis products. Since some triglycerides could be re-synthesized in the yeast cell after the absorption of free fatty acids (FFA), lipid extractions were also performed after the cells and the culture supernatant were separated by centrifugation. The lipid extraction from cells had to be adapted in order to avoid artifacts occurring upon cell lysis. When the cells were first broken using glass beads before adding the organic solvent, the level of FFA was obviously too high and not compatible with normal cell viability (data not shown). This finding suggested that some lipolysis was induced in cell homogenate when the lipase bound to cell wall was in contact with intracellular lipids. Such an artifactual lipolysis was previously observed with many tissues and cells, including pancreatic tissues (Meldolesi et al. 1971). The cells were then directly broken and homogenized in the presence of the chloroform–methanol mixture used for lipid extraction and the FFA levels were found to be much lower. Lipase activity was abolished under these conditions and the intracellular storage of triglycerides could then be observed.
In the complete YPO culture, the triglyceride (TG) levels decreased rapidly during the first 10 h of culture and then remained rather stable (1.5–2 mM TG, 4.5–6 mM acyl chain equivalents) up to 30 h (Fig. 6a). The TGs observed during the 10- to 30-h period were in fact stored inside the cells (Fig. 6c) with a maximum level of 0.16 ± 0.2 mmol TG (0.48 ± 0.6 mmol acyl chain equivalents) per gram of dry cells (14.3% w/w) at 10 h. Residual TGs in the culture medium were no more detectable at 15 h (Fig. 6b). Diglycerides and monoglycerides appeared transiently in the culture medium during the 5- to 10-h period (Fig. 6b), but their levels remained low, indicating that they were also degraded by lipolytic enzymes. The maximum levels of DG (2.4 ± 0.4 mM DG, 4.8 ± 0.8 mM acyl chain equivalents) and MG (1.5 ± 0.4 mM MG or acyl chain equivalents) were recorded at 5 and 10 h, respectively. DG were also observed transiently inside the cells at 10 h (0.11 ± 0.15 mmol DG (0.22 ± 0.3 mmol acyl chain equivalents) per gram of dry cells; 6.4% w/w; Fig. 6c), but not MG. FFA levels rose continuously during the first 15 h to reach 17.7 ± 7.1 mM in the complete YPO culture (Fig. 6a). The concentration in the culture medium however reached its maximum value (12.0 ± 0.5 mM) at 10 h (Fig. 6b). A peak of FFA was observed at the same time inside the cells (0.6 ± 0.5 mmol per gram of dry cells; 17.0% w/w; Fig. 6c). FFA then decreased in both the culture medium and the cells until they were no longer detected after 30 h. The contribution of total fat (TG + DG + FFA) storage to the yeast biomass reached a maximum of 37.8% w/w at 10 h.

Time-course hydrolysis of olive oil by Y. lipolytica and distribution of lipolysis products analyzed by TLC–FID. a Total culture in YPO, b YPO culture medium, c cells grown in YPO, d total culture in YPDO, e YPDO culture medium, f cells grown in YPDO. Each data point (mean ± SD) is the result of triplicate experiments
In the complete YPDO culture, the TG levels decreased rapidly during the first 5 h of culture but this decrease was then slowed down (Fig. 6d). TG at a concentration of 0.6 ± 0.3 mM TG (1.8 ± 0.9 mM acyl chain equivalents) were still observed in the culture supernatant at 66 h (Fig. 6e). Some TG were stored inside the cells with a peak of 0.07 ± 0.10 mmol TG (0.21 ± 0.3 mmol acyl chain equivalents) per gram of dry cells (6.3% w/w) transiently observed at 5 h, followed by a decrease in cellular TG and then a re-accumulation of TG reaching 0.05 ± 0.05 mmol TG (0.15 ± 0.15 mmol acyl chain equivalents) per gram of dry cells (4.3%w/w) at 66 h (Fig. 6f). Diglycerides and monoglycerides appeared in the YPDO culture medium during a longer period (5 to 46 h; Fig. 6e) than in the YPO medium (Fig. 6b). The maximum levels of DG (2.1 ± 0.9 mM DG, 4.2 ± 1.8 mM acyl chain equivalents) and MG (0.8 ± 1.1 mM MG or acyl chain equivalents) were recorded at 10 and 28 h, respectively. No DG and MG were detected inside the cells. The increase in FFA levels was slower in the YPDO medium than in the YPO medium, with a maximum concentration of 6.5 ± 4.7 mM reached at 28 h in the complete YPDO culture (Fig. 6d). The concentration in the culture supernatant reached its maximum value (4.4 ± 0.3 mM) at 15 h (Fig. 6e). As observed with TG, a peak of FFA was rapidly observed inside the cells at 5 h (0.46 ± 0.33 mmol per gram of dry cells; 9.4% w/w; Fig. 6f). FFA then decreased slowly in both the culture medium and the cells, where they were still detected after 66 h. The contribution of total fat (TG + FFA) storage to the yeast biomass reached a maximum of 15.7% w/w at 5 h.
The total amounts of FFA released upon lipolysis of olive oil were estimated from the quantitative analysis of residual TG and intermediate lipolysis products (DG, MG). The absorption rate of FFA by the yeast cells was inferred from the difference between the calculated and the experimentally measured FFA levels (Fig. 7). At 5 h, the mean level of FFA released upon lipolysis was higher in YPDO than in YPO medium (Fig. 7a, b), but the difference was not statistically significant (P > 0.05, Student t-test). From 10 h, the mean level of FFA released upon lipolysis in YPO medium however became much higher than in YPDO medium (Fig. 7a, b) and the difference was statistically significant (P < 0.005, Student t-test). The rate of FFA release in the YPO culture medium was found to be higher than the rate of FFA absorption by cells during the 5- to 15-h period (Fig. 7a), whereas in YPDO medium the rates of FFA release and absorption were similar (Fig. 7b).

Variations with time in the release of FFA upon olive oil lipolysis by Y. lipolytica and FFA absorption by yeast cells. a YPO culture medium, b YPDO culture medium. The total amounts FFA released upon lipolysis of olive oil at a given time t were estimated from the initial amounts of triglycerides (TG0) and the amounts of residual TG, DG, and MG at time t, as indicated in “Lipid extraction and analysis”. FFA absorption level at time t was deduced from the subtraction of the FFA amounts remaining in the culture medium at time t. Values are expressed in percentage (%) of total fatty acids from olive oil. Each data point (mean ± SD) is the result of triplicate experiments
Variations in the fatty acid composition of triglycerides and free fatty acids in Y. lipolytica cells
TG and FFAs from cell lipid extracts were separated and their fatty acid composition (% w/w) was analyzed by gas chromatography after derivatization into FAME (Fig. 8). When the cells were grown in YPO medium, the FFA present in the yeast cells were found to be enriched with time in saturated fatty acids (Fig. 8a). Palmitic acid (16:0; 10.6 ± 1.1% w/w in olive oil) rose up to 29.7 ± 5.0% w/w at 28 h. Stearic acid (18:0; 4.8 ± 1.4% w/w in olive oil) rose up to 24.2 ± 5.0% w/w at 28 h. Conversely, oleic acid (18:1; 79.4 ± 1.5% w/w in olive oil) decreased down to 34.6% w/w at 28 h. At the same time, TG from yeast cells became enriched in unsaturated fatty acids (16:1, 18:1, 18:2; Fig. 8b). The levels of 18:1 rose from 72.1 ± 6.7% w/w at 10 h to 83.4 ± 0.1% w/w at 28 h. Conversely, the levels of 16:0 decreased from 11.4 ± 2.2% w/w at 10 h to 3.2 ± 0.1% w/w at 28 h and those of 18:0 from 5.7 ± 8.0% w/w at 10 h to 0.8 ± 0.1% w/w at 28 h.

Variations with time in the fatty acid composition of triglycerides and free fatty acids in Y. lipolytica cells. a FFA in cells grown in YPO medium, b TG in cells grown in YPO medium, c FFA in cells grown in YPDO medium, d TG in cells grown in YPDO medium. Each data point is the mean value obtained from triplicate experiments
When the cells were grown in YPDO medium, the FFA present in the yeast cells were also found to become enriched in saturated fatty acids (16:0, 18:0) and lowered in 18:1 during the first 10 h of culture (Fig. 8c), but then no significant evolution in the FFA composition was noticed. The composition in fatty acids of intracellular TG did not evolved significantly with time and the unsaturated-to-saturated fatty acid ratio remained constant (Fig. 8d). A slight increase in 16:1 and 18:2 versus 18:1 was however noticed.
Discussion
Choice of optimum assays conditions for measuring the lipase activity produced by Y. lipolytica
In order to optimize the assay of lipase activity produced by Y. lipolytica, preliminary experiments were performed using two substrates, tributyrin and olive oil, and samples collected at various times from yeast cultures in YPO medium. Tributyrin is often used as a convenient substrate for measuring lipase activity because a fine and rather stable emulsion can be formed upon mechanical stirring, the butyric acid released upon lipolysis can be titrated directly by NaOH in a large pH range, and high specific activities can be recorded with most lipases. This short-chain triglyceride substrate however shows a low solubility in water and many esterases are also active on this substrate whereas they are unable to bind long-chain triglycerides and to display real lipase activities. Olive oil is probably the best substrate to detect lipase activities without ambiguity but this substrate requires the use of emulsifiers such as gum Arabic to obtain stable emulsions, and the direct titration of long-chain fatty acids is only possible at high pH values due to their high pKa.
From the experiments depicted in Fig. 1, it was found that the olive-oil-to-tributyrin-activity ratio varied in a narrow range and was similar to that previously estimated (0.85) for the purified YLLIP2 lipase (Aloulou et al. 2007a) and deduced from the specific activities of YLLIP2 on tributyrin (10,760 ± 115 U/mg at pH 6.0) and olive oil (9,180 ± 2,576 U/mg at pH 7.0) using the same assay conditions as those selected here. These findings indicate that most of the lipase activity measured in Y. lipolytica cultures could be due to YLLIP2 and that the activity measured with tributyrin reflects well the secretion of this lipase and not the presence of esterases.
Possible contribution of several lipases to the overall lipase activity measured in Y. lipolytica cultures
Two other enzymes (Lip7p and Lip8p) presenting sequence homologies with the extracellular fungal lipases were also identified as secreted lipases produced by Y. lipolytica (Fickers et al. 2005a) and these lipases might therefore contribute to the overall lipase activity measured in the present study. The growth of Y. lipolytica mutant strains in the presence of olive oil or tributyrin as the sole carbon source suggested that Lip7p and Lip8p were true lipases, but their substrate specificity was only investigated using para-nitrophenyl esters with various acyl chains so far (Fickers et al. 2005a; Song et al. 2006). From studies performed with a LIP2 knock-out Y. lipolytica strain, it was estimated that the lipolytic activity of these lipases might contribute to only 3% of the total lipase activity secreted by the wild-type strain of Y. lipolytica (Pignede et al. 2000). It was also proposed that Lip7p and Lip8p could be mainly associated to the cell wall and could correspond to the cell-bound lipase initially described by Ota et al. (1982), whereas YLLIP2 would be the main lipase released in the culture medium at the end of the growth phase (Fickers et al. 2004). Our data support this latter point since the olive-oil-to-tributyrin-activity ratio (0.84 ± 0.28) measured when most of the activity was released in the culture medium (t = 30 h) was almost identical to that measured with the purified YLLIP2 (0.85). This ratio slightly increased when most of the lipase activity was bound to yeast cells at the early and late stages of yeast cultures, suggesting that other enzymes were involved in the overall lipase activity during these periods. From the incubation of yeast cells with detergents and Western blot analysis, we however found that YLLIP2 was initially bound to the cell wall during the growth phase and probably contributed to a large extent to the cell-bound activity (Fig. 2). When the lipase time-course production in YPO medium was monitored by both total activity measurements and ELISA using anti-YLLIP2 antibodies, a very good correlation between these assays was observed (Fig. 5), and the complete loss of lipase activity (Fig. 3a) corresponded to the loss of YLLIP2 immunodetection by Western blot (Fig. 4). All these data support a major contribution of YLLIP2 in the overall lipase activity produced by Y. lipolytica.
Induction of lipase production
As shown in previous studies, it was observed here that olive oil was an inducer of lipase production when it was the sole source of carbon in the culture medium (Fig. 3a). When both olive oil and glucose were present in the culture medium, glucose acted as a repressor of lipase production and the lipase levels remained low until glucose was consumed and a delayed peak of lipase production was observed (Fig. 3c). In fact, the products of triglycerides digestion usually trigger lipase secretion better than triglycerides. Free fatty acids, particularly oleic acid, are the best inducers of enzyme secretion not only in Y. lipolytica (Destain et al. 1997; Pignede et al. 2000; Fickers et al. 2004) but also in humans. In yeast, there is a direct regulation of lipase gene expression by other genes specific for oleic acid (Desfougeres et al. 2009). In humans, oleic acid triggers first the secretion of the gastrointestinal hormone CCK which in turn triggers the secretion of the exocrine pancreas (Hildebrand et al. 1998). This latter process is not specific for lipase secretion but promotes the secretion of all pancreatic enzymes. Y. lipolytica also secreted other enzymes in the culture medium such as the alkaline protease (AEP), and the secretion of this enzyme is parallel to lipase secretion (Fig. 4). Lipase is degraded by AEP or other unidentified proteases when the pH becomes alkaline (Figs. 3a′ and 4) but TG have been already digested at that time (Fig. 6). In humans, digestive enzymes are also cleaved by proteases during the digestion process once they have digested their own substrates. Under normal conditions, the levels of residual pancreatic enzymes in the distal part of the intestine and in feces are extremely low.
Lipolysis of olive oil by Y. lipolytica and analogies with gastrointestinal lipolysis in humans
The extracellular lipases produced by Y. lipolytica, particularly YLLIP2, are involved in the release of fatty acids from an external source of fat and the uptake of these fatty acids by the yeast cell. This process is reminiscent of the gastrointestinal lipolysis process occurring in the human gut and that allows fat uptake after the digestion of dietary triglycerides (Lengsfeld et al. 2004). From the analysis by TLC–FID of the lipolysis products released in the YPO culture medium, we could estimate the amounts of FFA released from olive oil per unit time (1 U = 1 μmol FFA released per min) and thus the lipase turnover in its natural environment. In order to express this turnover in terms of specific activity (U per milligram of lipase), the mass concentration of YLLIP2 in the culture medium was either deduced from the lipase activity measurements performed with the pH-stat technique (see “Materials and methods”; Fig. 3) and the known specific activity of YLLIP2 under these conditions or as measured by ELISA. For the estimation of YLLIP2 mass concentration from activity measurements, it was considered that the lipase activity measured with the pH-stat technique was entirely due to YLLIP2 and the possible implication of other lipases was neglected based on the ELISA results (Fig. 5). The mean specific activity of Y. lipolytica lipase on the olive oil present in YPO medium was estimated to be 27 ± 11 U/mg during the first 10 h of culture. This specific activity is much lower (340-fold) than the specific activity of YLLIP2 on olive oil measured under optimized conditions with the pH-stat technique (9,180 ± 2,576 U/mg). This low enzyme turnover was however sufficient to hydrolyze all triglycerides in a few hours as shown in Fig. 6b. A similar finding was made previously with human digestive lipases in the course of meal digestion (Carriere et al. 2000). The specific activities of human gastric and pancreatic lipases on test meal triglycerides were found to be 33 and 12 U/mg, respectively, whereas these lipases showed much higher activities under optimized assay conditions. In our experiments with Y. lipolytica, the TG-to-lipase molar ratio in the YPO culture medium was also found to be in the same range (2.5-fold) as the TG-to-pancreatic-lipase ratio observed in human intestinal contents during a meal (Table 1). Proportionally, the yeast Y. lipolytica is therefore able to produce a lipolytic activity similar to that of the human pancreas.
Analogies between extracellular lipolysis by Y. lipolytica and lipolysis of lipoproteins in human vascular system
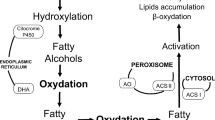
When Y. lipolytica is grown in the presence of olive oil as the sole carbon source, lipase secretion and lipolysis of triglycerides start rapidly after the initiation of the culture (Figs. 3a and 5). During the first hours of culture, most of the lipase activity is associated with the yeast cells (Fig. 1) as previously reported (Pereira-Meirelles et al. 2000). Triglycerides are largely degraded during that period (Fig. 6b), suggesting that the lipolysis process mainly occurs on the cell wall when olive oil droplets meet yeast cells (Fig. 9). Small protrusions on the surfaces of Y. lipolytica cells grown in the presence of fatty acids were observed by electron microscopy and it was hypothesized that these protusions are important for the uptake of neutral lipids by the cell (Mlickova et al. 2004). Lipid droplets (LDs) were found to be attached to the protrusions on the cell surfaces, and their number increased with the number of protrusions. The average diameter of LDs on the cell surface decreased from 310 ± 18 nm during the first hours of growth to 51 ± 30 nm at 40 h, suggesting that fatty acid uptake by the cell occurs on the protusions. Within this context, and assuming that olive oil droplets would bind similarly to the cell surface, the YLLIP2 lipase might be specifically localized at the surface of protusions where FFA would be released and directly absorbed by the cell (Fig. 9). This hypothesis however remains to be investigated by immunocytolocalization in future studies. The mechanism of YLLIP2 binding to the yeast cell wall and its further release in the culture medium remain unexplored, but the lipase can be easily released from the cells by using detergents (Fig. 2). This process is reminiscent of the lipolysis of lipoproteins occurring at the vascular side of blood vessels where lipoprotein lipase (LPL) is bound at the surface of the capillary endothelium through a non-covalent linkage by heparan sulfates (Carriere et al. 1995; Spillmann et al. 2006). The interaction between the lipase and lipid droplets occurs at the surface of the endothelial cells and free fatty acids can be directly absorbed through the cell membrane. Therefore, this lipolytic system seems to be tailored for producing free fatty acids at their site of absorption. The bioavailability of FFAs would certainly be less efficient if these FFAs were released in the bulk phase and had to diffuse/be transported towards the cells (Scow et al. 1979). In mammals, for instance, the intravenous injection of heparin decreases the uptake of serum lipoproteins by releasing LPL from the vascular endothelium (Engelberg 1980). Moreover, lipolysis products like FFAs are known to be cytotoxic and they are never found at high concentrations in biological systems, except in the presence of specific transporters such as bile salt micelles in the gut and serum albumin in the blood. It might therefore be essential for these FFAs to be taken up as soon as they are produced in the yeast culture medium.

Schematic representation of the putative mechanism for extracellular lipolysis and FFA uptake in Y. lipolytica cultures
Lipid absorption and storage by Y. lipolytica
The accumulation of FFA and TG storage by the yeast was found to be faster in YPDO medium (peak at 5 h; Fig. 6f) than in YPO medium (peak at 10 h; Fig. 6c), indicating that the glucose present in the YPDO medium was the preferred carbon source for cell growth during the first 5 h of culture. In YDO medium, no intra-cellular FFA and TG could be detected at that time and, in this case, all of the FFA released upon lipolysis were probably used for cell growth through β-oxidation and membrane biogenesis. Intra-cellular TG levels then increased while FFA appeared transiently inside the cells and TG storage remained rather stable from 10 to 30 h in cells growing in YPO medium (Fig. 6c). FFA were therefore used at the same time both for cell growth and energy storage via their re-esterification to form intra-cellular TG. The rate of FFA release in the culture medium was found to be higher than the rate of FFA absorption by cells during that period (Fig. 7a), suggesting a saturation of the FFA uptake by the cells. In YPDO medium, the rate of FFA release slowed down after 5 h and the absorption rate was found to be of the same level (Fig. 7b). A basal FFA level was always detected inside the cells under these conditions and intra-cellular TG slightly increased with time (Fig. 6f) until the culture was stopped. These findings support the use of glucose as the main source of carbon and energy for cell growth in YPDO medium, while the consumption of all FFA and stored TG (Fig. 6c) is required for increasing the cell biomass level in YPO medium (Fig. 3a) and to reach the same levels as those observed in the presence of glucose as the sole carbon source (Fig. 3c).
The maximum amounts of lipid detected inside the yeast cells (37.8% w/w of yeast dry mass at 10 h in YPO medium) were similar to those previously reported for Y. lipolytica (Montet et al. 1985; Papanikolaou et al. 2001; Ratledge and Wynn 2002), justifying the classification of Y. lipolytica among oleaginous yeasts (Beopoulos et al. 2009).
From the analysis of intracellular lipids (Fig. 8), it could be noticed that the FFA transiently observed in the yeast cells were enriched in saturated fatty acids (16:0, 18:0), whereas the TGs stored inside the cell were enriched in unsaturated fatty acids (18:1, 18:2). These features were more pronounced when the yeast cells were grown in YPO medium (Fig. 7a, b) and the metabolism of olive oil fatty acids was faster, suggesting that saturated fatty acids are preferentially oriented towards the β-oxidation pathway rather than towards their re-incorporation in TGs. This selectivity in fatty acid incorporation in yeast TGs was already observed: using various fat sources for the growth of Candida lipolytica (former name of Y. lipolytica), the unsaturated fatty acid contents of yeast cell TGs were always greater than that of the substrate (Montet et al. 1985). The fatty composition of intracellular lipids was however found to be highly dependent on the composition of the fat used as carbon source: the main intracellular fatty acid was stearic acid when the yeast was grown on stearin (Papanikolaou et al. 2001), linoleic acid contents were increased when the yeast was grown on rapeseed oil (Montet et al. 1985), and oleic acid was the main fatty acid in this study performed with olive oil. To explain the enrichment in unsaturated fatty acids of intracellular lipids, it was proposed that desaturases were actively involved in the conversion of palmitic and stearic acid into unsaturated fatty acids before their incorporation in the intracellular TGs (Montet et al. 1985). It was shown however in the present study that saturated fatty acids transiently accumulated as FFAs in the yeast cell before lipids were totally consumed. Fatty acid selectivity might therefore result from the specificity of the diacylglycerol O-acyltransferases involved in TG synthesis (Beopoulos et al. 2009).
So far, yeasts like Saccharomyces cerevisiae have been mainly used for studying the intracellular synthesis, storage, and mobilization of neutral lipids (Czabany et al. 2007; Daum et al. 2007), and it was shown that triglyceride lipolysis was functionally conserved from mammals to yeast (Kurat et al. 2006). The present study shows that yeasts are also interesting models for studying extracellular lipolysis and fat uptake and that interesting analogies with human gastrointestinal and vascular lipolysis can be made.
References
Alkasrawi M, Nandakumar R, Margesin R, Schinner F, Mattiasson B (1999) A microbial biosensor based on Yarrowia lipolytica for the off-line determination of middle-chain alkanes. Biosens Bioelectron 14:723–727
Aloulou A, Puccinelli D, De Caro A, Leblond Y, Carriere F (2007a) A comparative study on two fungal lipases from Thermomyces lanuginosus and Yarrowia lipolytica shows the combined effects of detergents and pH on lipase adsorption and activity. Biochim Biophys Acta 1771:1446–1456
Aloulou A, Rodriguez JA, Puccinelli D, Mouz N, Leclaire J, Leblond Y, Carriere F (2007b) Purification and biochemical characterization of the LIP2 lipase from Yarrowia lipolytica. Biochim Biophys Acta 1771:228–237
Barth G, Gaillardin C (1997) Physiology and genetics of the dimorphic fungus Yarrowia lipolytica. FEMS Microbiol Rev 19:219–237
Beopoulos A, Cescut J, Haddouche R, Uribelarrea JL, Molina-Jouve C, Nicaud JM (2009) Yarrowia lipolytica as a model for bio-oil production. Prog Lipid Res 48:375–387
Bertolini MC, Laramee L, Thomas DY, Cygler M, Schrag JD, Vernet T (1994) Polymorphism in the lipase genes of Geotrichum candidum strains. Eur J Biochem 219:119–125
Blum H, Beier H, Gross HJ (1987) Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8:93–99
Burnette WN (1981) “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate–polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem 112:195–203
Carriere F, Verger R, Lookene A, Olivecrona G (1995) Lipase structures at the interface between chemistry and biochemistry. EXS 73:3–26
Carriere F, Renou C, Lopez V, De Caro J, Ferrato F, Lengsfeld H, De Caro A, Laugier R, Verger R (2000) The specific activities of human digestive lipases measured from the in vivo and in vitro lipolysis of test meals. Gastroenterology 119:949–960
Cavalier JF, Lafont D, Boullanger P, Houisse D, Giallo J, Ballester JM, Carriere F (2009) Validation of lipolysis product extraction from aqueous/biological samples, separation and quantification by thin-layer chromatography with flame ionization detection analysis using O-cholesteryl ethylene glycol as a new internal standard. J Chromatogr A 1216:6543–6548
Czabany T, Athenstaedt K, Daum G (2007) Synthesis, storage and degradation of neutral lipids in yeast. Biochim Biophys Acta 1771:299–309
Daum G, Wagner A, Czabany T, Athenstaedt K (2007) Dynamics of neutral lipid storage and mobilization in yeast. Biochimie 89:243–248
Davidow LS, O’Donnell MM, Kaczmarek FS, Pereira DA, DeZeeuw JR, Franke AE (1987) Cloning and sequencing of the alkaline extracellular protease gene of Yarrowia lipolytica. J Bacteriol 169:4621–4629
Desfougeres T, Haddouche R, Fudalej F, Neuveglise C, Nicaud JM (2009) SOA genes encode proteins controlling lipase expression in response to triacylglycerol utilization in the yeast Yarrowia lipolytica. FEMS Yeast Res 10:93–103
Destain J, Roblain D, Thonart P (1997) Improvement of lipase production from Yarrowia lipolytica. Biotechnol Lett 19:105–107
Engelberg H (1980) Heparin and atherosclerosis. A review of old and recent findings. Am Heart J 99:359–372
Ferrer P, Montesinos JL, Valero F, Sola C (2001) Production of native and recombinant lipases by Candida rugosa: a review. Appl Biochem Biotechnol 95:221–255
Fickers P, Nicaud JM, Destain J, Thonart P (2003) Overproduction of lipase by Yarrowia lipolytica mutants. Appl Microbiol Biotechnol 63:136–142
Fickers P, Nicaud JM, Gaillardin C, Destain J, Thonart P (2004) Carbon and nitrogen sources modulate lipase production in the yeast Yarrowia lipolytica. J Appl Microbiol 96:742–749
Fickers P, Fudalej F, Le Dall MT, Casaregola S, Gaillardin C, Thonart P, Nicaud JM (2005a) Identification and characterisation of LIP7 and LIP8 genes encoding two extracellular triacylglycerol lipases in the yeast Yarrowia lipolytica. Fungal Genet Biol 42:264–274
Fickers P, Fudalej F, Nicaud JM, Destain J, Thonart P (2005b) Selection of new over-producing derivatives for the improvement of extracellular lipase production by the non-conventional yeast Yarrowia lipolytica. J Biotechnol 115:379–386
Finogenova TV, Morgunov IG, Kamzolova SV, Cherniavskaia OG (2005) Organic acid production by the yeast Yarrowia lipolytica (a review). Prikl Biokhim Mikrobiol 41:478–486
Guieysse D, Sandoval G, Faure L, Nicaud JM, Monsan P, Marty A (2004) New efficient lipase from Yarrowia lipolytica for the resolution of 2-bromo-arylacetic acid esters. Tetrahedron Asymmetr 15:3539–3543
Gurvitz A, Rottensteiner H (2006) The biochemistry of oleate induction: transcriptional upregulation and peroxisome proliferation. Biochim Biophys Acta 1763:1392–1402
Hadeball W (1991) Production of lipase by Yarrowia lipolytica. I. Lipases from yeasts. Acta Biotechnologica 11:159–167
Hildebrand P, Petrig C, Burckhardt B, Ketterer S, Lengsfeld H, Fleury A, Hadvary P, Beglinger C (1998) Hydrolysis of dietary fat by pancreatic lipase stimulates cholecystokinin release. Gastroenterology 114:123–129
Hou CT (1997) Characterization of new yeast lipases. J Am Oil Chem Soc 74:1391–1394
Hou CT, Johnston TM (1992) Screening of lipase activities with cultures from the agricultural research service culture collection. J Am Oil Chem Soc 69:1088–1097
Huge-Jensen B, Galluzzo DR, Jensen RG (1988) Studies on free and immobilized lipases from Mucor miehei. J Am Oil Chem Soc 65:905–910
Jolivet P, Bordes F, Fudalej F, Cancino M, Vignaud C, Dossat V, Burghoffer C, Marty A, Chardot T, Nicaud JM (2007) Analysis of Yarrowia lipolytica extracellular lipase Lip2p glycosylation. FEMS Yeast Res 7:1317–1327
Klose J (1990) Blotting and sequencing. Electrophoresis 11:517–593
Kurat CF, Natter K, Petschnigg J, Wolinski H, Scheuringer K, Scholz H, Zimmermann R, Leber R, Zechner R, Kohlwein SD (2006) Obese yeast: triglyceride lipolysis is functionally conserved from mammals to yeast. J Biol Chem 281:491–500
Lanciotti R, Gianotti A, Baldi D, Angrisani R, Suzzi G, Mastrocola D, Guerzoni ME (2005) Use of Yarrowia lipolytica strains for the treatment of olive mill wastewater. Bioresour Technol 96:317–322
Lengsfeld H, Beaumier-Gallon G, Chahinian H, De Caro A, Verger R, Laugier R, Carrière F (2004) Physiology of gastrointestinal lipolysis and therapeutical use of lipases and digestive lipase inhibitors. In: Müller G, Petry S (eds) Lipases and phospholipases in drug development. Wiley-VCH, Weinheim, pp 195–229
Madzak C, Gaillardin C, Beckerich JM (2004) Heterologous protein expression and secretion in the non-conventional yeast Yarrowia lipolytica: a review. J Biotechnol 109:63–81
Meldolesi J, Jamieson JD, Palade GE (1971) Composition of cellular membranes in the pancreas of the guinea pig. II. Lipids. J Cell Biol 49:130–149
Mlickova K, Roux E, Athenstaedt K, d’Andrea S, Daum G, Chardot T, Nicaud JM (2004) Lipid accumulation, lipid body formation, and acyl coenzyme A oxidases of the yeast Yarrowia lipolytica. Appl Environ Microbiol 70:3918–3924
Montet D, Ratomahenina R, Galzy P, Pina M, Graille J (1985) A study of the influence of the growth media on the fatty acid composition in Candida lipolytica diddens and lodder. Biotechnol Lett 7:733–736
Oswal N, Sarma PM, Zinjarde SS, Pant A (2002) Palm oil mill effluent treatment by a tropical marine yeast. Bioresour Technol 85:35–37
Ota Y, Oikawa S, Morimoto Y, Minoda Y (1982) Purification and some properties of cell-bound lipase from Saccharomycopsis lipolytica. Agric Biol Chem 46:2885–2893
Papanikolaou S, Chevalot I, Komaitis M, Aggelis G, Marc I (2001) Kinetic profile of the cellular lipid composition in an oleaginous Yarrowia lipolytica capable of producing a cocoa-butter substitute from industrial fats. Antonie Leeuwenhoek 80:215–224
Papanikolaou S, Chevalot I, Komaitis M, Marc I, Aggelis G (2002) Single cell oil production by Yarrowia lipolytica growing on an industrial derivative of animal fat in batch cultures. Appl Microbiol Biotechnol 58:308–312
Pereira-Meirelles FV, Rocha-Leão MHM, Sant’Anna GL Jr (2000) Lipase location in Yarrowia lipolytica cells. Biotechnol Lett 22:71–75
Pignede G, Wang H, Fudalej F, Gaillardin C, Seman M, Nicaud JM (2000) Characterization of an extracellular lipase encoded by LIP2 in Yarrowia lipolytica. J Bacteriol 182:2802–2810
Ratledge C, Wynn JP (2002) The biochemistry and molecular biology of lipid accumulation in oleaginous microorganisms. Adv Appl Microbiol 51:1–51
Scioli C, Vollaro L (1997) The use of Yarrowia lipolytica to reduce pollution in olive mill wastewaters. Wat Res 31:2520–2524
Scow RO, Desnuelle P, Verger R (1979) Lipolysis and lipid movement in a membrane model. Action of lipoprotein lipase. J Biol Chem 254:6456–6463
Senez JC (1979) Single cell proteins cultivated on hydrocarbons. Bibl Nutr Dieta 28:30–39
Song HT, Jiang ZB, Ma LX (2006) Expression and purification of two lipases from Yarrowia lipolytica AS 2.1216. Protein Expr Purif 47:393–397
Spillmann D, Lookene A, Olivecrona G (2006) Isolation and characterization of low sulfated heparan sulfate sequences with affinity for lipoprotein lipase. J Biol Chem 281:23405–23413
Turki S, Kraeim IB, Weeckers F, Thonart P, Kallel H (2009) Isolation of bioactive peptides from tryptone that modulate lipase production in Yarrowia lipolytica. Bioresour Technol 100:2724–2731
Wache Y, Aguedo M, Nicaud JM, Belin JM (2003) Catabolism of hydroxyacids and biotechnological production of lactones by Yarrowia lipolytica. Appl Microbiol Biotechnol 61:393–404
Wessel D, Flugge UI (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138:141–143
Acknowledgements
The Ph.D. work of Mrs. Amal Najjar received financial support from El-Sabat Association, Lebanon. We are grateful to Dr. Régine Lebrun and Sabrina Lignon (IMM, Marseille, France) for performing the N-terminal protein sequencing and to Dr. Mathieu Schue (CNRS-UPR9025, Marseille) for the validation of the ELISA for measuring YLLIP2 in Y. lipolytica cultures.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is dedicated to the memory of our colleague and friend, Dr. Michèle Violet-Asther, who was killed on September 25, 2009.
Rights and permissions
About this article
Cite this article
Najjar, A., Robert, S., Guérin, C. et al. Quantitative study of lipase secretion, extracellular lipolysis, and lipid storage in the yeast Yarrowia lipolytica grown in the presence of olive oil: analogies with lipolysis in humans. Appl Microbiol Biotechnol 89, 1947–1962 (2011). https://doi.org/10.1007/s00253-010-2993-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2993-5