
Abstract
The purpose of this study is to evaluate whether sample preservation can affect the yield of nucleic acid extracts from environmental samples. Storage of microbial samples was studied using three sediment types of varying carbon contents (10–57% carbon of dry weight). Four different storage solutions were tested at three temperatures. Freezing of samples at −20 °C or −80 °C, either without preservative or in phenol–chloroform solution, retained nucleic acid quantities very efficiently. Storage of samples in phenol–chloroform solution at +4 °C also gave good yields except for sediment with extremely high-carbon content. Ethanol and RNAlater® preservation decreased nucleic acid yields drastically at all temperatures. To study how sample preservation may affect the result of microbial community analysis, one type of sediment was selected for length heterogeneity-PCR analysis and PCR cloning of the 16S rRNA genes. Ethanol and RNAlater® preservation caused a slight bias towards certain microbial types in the community analyses shown by underrepresentation of Bacteroidetes, Betaproteobacteria and Gammaproteobacteria-affiliated peak sizes and overrepresentation of Actinobacteria, Chloroflexi and Alphaproteobacteria-affiliated peak sizes. Based on the results of this study, preservation in phenol–chloroform solution can be recommended as an alternative storage method when freezing is not possible such as during extended field sampling; however, ethanol and RNAlater® may cause serious problems when used as preservatives for environmental samples containing humic acids.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The study of molecular microbial ecology relies on the quantity and the quality of the nucleic acid extractions, both of which can be greatly affected by the methods used in the handling and preservation of the samples (e.g. Rochelle et al. 1994; Sekar et al. 2009) and in the extraction of the nucleic acids (e.g. Luna et al. 2006). During the last two decades, a variety of methods have been developed and studied for the extraction of nucleic acids from difficult materials, such as sediments and soils containing high amounts of humic acids (Carrigg et al. 2007; Griffiths et al. 2000; Hurt et al. 2001; Leff et al. 1995; Martin-Laurent et al. 2001; Miller et al. 1999; Niemi et al. 2001; Robe et al. 2003; Roose-Amsaleg et al. 2001; Sessitsch et al. 2002; Stach et al. 2001). Extraction of nucleic acids from samples containing humic acids is not a trivial task since co-eluted humic substances can inhibit enzymatic reactions, such as PCR, and coloured samples can interfere in hybridization experiments.
The main focus has been on the extraction process, and few studies have considered sample preservation before nucleic acid extraction. To our knowledge, only a few studies have addressed the effects of storage on soil samples (Lauber et al. 2010; Sessitsch et al. 2002; Wallenius et al. 2010) and only one study on sediment samples (Rochelle et al. 1994); however, none of the above studies analysed the effect of sample preservation solutions. Storage of DNA samples at −20 °C and RNA samples at −70 to −80 °C is usually considered to be the most reliable method of preservation and is therefore most widely used; however, in many cases (e.g. during extended field sampling) immediate freezing is often not possible. Furthermore, adequate preservation solution would protect labile messenger RNA molecules from degradation during the critical moments between sample melting and RNA extraction. Different storage solutions such as ethanol (Harry et al. 2000) and RNAlater® (Ambion) (Foti et al. 2008) have been employed for preservation of environmental samples; however, thorough investigations of the effects of freezing and storage solutions on nucleic acid quantities and qualities are lacking.
The objective of this study was to evaluate the effect of common sample preservation methods, including freezing and different storage solutions, on both the yield and quality of DNA and RNA acquired from sediment samples. Nucleic acid quantities were assessed from three types of sediments using a fluorometric method. Qualities were assessed by studying whether different storage conditions have effects on results that display community structure and community member abundances. This was done from one of the samples using length heterogeneity-PCR (LH-PCR) (Suzuki et al. 1998). LH-PCR was chosen because it has been shown to be a far more reproducible and technically less complex method than the more commonly used terminal restriction fragment length polymorphism while still having a very good resolution of the whole community structure of complex microbial systems (Mills et al. 2003). LH-PCR analysis is based on the natural length variation of the 16S rRNA gene. In this study, we used the first third of the gene (varying between the positions 465 to 565 in Escherichia coli), within which some phylogenetic groups have relatively conserved fragment sizes facilitating some predictive power for taxonomic analysis of the data (Tiirola et al. 2003). In addition, a clone library was created to link LH-PCR peak sizes to the taxonomic groups to aid the interpretation of the observed community changes in the LH-PCR profiles. The results revealed that selection of the sample preservation method can have a strong effect on the yield of nucleic acid extracts. Further, the PCR-based analyses of differently preserved samples reflected slightly different profiles of the bacterial community.
Materials and methods
Sample collection and preservation
Sediment samples representing three different sediment types (Table 1) were collected using a Kajak-type sediment corer. The upper two centimeters of two sediment cores were pooled from each site and brought to the laboratory in an airtight plastic bag. Samples were stored at +4 °C until the experiments were started within 1 day to 1 week of the sample collection. The sediment was slurried with a Vortex mixer and divided into 200-μl aliquots in 2-ml screw-cap tubes. Aliquots were stored (1) without preservatives, (2) in absolute ethanol (1 ml), (3) in RNAlater® solution (Ambion) (1.6 ml) or (4) in phenol–chloroform–isoamyl alcohol (PCIAA) (25:24:1) (0.4 ml). In cases 2–4 the sediment sample was mixed with the preservative by vortexing the sample briefly. Four temperature treatments were followed for each preservative for the Lake Jyväsjärvi sediment: (A) −20 °C for 24 h, (B) +4 °C for 1 month, (C) −20 °C for 1 month and (D) snap freezing in liquid nitrogen followed by storage at −80 °C for 1 month. As no particular differences were observed between nucleic acid preservation at −20 °C and −80 °C, the last treatment D was replaced by treatment E (+4 °C for 24 h) for the Baltic Sea and Lake Mekkojärvi sediments. Three replicate aliquots were stored per treatment pair together with three replicate aliquots of the fresh control samples, which were subjected to nucleic acid extraction immediately after application of the storage treatments. Because of the short temporary storage period after sample collection (see above), the fresh control samples may not represent the in situ situation. This, however, does not affect the outcome of this study because all the storage treatments including the extraction of nucleic acids of fresh control samples were started simultaneously for each sediment type.
Organic content of the sediment was determined as the ratio of the loss of mass after combustion at a high temperature (550 °C, 2 h) to the dry mass (determined by drying in 105 °C, 24 h) of the sediment.
Nucleic acid extraction
After storage, both ethanol and RNAlater® solutions were rapidly separated from samples by centrifugation (13,000 g, +4 °C, 5 min) and removal of the supernatant. PCIAA (25:24:1, 400 μl) was then added to those samples which had not been stored in that preservative. Thereafter, all the samples were subjected to identical nucleic acid extraction procedures utilizing a slightly modified version of the protocol of Griffiths et al. (2000) for simultaneous extraction of DNA and RNA. This started by the addition of 0.6 g of glass beads (0.1 mm) and homogenization in a FastPrep machine (FP120) (Bio 101) (30 s, speed 5.5). After homogenization, 400 μl of hexadecyltrimethylammonium–NaCl extraction buffer (5% CTAB, 0.85 M NaCl, 120 mM of potassium phosphate buffer) was added. After centrifugation (13,000 g, +4 °C, 5 min), the clear nucleic acid phase of the supernatant was carefully taken away and washed with chloroform–isoamyl alcohol (24:1) to remove phenol residues. Nucleic acids were precipitated using polyethylene glycol 6000 (PEG)–NaCl mixture (30% PEG, 1.6 M NaCl) at room temperature for 2 h. The precipitate was pelleted by centrifugation (13,000 g, 30 min), and the pellet was washed with ice-cold 70% ethanol. After washing, the pellet was dried and dissolved in 100 μl of diethylpyrocarbonate (DEPC)-treated water and stored at −20 °C. The quantities of extracted DNA and RNA were determined fluorometrically with a Qubit fluorometer (Invitrogen) using a Quant-iT DNA Assay Kit and a Quant-iT RNA Assay Kit (Invitrogen), respectively, according to the manufacturer’s protocols.
Analysis of the bacterial communities
Lake Jyväsjärvi sediment was selected for the microbial community analysis as it had a median carbon content and represented a sediment type common in boreal mesohumic lakes. LH-PCR was performed separately for both 16S ribosomal RNA and 16S rRNA genes. For the 16S rRNA-based LH-PCR analysis, total RNA was reverse-transcribed to cDNA using Omniscript reverse transcriptase (RT) enzyme (Qiagen) on Deoxyribonuclease I (Fermentas)-treated nucleic acid extracts. For DNase treatment, 1 μl of nucleic acid extract was incubated in a 5.0-μl mixture containing 1x DNase reaction buffer, 1.2 U of RNase inhibitor and 0.5 U of Deoxyribonuclease I at 37 °C for 30 min followed by an addition of 0.5 μl of 25 mM of EDTA and incubation at 65 °C for 10 min. In reverse transcription, 0.5 μl of DNase-treated nucleic acid extraction (0.2–1.2 ng of RNA) was incubated in a 2.0-μl mixture containing 1x Omniscript reaction buffer, 10 μM of random hexamers (Promega), 0.5 mM of dNTPs, 0.1 U of RT enzyme and 1 U of RNAse inhibitor at 37 °C for 60 min.
For LH-PCR analysis, PCR was performed for both DNA and cDNA using the following primers: F8 (5′-AGAGTTTGATCMTGGCTCAG-3′) (Weisburg et al. 1991) (1:4 IRD700-labelled) and PRUN518r (5′-ATTACCGCGGCTGCTGG-3′) (Muyzer et al. 1993) in a GeneAmp PCR system 9600 (Perkin Elmer). In the PCR reaction for DNA, 1 μl of nucleic acid extraction (2–20 ng of DNA) was used as a template in a 20-μl PCR mixture containing 0.2 mM of dNTPs, 0.3 μM of each primer, 1x Biotools reaction buffer, 1 mg ml−1 of bovine serum albumin (BSA) and 0.2 U of Biotools polymerase. In the PCR reaction for cDNA, 2 μl of RT product was used as a template in a 30-μl PCR mixture containing 0.2 mM of dNTPs, 0.3 μM of each primer, 1x Biotools reaction buffer, 1 mg ml−1 of BSA and 0.3 U of Biotools polymerase. The PCR procedure for DNA included an initial denaturation step at 94 °C for 10 min and 35 cycles of amplification (94 °C for 30 s, 50 °C for 30 s, 72 °C for 3 min). For cDNA, PCR was performed with an initial denaturation step at 95 °C for 5 min and 30 cycles of amplification (94 °C for 30 s, 50 °C for 30 s, 72 °C for 3 min). The effect of varying PCR-template concentrations was studied in a separate community profile analysis. The templates of samples with low yields of DNA were concentrated to the concentration of high-yield samples (2:1 to 20:1) and the templates of samples with high yields of DNA were diluted 1:10 before PCR, and these samples as well as the original samples were analysed by LH-PCR. Along with the PCR of actual samples, two types of negative controls were also amplified: one to test the purity of PCR reagents where DEPC-treated water instead of samples was used as a template, and another to test the success of DNase treatment in removing DNA completely where DNase-treated nucleic acid samples were used as a template.
Gel electrophoresis for LH-PCR was performed with an automated LI-COR 4200 sequencer (LI-COR Biotech, Lincoln, NE) using a 6% long-range denaturing polyacrylamide gel (FMC Bioproducts, Rockland, ME). A mixture of size standards of 470 bp, 527 bp and 553 bp (Tiirola et al. 2003) were run in separate lanes after every three or seven samples. Density profiles of samples were obtained and converted into table format using Quantity One software package (BioRad) after a rolling disc background subtraction.
A clone library analysis of 16S rRNA gene amplicons of fresh control samples was performed to couple LH-PCR peaks with the taxonomic information. PCR for this analysis was performed as explained above for 16S rRNA gene-based LH-PCR analysis, except that non-labelled primers were used. PCR products of replicate control samples were pooled for subsequent cloning. The 16S rRNA gene amplicons were cloned using EZCells (Qiagen) and TOPO TA Cloning Kit (Invitrogen) for sequencing according to manufacturer’s instructions. Clones were sequenced using a T7-plasmid primer, BigDye 3.1 kit and an ABI3700 capillary sequencer (Applied Biosystems). Sequences were edited using a Sequence Scanner 1.0 (Applied Biosystems) and the lengths of the resulting 91 partial 16S rRNA gene sequences were calculated using the program BioEdit. Taxonomic classification of the sequences was performed using the SeqMatch tool of the RDP database web site (http://rdp.cme.msu.edu/).
Nucleotide sequence accession numbers
The 16S rRNA gene sequences determined in this study were deposited in the EMBL database under accession numbers FN995003-FN995093.
Statistical analyses
Statistical analyses of the DNA and RNA concentrations of sediment nucleic acid extractions were performed using one-way analysis of variance (α = 0.05) with control and each storage solution/temperature treatment combination as factor levels. This was followed by pairwise comparisons of sample concentrations in each storage solution/temperature treatment combination to those of control samples by planned contrasts with Hochberg–Bonferroni-corrected (Hochberg 1988) α values in each partial test. ANOVA and pairwise analyses were performed using SPSS 17.0 (SPSS Inc., Chicago, IL). Analysis of the LH-PCR data from Lake Jyväsjärvi was performed in MATLAB® environment (MathWorks, Inc.). LH-PCR profiles were aligned using their cross-correlations (Kay 1993) and pairwise similarities were calculated using linear Pearson correlation. Using the correlation matrices generated, principal coordinate analysis (PCoA) was performed and was used as a graphical means to show relatedness among profiles. Correlation matrices were transformed into distance matrices using the equation \( d = \left( {{1} - r} \right)/{2} \), where d and r denote distance value and Pearson’s correlation coefficient, respectively. Distance matrices were also generated from the measured DNA and RNA quantities using Euclidean distance. To test the relationships between different distance matrices, Mantel’s test with 9,999 permutations was performed using PC-ORD 4.17 for Windows (MjM Software, Gleneden Beach, OR, USA).
Results
Nucleic acid yields after sample preservation treatments
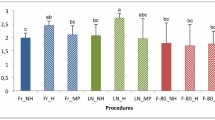
Considerable differences were observed in nucleic acid yields between the studied sediment types. The yields of fresh control samples from Lake Jyväsjärvi were 582 ± 121 μg and 364 ± 27 μg, from the Baltic Sea 326 ± 235 μg and 151 ± 98 μg, and from Lake Mekkojärvi 80 ± 24 μg and 29 ± 6 μg of DNA and RNA per gram of organic matter, respectively. The yields of extracted DNA and RNA varied considerably between different treatments (Fig. 1); this variation was mainly caused by the storage solution and less by the temperature or time. During nucleic acid extraction, it was noticed that sediment samples stored in ethanol and RNAlater® were more brownish, indicating that humic acids were more often present in these nucleic acid extracts. The storage effects in general seemed to be similar for samples from Lake Jyväsjärvi and the Baltic Sea (Fig. 1a, b); DNA and RNA yields of samples preserved in PCIAA and without storage solutions irrespective of the storage temperature were comparable to those of fresh control samples, but the yields from samples stored in ethanol and in RNAlater® were much lower. The only moderately good acquisitions from these samples were obtained from the Lake Jyväsjärvi sediment stored in ethanol at −80 °C, which did not statistically differ from the control samples (after stringent Bonferroni corrections of α values) (Fig. 1a). In contrast, the nucleic acid yields of samples from Lake Mekkojärvi stored in PCIAA and without storage solution at −20 °C did not differ from those of control samples, but negligible or moderately reduced yields of DNA and RNA were obtained from samples stored at +4 °C without storage solution or in PCIAA (Fig. 1c). As with samples from Lake Jyväsjärvi and the Baltic Sea, the nucleic acid yields of samples stored in ethanol and in RNAlater® were much lower than those of control samples, except for samples stored short-term in ethanol.

DNA and RNA yields of nucleic acid extracts from sediment samples from a Lake Jyväsjärvi, b a coastal station of the Northern Baltic Sea and c Lake Mekkojärvi, stored under different preservative/temperature combinations. Yields are shown as percentages (±SD) relative to those of freshly treated control samples. Statistically significant (p < 0.05) deviations from control values are marked with stars. N = 3 in each treatment
Effects on the outcome of bacterial community structure analysis
The 16S rRNA PCR products clearly visible in the agarose gel electrophoresis (approximately 10 ng μl−1) were obtained from all the DNA and reverse-transcribed cDNA templates, except for all RNAlater®-treated and some ethanol-treated DNA samples, which gave only weak products. Furthermore, after a year of post-experimental storage of nucleic acid extracts at −20 °C, PCR amplification was completely unsuccessful from the RNAlater®-treated DNA samples (results not shown). Negative PCR controls (sterile water instead of sample) and RT-PCR controls (DNase-treated sample) remained clean.
The microbial community of the Lake Jyväsjärvi sediment was very complex as shown by the LH-PCR electropherograms of DNA and RNA samples (Supplementary data, Figs. S1, S2). Peaks representing different amplicons were scattered making the normal procedure of peak-size recognition and single-peak area calculation difficult. Therefore, a correlation-based procedure was adopted for comparison of the profiles. Both the DNA (16S rRNA gene) and RNA (16S rRNA)-based LH-PCR profiles were separated in the PCoA plot by the storage solution used and by the temperature treatment (Fig. 2). Profiles of samples of most of the ethanol and RNAlater® treatments differed slightly from the control profiles (Figs. 2, S1, S2). In contrast, the profiles of samples stored in PCIAA and without preservative did not vary or varied only very slightly from the control profiles (Figs. 2, S1, S2). These samples also contained less variability between replicates within each storage solution/temperature treatment combination than profiles of samples stored in ethanol and in RNAlater® (Fig. 2). Both the 16S rRNA and 16S rRNA gene-based community structure were significantly related to RNA and DNA yields, respectively (Mantel’s test: RNA, r = 0.33, p < 0.05; DNA, r = 0.37, p < 0.05).

Graphical representation of principal coordinate analyses (first two axes shown) of the correlation-based comparison of a the 16S rRNA gene and b the 16S rRNA-based LH-PCR profiles for Lake Jyväsjärvi sediment samples stored under different preservative/temperature conditions
Sequencing the 16S rRNA gene clone library (91 clones) of Lake Jyväsjärvi showed the dominance of Proteobacteria (35%), Bacteroidetes (15%) and unclassified bacteria (16%). Linking the LH-PCR peaks with clone library sequences of corresponding size indicated that storage in ethanol or RNAlater® could lead to slight changes in the relative representation of major taxonomic phyla and classes, shown by clear underrepresentation of Bacteroidetes, Betaproteobacteria and Gammaproteobacteria-affiliated peaks and overrepresentation of Actinobacteria, Chloroflexi and Alphaproteobacteria-affiliated peaks (Fig. S1).
To test whether results of the LH-PCR analyses could be connected to differences in the template concentrations in the PCR reaction (Chandler et al. 1997), low-yield samples were amplified in concentrated template concentrations and high-yield samples in 1:10 dilutions followed by LH-PCR analysis, as explained above. PCoA results from this analysis did not, however, reveal any significant effect on the general pattern shown in Fig. 2 (Fig. S3). This notion was verified by significant results from Mantel’s test comparing the relationship between distance matrices of original versus template dilutions/concentrations (r = 0.69; p < 0.05). Specific comparison of LH-PCR profiles indicated that increasing the amount of DNA in PCR reactions of low-yield samples could lead to more underestimation of Bacteroidetes, Betaproteobacteria, and Gammaproteobacteria and overestimation of Alphaproteobacteria than with the original amount of DNA applied (Fig. S2).
Discussion
The highest nucleic acid yields and microbiological fingerprints most similar to those of non-preserved control profiles were obtained from samples which were stored without storage solutions or in PCIAA. The community analysis results of the samples stored frozen without storage solutions are in accordance with the previous studies on soil samples (Sessitsch et al. 2002; Lauber et al. 2010; Wallenius et al. 2010). Superior yields from frozen samples observed in some of the samples can stem from the addition of an extra freeze–thaw cycle which aids in effective lysis of microbial cells (Sessitsch et al. 2002). The positive effect of PCIAA is most likely due to denaturation of nucleases during storage and sample thawing. Very promising results were also obtained in the assessment of the ability of PCIAA to act as a field storage agent, i.e. to preserve nucleic acid quantities and qualities of samples stored unfrozen at +4 °C. This ability seems, however, to depend on the sediment type. Reduced nucleic acid yields of extremely organic sediment samples from Lake Mekkojärvi stored in PCIAA can be related to lower pH, anoxia or overloading the PCIAA inactivation capacity due to the extremely high content of organic matter consisting mostly of humic substances. The very promising but variable results of the preservation effects of PCIAA definitely call for more thorough testing with a wider variety of environmental samples.
Ethanol and RNAlater® were not useful solutions for the preservation of nucleic acid quantities of sediment samples. The use of these solutions also slightly affected the representation of the microbial fingerprints, which can have some consequences for microbial community studies. The poor performance of RNAlater® was very surprising since RNAlater® has been suggested for RNA preservation of many types of samples, such as tissues, cultured cells, yeasts and bacteria (Ambion, Manufacturer protocol), as well as for DNA and RNA preservation of microcrustaceans (Gorokhova 2005) and sediment samples (Foti et al. 2008). In our preliminary testing, RNAlater® seemed to work satisfactorily for bacterial and yeast monoculture samples, but nucleic acid yields decreased drastically when the samples contained humic soil material (Fig. S4). The manufacturer’s protocol for RNAlater® suggests a stabilization period of samples for 1 day at +4 °C in the solution before freezing, but we suggest that this could have led to even more drastic reductions in the nucleic acid yields of frozen sediment samples because the storage effects were already seen after 1 day of incubation. Harry et al. (2000) suggested ethanol for preservation of soil samples to ensure good bacterial DNA yields in cases where freezing is impossible, and in their study, bacterial DNA was preserved in ethanol without significant reductions for over a year. Contradictory results between our study and that of Harry et al. (2000) might arise from differences in the characteristics (e.g. texture and physicochemical properties) of the matrices studied.
According to the patent (US patent no. 6528641), RNAlater® contains a high concentration of ammonium sulphate, which is commonly used for protein precipitation. Upon application to the sample, RNAlater® and ethanol might therefore have caused precipitation and fixation of proteins and other organic compounds, such as humic substances, onto nucleic acids. This would lead to the drastic reductions in nucleic acid yields via the loss of most nucleic acid molecules either during the sample storage or during partitioning and separation of the organic phase in the nucleic acid extraction procedure. In addition, fixation and co-elution of organic compounds may have caused the observed slight biases in the community analyses. This might have taken place through partial inhibition of PCR by humic substances. It is possible that fixation of proteins and other organic matter onto nucleic acid strands lowered the “effective quantity” of DNA and RNA suitable for PCR amplification and reverse transcription by stopping the elongation of complementary strands, and this possibly led to the formation of PCR chimeras and other artifacts. On the other hand, the differences in community profiles between samples stored in ethanol/RNAlater® and other samples can also be explained by selective loss of nucleic acids during nucleic acid extraction. If some of the bacterial groups have more proteins in their cells than others, fixation of nucleic acids onto proteins would facilitate selective discarding of nucleic acids of more protein-rich bacteria. Another possible reason for the varying PCR results in low-yield DNA and RNA samples could be connected to differences in the (effective) template concentrations in the PCR amplification (Chandler et al. 1997). This hypothesis, however, was tested and rejected in additional LH-PCR analysis (dilution/concentration tests).
Although the 16S rRNA gene profiles of the samples of Lake Jyväsjärvi stored unpreserved at +4 °C were similar to control profiles, very slight deviations from the control profiles were observed for rRNA profiles. In addition, the drastic reduction of nucleic acids in the samples from Lake Mekkojärvi during storage at +4 °C for 1 month implies that microbes that adapted to low-redox conditions in the sediment were almost completely destroyed during storage, presumably due to oxygen contact. In previous studies, Rochelle et al. (1994) have also detected very rapid changes in the 16S rRNA gene-based denaturing gradient gel electrophoresis fingerprints of deep marine sediment samples and recommended immediate freezing of samples after sampling if molecular biological characterizations of bacterial diversity will be made. Thus, storage of sediment samples unpreserved and unfrozen cannot be recommended.
Our study shows that different sample preservation methods have varying effects on both the quantity and quality of nucleic acid extracts. These effects can have some consequences for subsequent molecular microbiological studies and therefore we encourage researchers working in the field to be careful in choosing the sample storage method. Freezing of samples in aliquots without storage solutions or in PCIAA seem to be the preferred storage methods when good DNA and RNA yields are required. These storage methods also perform best when reliable 16S rRNA gene and 16S rRNA-based pictures of the in situ potential and active microbial community are required. For conditions where freezing is not possible, such as during extended field sampling, PCIAA solution can offer a satisfactory alternative, except for extremely organic samples; however, we suggest that in cases of storage in PCIAA and if the samples cannot be frozen after the field sampling, the nucleic acid extraction should be conducted within 1 month from sampling using a PCIAA-aided protein precipitation and separation protocol. For environmental samples containing humic acids, storage in ethanol or RNAlater® should be avoided. In the future, it would be important to study whether the loss of nucleic acids happens during sample storage or by the preservative interference during the extraction process.
References
Carrigg C, Rice O, Kavanagh S, Collins G, O’Flaherty V (2007) DNA extraction method affects microbial community profiles from soils and sediment. Appl Microbiol Biotechnol 77:955–964
Chandler DP, Fredrickson JK, Brockman FJ (1997) Effect of PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries. Mol Ecol 6:475–482
Foti MJ, Sorokin DY, Zacharova EE, Pimenov NV, Kuenen JG, Muyzer G (2008) Bacterial diversity and activity along a salinity gradient in soda lakes of the Kulunda Steppe (Altai, Russia). Extremophiles 12:133–145
Gorokhova E (2005) Effects of preservation and storage of microcrustaceans in RNAlater on RNA and DNA degradation. Limnol Oceanogr Meth 3:143–148
Griffiths RI, Whiteley AS, O’Donnell AG, Bailey MJ (2000) Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol 66:5488–5491
Harry M, Gambier B, Garnier-Sillam E (2000) Soil conservation for DNA preservation for bacterial molecular studies. Eur J Soil Biol 36:51–55
Hochberg Y (1988) A sharper Bonferroni procedure for multiple tests of significance. Biometrika 75:800–802
Hurt RA, Qiu X, Wu L, Roh Y, Palumbo AV, Tiedje JM, Zhou J (2001) Simultaneous recovery of RNA and DNA from soils and sediments. Appl Environ Microbiol 67:4495–4503
Kay SM (1993) Fundamentals of statistical signal processing. Prentice Hall Signal Processing Series. Prentice Hall, New Jersey
Lauber CL, Zhou N, Gordon JI, Knight R, Fierer N (2010) Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol Lett 307:80–86
Leff LG, Dana JR, McArthur JV, Shimkets LJ (1995) Comparison of methods of DNA extraction from stream sediments. Appl Environ Microbiol 61:1141–1143
Luna GM, Dell’Anno A, Danovaro R (2006) DNA extraction procedure: a critical issue for bacterial diversity assessment in marine sediments. Environ Microbiol 8:308–320
Martin-Laurent F, Phillippot L, Hallet S, Chaussod R, Germon JC, Soulas G, Catroux G (2001) DNA extraction from soils: old bias for new microbial diversity analysis methods. Appl Environ Microbiol 67:2354–2359
Miller DN, Bryant JE, Madsen EL, Ghiorse WC (1999) Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl Environ Microbiol 65:4715–4724
Mills DK, Fitzgerald K, Litchfield CD, Gillevet PM (2003) A comparison of DNA profiling techniques for monitoring nutrient impact on microbial community composition during bioremediation of petroleum-contaminated soils. J Microbiol Meth 54:57–74
Muyzer G, Dewaal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturating gel electrophoresis of polymerase chain reaction amplified genes coding for 16S ribosomal RNA. Appl Environ Microbiol 59:695–700
Niemi RM, Heiskanen I, Wallenius K, Lindström K (2001) Extraction and purification of DNA in rhizosphere soil samples for PCR-DGGE analysis of bacterial consortia. J Microbiol Meth 45:155–165
Robe P, Nalin R, Capellano C, Vogel T, Simonet P (2003) Extraction of DNA from soil. Eur J Soil Biol 39:183–190
Rochelle PA, Cragg BA, Fry JC, Parkes RJ, Weightman AJ (1994) Effect of sample handling on estimation of bacterial diversity in marine sediments by 16S rRNA gene sequence analysis. FEMS Microbiol Ecol 15:215–225
Roose-Amsaleg CL, Garnier-Sillam E, Harry M (2001) Extraction and purification of microbial DNA from soil and sediment samples. Appl Soil Ecol 18:47–60
Sekar R, Kaczmarsky LT, Richardson LL (2009) Effects of freezing on PCR amplification of 16S rRNA genes from microbes associated with black band disease of corals. Appl Environ Microbiol 75:2581–2584
Sessitsch A, Gyamfi S, Stralis-Pavese N, Weilharter A, Pfeifer U (2002) RNA isolation from soil for bacterial community and functional analysis: evaluation of different extraction and soil conservation protocols. J Microbiol Meth 51:171–179
Stach JE, Bathe S, Clapp JP, Burns RG (2001) PCR-SSCP comparison of 16S rDNA sequence diversity in soil DNA obtained using different isolation and purification methods. FEMS Microbiol Ecol 36:139–151
Suzuki M, Rappe MS, Giovannoni SJ (1998) Kinetic bias in estimates of coastal picoplankton community structure obtained by measurements of small-subunit rRNA gene PCR amplicon length heterogeneity. Appl Environ Microbiol 64:4522–4529
Tiirola MA, Suvilampi JE, Kulomaa MS, Rintala JA (2003) Microbial diversity in a thermophilic aerobic biofilm process: analysis by length-heterogeneity-PCR (LH-PCR). Wat Res 37:2259–2268
Wallenius K, Rita H, Simpanen S, Mikkonen A, Niemi RM (2010) Sample storage for soil enzyme activity and bacterial community profiles. J Microbiol Meth 81:48–55
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Acknowledgements
This study was funded by grants from the Finnish Cultural Foundation and Academy of Finland (grants 120089 and 105860). We thank Olli Yli-Harja and Matti Nykter for introducing us to signal processing and Nina Vehniäinen for the valuable help in the laboratory. We also thank Roger Jones for his helpful comments on our manuscript. Furthermore, we would like to thank the two anonymous reviewers and the editor for their helpful and constructive comments.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 439 kb)
Rights and permissions
About this article
Cite this article
Rissanen, A.J., Kurhela, E., Aho, T. et al. Storage of environmental samples for guaranteeing nucleic acid yields for molecular microbiological studies. Appl Microbiol Biotechnol 88, 977–984 (2010). https://doi.org/10.1007/s00253-010-2838-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2838-2