
Abstract
Two laccase isoenzymes were purified and characterized from the basidiomycete Coriolopsis rigida during transformation of the water-soluble fraction of “alpeorujo” (WSFA), a solid residue derived from the olive oil production containing high levels of toxic compounds. Zymogram assays of laccases secreted by the fungus growing on WSFA and WSFA supplemented with glucose showed two bands with isoelectric points of 3.3 and 3.4. The kinetic studies of the two purified isoenzymes showed similar affinity on 2,6-dimethoxyphenol and 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulfonic acid), used as phenolic and non-phenolic model substrate, respectively. The molecular mass of both proteins was 66 kDa with 9% N-linked carbohydrate. Physico-chemical properties of the purified laccases from media containing WSFA were similar to those obtained from medium with glucose as the main carbon source. In-vitro studies performed with the purified laccases revealed a 42% phenol reduction of WSFA, as well as changes in the molecular mass distribution. These findings indicate that these laccases are involved in the process of transformation, via polymerization by the oxidation of phenolic compounds present in WSFA. A single laccase gene, containing an open reading frame of 1,488 bp, was obtained in PCR amplifications performed with cDNA extracted from mycelia grown on WSFA. The product of the gene shares 90% identity (95% similarity) with a laccase from Trametes trogii and 89% identity (95% similarity) with a laccase from Coriolopsis gallica. This is the first report on purification and molecular characterization of laccases directly involved in the transformation of olive oil residues.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The commercial olive oil production by a two-phase extraction system generates a semi-solid organic waste, named “alpeorujo”. This by-product is further dried and extracted with solvents to obtain an extra yield of oil and a dry olive-mill residue (Vlyssides et al. 1998), which has a great potential as fertilizer or amendment due to high content in both organic and inorganic nutrients (Sampedro et al. 2007). However, the main technical constraint to the biological upgrading of this residue is due to the presence of toxic compounds, including a water-soluble phenolic fraction, which has been shown to have phytotoxic (Sampedro et al. 2005; Bonanomi et al. 2006) and antimicrobial properties (Kotsou et al. 2004; Saparrat et al. 2010). Although many approaches have been explored to detoxify both solid and liquid residues from the olive oil industry, including thermal processes, electrolysis, ozonation, and evaporation, among others, bioremediation has gained increasing importance in recent years. The treatment with fungi such as Pleurotus spp., Chalara paradoxa, and Lentinula edodes has been shown to be useful in the decolorization and detoxification of olive oil mill wastewaters (Robles et al. 2000; Tsioulpas et al. 2002; Dias et al. 2004).
Coriolopsis rigida is a white-rot fungus which has been extensively studied for its ability to degrade lignin, polycyclic aromatic hydrocarbons, and dyes (Capelari and Zadrazil 1997; Gómez et al. 2006; Sánchez-López et al. 2008). In addition, this fungus is able to detoxify the water-soluble fraction of “alpeorujo” (WSFA), which contains most of the toxic phenolic compounds from the dry residue, and thus reducing its phyto- and microtoxicity through phenol polymerization (Sampedro et al. 2004; Aranda et al. 2007; Saparrat et al. 2010). Although the mechanisms of detoxification of olive oil by-products and effluents remained controversial (de la Rubia et al. 2008), it has been recently demonstrated that at least in the case of C. rigida, the action of its extracellular laccases on phenol content is involved in the reduction of the residue toxicity as revealed against Azospirillum brasiliense, a N2-fixing soil rhizobacterium which promotes plant growth (Saparrat et al. 2010).
Laccases (p-diphenol: oxygen oxidoreductase; EC 1.10.3.2) are phenol-oxidases which catalyze the oxidation of a great variety of phenolic compounds and aromatic amines using molecular oxygen as electron acceptor (Baldrian 2006), and due to their properties, they have been widely used for industrial applications, including pulp bleaching in paper industry, dye decolorization, and detoxification of environmental pollutants (Mayer and Staples 2002; Saparrat et al. 2008). The aim of this work was to understand and identify the mechanisms of transformation of WSFA by C. rigida. We have hence purified and characterized two laccase isoenzymes secreted by C. rigida grown on WSFA and demonstrated their direct effect in the transformation of “alpeorujo” via polymerization of soluble compounds present in the residue. In addition, a molecular characterization of the gene encoding both isoenzymes is provided.
Methods
Water-soluble fraction of “alpeorujo”
Olive-mill dry residue was collected from an olive oil manufacturer (Aceites Sierra Sur S.L., Granada, Spain), autoclaved, and stored at 4°C until use. WSFA was obtained by Soxhlet extraction of olive-mill dry residue with water by using a solid/liquid ratio of 1:8 (w v −1) for 16 h, and then filtered by vacuum through a 0.22-μm membrane and stored at −20°C until use.
C. rigida cultures
A dikaryon of C. rigida strain LPSC No. 232, from the culture collection of the La Plata Spegazzini Institute (= Spanish Type Culture Collection, CECT, nº 20449), was grown on peptone–yeast extract medium (Saparrat et al. 2002), where glucose was substituted by WSFA as the main carbon source at different concentrations (25%, 50%, and 100%) and pH adjusted to 5.0 with HCl. C. rigida was also grown on this peptone–yeast extract medium containing both 50% WSFA and glucose (either 5 or 10 g L−1) in order to increase the enzyme production for further purification studies, as well as on the basal medium, with glucose–peptone–yeast extract, supplemented with 150 μM CuSO4 (Saparrat et al. 2002), in order to compare the zymogram profiles of laccases secreted in this medium with those produced on cultures with WSFA. In all cases, a mycelial suspension (1%, v v −1) was inoculated in 1 L Erlenmeyer flasks containing 200 mL of each medium. The mycelial suspensions were obtained from homogenized pellets from 5-day-old shaken cultures, grown on the basal medium with glucose and 150 μM CuSO4 (Saparrat et al. 2002). Three replicate cultures of each media were grown at 150 rpm and 28 ± 1.5°C in the darkness for 57 days in a chamber with controlled humidity to minimize the evaporation.
Analytical procedures
Samples (2 mL) were taken periodically from the cultures, and the supernatants, separated from mycelia by centrifugation at 8,000×g for 10 min, were analyzed for different parameters. Phenol content was determined by the Folin-Ciocalteu reagent (Singleton and Rossi 1965), using tannic acid as the standard. Laccase activity was assayed by the oxidation of 5 mM 2,6-dimethoxyphenol (DMP) to coerulignone (ε 469 = 27,500 M−1 cm−1) in 100 mM sodium acetate buffer (pH 5.0). Peroxidase (EC 1.11.1.7) activity was assayed as laccase activity in the presence of 0.1 mM H2O2 (Saparrat and Guillén 2005). Manganese peroxidase (EC 1.11.1.13) activity was estimated by measuring the formation of Mn+3–tartrate complex (ε 238 = 6,500 M−1 cm−1) during the oxidation of 0.1 mM MnSO4 in 100 mM sodium tartrate buffer (pH 5.0) and 0.1 mM H2O2 (Saparrat and Guillén 2005). International enzymatic units (micromoles per min) were used. Proteins were determined according to Bradford method, using bovine albumin as standard and Bio-Rad kit assay.
Laccase purification
The purification process was carried out with extracellular crude extracts obtained from 57-day-old cultures growing on 50% WSFA and 10 g L−1 glucose, which showed the highest laccase activity levels. The liquid culture was separated from mycelia by centrifugation at 20,000×g for 20 min, and the supernatant was collected and concentrated by ultrafiltration (Pall Filtron, 3-kDa cutoff membrane) and then dialyzed against 10 mM sodium acetate (pH 5.0).
To design the purification process, the pH stability of laccase activity was studied in 100 mM borate–citrate–phosphate buffer (pH 3 to 7) at room temperature for 72 h. Crude enzyme preparation was incubated in the same buffer (pH 7.0) at 4°C and room temperature for 72 h to analyze thermal stability.
The crude enzyme preparation was applied to a HiTrap-Q (Amersham Biosciences) column equilibrated with 10 mM sodium acetate buffer pH 5.0 at a flow rate of 1.5 mL min-1. Retained proteins were eluted for 160 min with a NaCl gradient from 0 to 1 M NaCl and maintaining this salt concentration for 10 min to elute proteins and pigments retained. Fractions of 4.5 mL were collected in tubes containing 0.45 mL of 500 mM phosphate buffer (pH 7.0). Fractions with the laccase activity were pooled and concentrated (Filtron Microsep; 3-kDa cutoff), and samples of 1 mL were applied to a Superdex 200 (Amersham Pharmacia Biotech HR 16/60) column equilibrated with 50 mM phosphate buffer (pH 7.0) containing 150 mM NaCl at a flow rate of 0.4 mL min−1. The laccase peak was pooled, concentrated (Filtron Microsep, 3-kDa cutoff), and dialyzed against 10 mM sodium acetate (pH 5.0) by a PD-10 desalting column (Amersham Biosciences). Then, 1 mL samples were applied to a Mono-Q anion-exchange column (Pharmacia HR 5/5) equilibrated with the same buffer. Both laccase isoenzymes were eluted with a linear NaCl gradient from 0 to 250 mM for 42 min and from 250 mM to 1 M for 5 min at a flow rate of 0.8 mL min−1. Fractions of 2 mL were collected, and laccase peaks were pooled, concentrated, and stored with 10% (w v −1) glycerol at −4°C.
The laccase isoenzymes secreted in basal medium with glucose and copper were also purified but using the previously reported protocol (Saparrat et al. 2002).
Enzyme characterization
A preliminary characterization of laccase was performed by isoelectric focusing using the extracellular crude extracts from cultures grown on WSFA and WSFA supplemented with glucose. The isoelectric point (pI) was determined by zymograms performed on 5% polyacrylamide gels with a thickness of 1 mm by using a pH range from 3 to 10 (Bio-Rad Ampholine). The sample (20 μl) contained 5–10 mU of laccase. The anode and cathode solutions were 1 M phosphoric acid and 1 M sodium hydroxide, respectively. The pH gradient was measured on the gel by means of a contact electrode. Protein bands with laccase activity were detected by using 5 mM DMP in 100 mM sodium acetate buffer (pH 5.0) after the gels were washed for 10 min with the same buffer.
The molecular mass of isoenzymes was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and size-exclusion chromatography. SDS-PAGE was performed with 7.5% polyacrylamide gels by using low- and high-molecular-mass standards (Bio-Rad). Size exclusion chromatography was carried out on Superdex 200 column as described above. The column was calibrated with blue dextran (2,000 kDa), albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen A (25 kDa), and ribonuclease A (13.7 kDa). The N-glycan content of purified laccases was determined by the difference in molecular mass (estimated by SDS-PAGE) found before and after treatment of laccase isoenzymes with endo-N-acetylglucosaminidase.
The kinetic constant K m was calculated for LacI and LacII from cultures grown on WSFA and glucose, as well as for laccases obtained from the basal medium with glucose and copper, using DMP as model phenolic substrate and 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) as non-phenolic substrate.
WSFA treatment with laccase
Enzymatic treatment was carried out for 24 h at room temperature on 40 μL of WSFA diluted 1:10 with 500 mM sodium phosphate buffer at pH 7.0, since although optimum pH for both LacI and LacII was 2.0, highest enzyme stability in presence of WSFA after 24 h was achieved at pH 7.0 (95% of residual activity). The laccase used (1 U mL−1) was a mixture of LacI and LacII (obtained from WSFA media, after the size-exclusion chromatography). At the end of incubation, phenolic content on liquid fraction was analyzed. Changes in the molecular mass distribution of the WSFA fractions after enzymatic treatment were also analyzed as described by Jaouani et al. (2005) through size-exclusion chromatography on Sephadex G-100 column (Pharmacia, 1 × 48 cm) equilibrated with 50 mM NaOH and 25 mM LiCl2, at a flow rate of 0.4 mL min−1, according to Sarkanen et al. (1982). Blue dextran and syringic acid were used as low- and high-molecular mass standards, respectively.
Preparation of genomic DNA, total RNA, and reverse transcription
The mycelium was collected from 57-day-old cultures with 50% WSFA and 10 g L−1 glucose, corresponding to the maxima levels of laccase activity, separated from the culture liquid by centrifugation at 20,000×g for 20 min, washed with water treated with diethylpyrocarbonate to inactivate RNAases, and kept at −80°C for RNA extraction. Likewise, 5-day-old mycelium, grown in basal medium with glucose medium, was used for genomic DNA extraction as described below.
Total RNA was isolated using Ultraspec RNA (Biotecx Laboratories, Inc.) and treated with DNAse I using the “Deoxyribonuclease I, Amplification Grade” (Invitrogen, UK), to remove the chromosomal DNA contamination from the samples. First-strand cDNA was synthesized using the “GeneAmp Gold RNA PCR Reagent Kit” (Applied Biosystems, USA). This cDNA was used as template in polymerase chain reactions (PCR) described below. Genomic DNA was obtained using Genomix DNA extraction kit (Talent, Italy) according to manufacturer's instructions.
PCR amplification and cloning of the laccase gene fragment
The cDNA obtained as described above, was used as template for PCR amplification. Degenerate primers CR1 (5′ GCNATHGGNCCNAARGC 3′) designed on the basis of the N-terminal sequence of laccase from C. rigida purified from basal medium with glucose and copper (Saparrat et al. 2002), and PCu4 (5′ TGRAARTCDATRTGRCARTG 3′), based on the conserved sequence of the copper-binding region IV (HCHIDFH) in fungal laccases (Hong et al. 2007), were used to amplify laccase genes from C. rigida growing on WSFA. Amplified fragments were inserted into pGEM-T easy cloning vector (Promega). After transformation of the recombinant vectors into the Escherichia coli DH5α strain, the clones containing the inserted fragments were isolated and verified by DNA sequencing using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) and the automated ABI Prism 3730 DNA sequencer (Applied Biosystems). The sequence was compared by BLAST search in GenBank (Altschul et al. 1997).
Amplification of the flanking sequences of the laccase gene
In order to obtain the flanking sequence of the laccase gene corresponding to the C-terminus of the protein, primer CR2 (5′ AACGACGCCATAAGCCCAAAC 3′) was designed based on 3′ extreme from fungal laccase sequences with the highest homology (see “Results” section), and primer CR3 (5′ GCCATCGGGCCCAAGG 3′) was designed on the 5′ extreme from the laccase gene fragment obtained as described above.
PCR reaction was carried out with these two primers using 0.2 μg of either cDNA or DNA as template. Cycling parameters were 95°C for 3 min followed by 35 cycles of 94°C for 1 min, 52°C for 40 s, and 72°C for 1 min and the final extension at 72°C for 10 min.
In-gel trypsin digestion
Digestion of protein bands corresponding to LacI and LacII was performed with trypsin by using the DigestPro MS system (Intavis AG, Germany) following the standard protocol. The elution mixture was then dried down in a speed vacuum and resuspended in 4 μL 30% acetonitrile and 0.1% TFA.
MS and MS/MS analysis were performed with an Autoflex Smartbeam TOF/TOF (Bruker Daltonics, Germany) spectrometer equipped with a LIFT ion selector and a Reflectron ion reflector. Data collection was performed in fully automated, fuzzy-logic mode. Typically, 1,000 scans for peptide mass fingerprint and 200 scans for MS/MS were collected. Automated analysis of mass data was performed using flexAnalysis 3.0 software (Bruker-Daltonics, Germany). Internal calibration of MALDI-TOF mass spectra was performed using two trypsin autolysis ions with m/z 842.510 and m/z 2,211.105; for MALDI-MS/MS, calibrations were performed with fragment ion spectra obtained for the proton adducts of a peptide mixture covering the m/z 800–3,200 region.
MALDI-MS and MS/MS data were combined through the Biotools 3.0 software (Bruker-Daltonics, Germany) to search a nrNCBI database using MASCOT software (Matrix Science, UK). The following parameters were used for MASCOT searching: enzyme, trypsin; fixed modifications, carbamidomethyl (C); allow up to 1 missed cleavage; peptide tolerance 20 ppm, MS/MS tolerance 0.5 Da.
Results
Enzyme activity on WSFA
Activities of ligninolytic enzymes including laccase, peroxidase, and manganese peroxidase were assayed in the supernatant of cultures with different WSFA concentrations. Laccase was the single extracellular ligninolytic activity detected at the three concentrations of WSFA tested (25%, 50%, and 100%), reaching the highest activity levels after 21 days of incubation (Fig. 1a). Since no difference was found between 21-day-old cultures containing 100% or 50% WSFA, the effect of glucose (5 and 10 g L−1) was only evaluated on cultures with 50% WSFA (Fig. 1b). The highest activity levels were achieved on 57-day-old cultures in the presence of 10 g L−1 of glucose (1,466 mU mL−1).

a Laccase activity (mU mL−1) from C. rigida cultures grown in basal medium, without glucose, with 25% (filled circle), 50% (filled square), or 100% WSFA (filled triangle). b Laccase activity (mU mL−1) of C. rigida cultures grown in basal medium with 50% WSFA supplemented with 5 (filled circle) or 10 g L−1 of glucose (filled square)
A preliminary characterization of an extracellular laccase preparation from cultures of C. rigida grown on 50% WSFA, 50% WSFA and glucose (10 g L−1) as well as on glucose (10 g L−1), and 150 μM CuSO4 was carried out to design the purification process. Zymograms revealed that two laccase isoenzymes produced in the medium containing only WSFA were similar to those produced in WFSA supplemented with glucose (Fig. 2). The pI of laccase activity bands were 3.3 and 3.4, similar to those observed in a previous report in cultures of C. rigida grown in the basal medium with copper (Saparrat et al. 2002). Thus, the full characterization and purification of laccases was carried out using extracellular crude extracts produced in WSFA medium with 10 g L−1 glucose, since it provided slightly higher activity levels and less WFSA concentration, and also to reduce interferences during the purification process associated with waste components. Laccase activity was stable at both 4°C and 25°C, retaining 81% of activity after 72 h in 100 mM borate–citrate–phosphate buffer pH 7.0. A dramatic decrease in laccase activity was observed at acidic pH (88% of lost activity after 72 h incubation at pH 3.0).

Zymograms in isoelectrofocusing gels of C. rigida laccases secreted in different culture conditions: 50% WSFA (lane 1), 50% WSFA and glucose (lane 2), glucose and 150 μM CuSO4 (lane 3). pI values are given to the left
Purification of laccase
Table 1 summarizes the purification process of C. rigida laccases secreted in 50% WSFA medium supplemented with glucose. During the first chromatography step (Q-Cartridge), the laccase activity was separated from most of impurities, although color was still present due to the chromophores from WSFA which showed high absorbance at 280 nm (Fig. 3a). A major protein peak with laccase activity was obtained with the size-exclusion chromatography (Superdex 200) (Fig. 3b). The last chromatographic step involving a high-resolution ion-exchange column (Mono-Q) was necessary to resolve two laccase activity peaks: LacI and LacII (Fig. 3c). At the end of the process, LacI and LacII were purified 32.7- and 31.2-fold, respectively. The overall yield of the purification was 2.2% and 2.6% for LacI and LacII, respectively (Table 1).

Purification of laccases from C. rigida secreted in medium with WSFA and glucose: HiTrap-Q (a), Superdex 200 (b), and Mono-Q (c) chromatography. Absorbance at 280 nm (solid line), NaCl gradient (dashed line), and laccase activity (filled circle) are indicated
To compare the catalytic properties of two laccase isoenzymes produced by C. rigida on WSFA with those reported previously from a basal medium with copper (Saparrat et al. 2002), they were also purified to homogeneity, obtaining similar efficiency of purification to those reported in the mentioned study.
Enzyme characterization
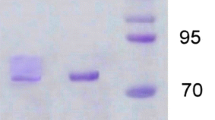
The molecular masses of both LacI and LacII obtained from cultures with WSFA were 60 kDa, estimated by size-exclusion chromatography, and 66 kDa determined by SDS-PAGE (Fig. 4). The difference between the molecular mass of each laccase and its corresponding deglycosylated form (after SDS-PAGE) revealed that both LacI and LacII are glycoproteins with 9% N-linked carbohydrate (Fig. 4). The same properties were obtained from the isoenzymes purified from basal medium with glucose and copper (Saparrat et al. 2002).

Electrophoresis gel of purified C. rigida laccases secreted in medium with WSFA and glucose: LacI (lane 1), deglycosylated LacI (lane 2), LacII (lane 3), deglycosylated LacII (lane 4), and molecular mass standard (lane 5)
The K m of both LacI and LacII isoenzymes purified from media supplemented with WSFA and glucose were respectively 14 μM for ABTS and 21.2 and 18.5 μM for DMP. In the case of laccases from the basal medium with glucose and copper, the K m values of both LacI and LacII were 12 and 11 μM for ABTS, and 15.3 and 17.5 μM for DMP, respectively (Saparrat et al. 2002).
WSFA transformation by laccase
The incubation of WSFA for 24 h with a laccase preparation (LacI + LacII), revealed a 42% free phenol reduction (from 368.9 to 214.4 μg mL−1), estimated by Folin method. The analysis of these samples in a size-exclusion column showed that the main peak, corresponding to low molecular mass, decreased, while the peak corresponding to higher molecular mass increased (Fig. 5).

Molecular mass distribution of phenols from WSFA estimated as profiles of absorbance at 280 nm by size-exclusion chromatography, before (solid line) and after treatment for 24 h with a C. rigida laccase preparation (dashed line)
Identification of C. rigida laccase gene expressed in presence of WSFA
In order to obtain partial genomic sequences encoding the laccase isoenzymes involved in transformation of WSFA, PCR assays were carried out following the protocols described above using cDNA as template, and a single 1.2 kb band was obtained. More than 10 clones containing the 1.2 kb insert were sequenced, resulting all of them in the same sequence. Thus, a single sequence was obtained, which showed the highest homology (87%) with the lcc1 laccase gene from Trametes sp. C 30 (GenBank accession number no. AF491759.1) and lac1 and lacA laccase genes from C. gallica (86% and 85% of homology) (GenBank accession number no. DQ431716.1 and AY875867.1, respectively). The sequence obtained, which included the N-terminus coding region, was used to design the primer CR3, corresponding to the exact sequence of the N-terminus, and the sequences showing highest homologies were then used to design the primer CR2 to obtain the flanking region corresponding to the C-terminus. A single band was obtained in PCR amplifications performed with either genomic DNA (∼2.1 kb) or cDNA (∼1.4 kb) and primers CR2 and CR3. Alignment of the genomic fragment and the corresponding cDNA laccase gene of C. rigida, named lcc1, showed a complete nucleotide match in their overlapping regions and indicated the presence of 11 exons. The gene was deposited in GenBank (accession number GQ377839).
The corresponding amino-acidic sequence of the cDNA showed the highest identity with lac1 from T. trogii (90% identity and 94% similarity) (accession number CAC13040) and an identity of 89% with C. gallica laccase (94% similarity) (accession number ABD93940). The deduced protein presents 496 amino acids with a molecular mass of 66 kDa. The four copper domains, which are highly conserved in laccases (Hong et al. 2007), as well as the pattern of His-X-His repeats involved in the coordination of the trinuclear type-2/type-3 copper site, were found. The expected residues that coordinate the type-1 copper site were also observed: His 394, His 455, and Cys 450, involved in the trigonal coordination of type copper and Phe 460. Four potential N-glycosylation sites were also identified at positions 51, 54, 207, and 433, following the pattern of Asn–X–Ser/Thr.
Protein identification
The results of the digested LacI and LacII bands, analyzed by MALDI-TOF/TOF, showed no differences between tryptic fingerprints of both LacI and LacII isoenzymes. In addition, the sequencing of seven different peptides from each isoenzyme, which matched with the sequence deduced form lcc1 cDNA, did not reveal any amino-acidic differences (data not shown).
Discussion
White-rot fungi have developed a non-specific oxidative system, including different oxidoreductases such as laccases and peroxidases, low molecular mass metabolites, and activated oxygen species, to degrade lignin and other aromatic pollutants causing environmental problems (Schoemaker et al. 1991). During transformation of WSFA, laccase was the sole ligninolytic enzyme activity detected. This enzyme is present in most of basidiomycetes, having a significant role especially when they are secreted as the sole ligninolytic enzyme (Eggert et al. 1997; Saparrat et al. 2002; Jaouani et al. 2005). The activity levels of laccases produced by C. rigida in the cultures supplemented with WSFA were lower than those detected in a previous report, where the fungus was grown in basal medium with glucose and copper (Saparrat et al. 2002). This could be due either to the presence of high levels of free phenols in WSFA which might affect enzyme secretion or reduce its activity (Tomati et al. 1991; Tsioulpas et al. 2002) or to the absence of copper which could act as enzyme inductor.
Analytical isoelectric focusing of extracellular crude extracts from cultures of C. rigida growing in the presence of WSFA showed two laccase bands (Fig. 2). The isoenzymes secreted in WSFA supplemented with glucose were similar, although the presence of glucose in the media delayed the secretion of laccase activity. This delay could be due to glucose repression, since CreA consensus sequences have been found in the non-coding regions of laccases from other basidiomycetes (Mansur et al. 1998; Galhaup et al. 2002). All the laccase isoenzymes detected showed similar acidic pI to those reported in other white-rot fungi, including Grammothele subargentea (pI 3.5), Cerrena unicolor (pI 3.6 and 3.7 for LacI and LacII, respectively), and Panus tigrinus (pI 3.15) (Michniewicz et al. 2006; Quaratino et al. 2007).
It is well known that culture media composition may affect the secretion of laccases (Leontievsky et al. 1997). For example, different laccase isoenzymes were induced in Pycnoporus sanguineus growing in the presence of high concentrations of starch and other sugars (Dantán-González et al. 2008). LacII from Pleurotus eryngii was induced by lignin and derived compounds added to basal medium with glucose (Muñoz et al. 1997). Botryosphaeria rhodina laccases were induced by soybean oil, Tween 80, or copper (Dekker et al. 2007). Trametes versicolor and P. tigrinus laccases were induced by xylidine (Minussi et al. 2007; Quaratino et al. 2008). Although in the case of C. rigida, a previous report had shown that two laccase isoenzymes were produced in the basal medium with glucose and copper (Saparrat et al. 2002), one of the main objectives of this study was to identify the enzymes induced under transformation of WSFA. Most of laccases are monomeric glycoproteins with a molecular mass between 50 and 80 kDa (Thurston 1994; Baldrian 2006). Two C. rigida laccases purified from the cultures with WSFA are monomeric proteins, as deduced by their similar molecular mass determined by SDS-PAGE and size-exclusion chromatography. A slight difference in the pI values permitted their separation in Mono-Q column. Although the same isoenzymes had been also obtained from basal medium with glucose and copper (Saparrat et al. 2002), the yield of purification process in the media with WSFA was lower (2.2% and 2.6% final yield for LacI and LacII, respectively), possibly due to the low-activity level obtained in this medium. The laccase isoenzymes from WSFA cultures showed similar affinity for both DMP and ABTS, which were used as phenolic and non-phenolic model compound respectively, when compared to the respective values of laccases obtained from the basal medium with glucose and copper. Both DMP and ABTS are typical laccase substrates, but DMP was especially chosen since it is the most abundant volatile phenol found after thermal treatment of olive stones and solvent-extracted olive pulp (Petrov et al. 2008). Similar K m values were also described for laccases from Pycnoporus coccineus, 27 and 36 μM for DMP and ABTS, respectively (Jaouani et al. 2005), and Daedalea quercina, 48 and 38 μM for DMP and ABTS, respectively (Baldrian 2004). Zymograms carried out by isoelectrofocusing with the extracellular crude extracts from WSFA cultures suggested that the isoenzymes secreted in these conditions were the same to those secreted in the basal medium with glucose and copper. This was also supported by the kinetic constant values as well as the data regarding molecular mass described above.
Since toxicity of the “alpeorujo” is mainly due to hydrosoluble compounds which include free phenols as well as glycosylated phenols, secoiridoids, and flavonoids (Capasso et al. 1995), we analyzed the effect upon WSFA of the extracellular laccases secreted by C. rigida on WSFA. Aranda et al. (2006) previously reported the reduction in the levels of the main phenolic compounds from “alpeorujo” such as tyrosol and hydroxytyrosol by C. rigida, suggesting the possible participation of its laccase activity in the process. The results obtained in this work indicate that laccases reduced levels of soluble aromatics including free phenols via polymerization of these compounds, as deduced from gel permeation profiles of treated WSFA, which showed a reduction of the peak area corresponding to low molecular mass and an increase of the higher molecular mass peak (Fig. 5). Similar results regarding molecular mass profiles have been reported by using free or immobilized crude laccase from P. coccineus or L. edodes using other effluent from the olive oil production (D'Annibale et al. 2000; Jaouani et al. 2005; Berrio et al. 2007). Since reduction of phytotoxicity and microtoxicity of “alpeorujo” is directly related to polymerization of phenols present in this residue (Aranda et al. 2007; Saparrat et al. 2010), we can conclude that the isozymes purified in this work are directly involved in the process of transformation of WSFA, and therefore they might be, at least at some extent, related to its detoxification.
Although two laccase isoenzymes have been isolated from C. rigida grown in presence of WSFA, a single laccase cDNA could be identified and cloned. The C. rigida laccase amino-acidic sequence deduced from this cDNA sequence showed highest homology with T. trogii laccase, and the deduced amino-acidic sequence showed all the domains observed for blue laccase from other basidiomycetes (Colao et al. 2003). The presence of two different laccase bands cannot be due to variants of the same enzyme with distinct glycosylation, since after deglycosylation of LacI and LacII, two bands were still visible in the gel (data not shown). Thus, the most plausible hypothesis is that each band would correspond to different allelic variants, since we are studying a dikaryotic fungal strain, although only one could be recovered by PCR. This would be supported by the physico-chemical properties of both LacI and LacII, the same amino-acidic N-terminus determined in both isoenzymes in the basal medium with glucose and copper (Saparrat et al. 2002) and the same peptide mass fingerprint obtained for both isoenzymes by MALDI-TOF MS. Although no differences were found in the sequenced peptides analyzed in both isoenzymes, small changes in their electrophoretic mobility could have been explained if the two protein bands corresponded to allelic variants, according to the results reported in other fungal species where a substitution in only three amino acids changed the isoelectric point (Ruiz-Dueñas et al. 1999).
In summary, we have identified two laccase isoenzymes from C. rigida directly involved in the transformation of “alpeorujo” via polymerization of phenols. The isoenzymes are identical to those produced by C. rigida in the basal medium with glucose and copper, and are encoded by the lcc1 gene, which is expressed during “alpeorujo” transformation. Since further research is needed to completely understand the mechanisms of detoxification, as well as to optimize the process to be integrated at the industrial level, we have recently initiated attempts to explore the transcriptional response to different environmental conditions of lcc1 gene.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Aranda E, Sampedro I, Ocampo JA, García-Romera I (2006) Phenolic removal of olive-mill dry residues by laccase activity of white-rot fungi and its impact on tomato plant growth. Int Biodeter Biodegr 58:176–179
Aranda E, García-Romera I, Ocampo JA, Carbone V, Mari A, Malorni A, Sannino F, De Martino A, Capasso R (2007) Chemical characterization and effects on Lepidium sativum of the native and bioremediated components of dry olive mill residue. Chemosphere 69:229–239
Baldrian P (2004) Purification and characterization of laccase from the white-rot fungus Daedalea quercina and decolorization of synthetic dyes by the enzyme. Appl Microbiol Biotechnol 63:560–563
Baldrian P (2006) Fungal laccases: occurrences and properties. FEMS Microbiol Rev 30:215–242
Berrio J, Plou FJ, Ballesteros A, Martínez AT, Martínez MJ (2007) Immobilization of Pycnoporus coccineus laccase on Eupergit C: stabilization and treatment of olive oil mill wastewaters. Biocatal Biotransform 25:130–134
Bonanomi G, Sicurezza MG, Caporaso S, Esposito A, Mazzoleni S (2006) Phytotoxicity dynamics of decaying plant materials. New Phytol 169:571–578
Capasso R, Evidente A, Schivo I, Orru G, Marciallis MA, Cristinzo G (1995) Antibacterial polyphenols from olive oil mill waste waters. J Appl Bacteriol 79:393–398
Capelari M, Zadrazil F (1997) Lignin degradation and in-vitro digestibility of wheat straw treated with Brazilian tropical species of white rot fungi. Folia Microbiol 42:481–487
Colao MC, Garzillo AM, Buonocore V, Schiesser A, Ruzzi M (2003) Primary structure and transcription analysis of a laccase-encoding gene from the basidiomycete Trametes trogii. Appl Microbiol Biotechnol 63:153–158
D'Annibale A, Stazi SR, Vinciguerra V, Sermanni GG (2000) Oxirane-immobilized Lentinula edodes laccase: stability and phenolics removal efficiency in olive mill wastewater. J Biotechnol 77:265–273
Dantán-González E, Vite-Vallejo O, Martínez-Anaya C, Méndez-Sánchez M, González MC, Palomares LA, Folch-Mallol J (2008) Production of two novel laccase isoforms by a thermotolerant strain of Pycnoporus sanguineus isolated from an oil-polluted tropical habitat. Int Microbiol 11:163–169
de la Rubia T, Lucas M, Martínez J (2008) Controversial role of fungal laccases in decreasing the antibacterial effect of olive mill waste-waters. Bioresource Technol 99:1018–1025
Dekker RFH, Barbosa AM, Giese EC, Godoy SDS, Covizzi LG (2007) Influence of nutrients on enhancing laccase production by Botryosphaeria rhodina MAMB-05. Int Microbiol 10:177–185
Dias AA, Bezerra RM, Pereira AN (2004) Activity and elution profile of laccase during biological decolorization and dephenolization of olive mill wastewater. Bioresource Technol 92:7–13
Eggert C, Temp U, Eriksson KEL (1997) Laccase is essential for lignin degradation by the white-rot fungus Pycnoporus cinnabarinus. FEBS Lett 407:89–92
Galhaup C, Wagner H, Hinterstoisser B, Haltrich D (2002) Increased production of laccase by the wood-degrading basidiomycete Trametes pubescens. Enzyme Microb Technol 30:529–536
Gómez J, Solar DR, Pazos M, Sanroman MA (2006) Applicability of Coriolopsis rigida for biodegradation of polycyclic aromatic hydrocarbons. Biotechnol Lett 28:1013–1017
Hong YZ, Zhou HM, Tu XM, Li JF, Xiao YZ (2007) Cloning of a laccase gene from a novel basidiomycete Trametes sp. 420 and its heterologous expression in Pichia pastoris. Curr Microbiol 54:260–265
Jaouani A, Guillén F, Penninckx MJ, Martínez AT, Martínez MJ (2005) Role of Pycnoporus coccineus laccase in the degradation of aromatic compounds in olive oil mill wastewater. Enzyme Microb Technol 36:478–486
Kotsou M, Mari L, Lasaridi K, Chatzipavlidis L, Balis C, Kyriacou A (2004) The effect of olive oil mill wastewater (OMW) on soil microbial communities and suppressiveness against Rhizoctonia solani. Appl Soil Ecol 26:113–121
Leontievsky AA, Vares T, Lankinen P, Shergill JK, Pozdnyakova NN, Myasoedova NM, Kalkkinen N, Golovleva LA, Cammack R, Thurston CF, Hatakka A (1997) Blue and yellow laccases of ligninolytic fungi. FEMS Microbiol Lett 156:9–14
Mansur M, Suárez T, González AE (1998) Differential gene expression in the laccase gene family from basidiomycete I-62 (CECT 20197). Appl Environ Microbiol 64:771–774
Mayer AM, Staples RC (2002) Laccase: new functions for an old enzyme. Phytochemistry 60:551–565
Michniewicz A, Ullrich R, Ledakowicz S, Hofrichter M (2006) The white-rot fungus Cerrena unicolor strain 137 produces two laccase isoforms with different physico-chemical and catalytic properties. Appl Microbiol Biot 69:682–688
Minussi RC, Miranda MA, Silva JA, Ferreira CV, Aoyama H, Marangoni S, Rotilio D, Pastore GM, Duran N (2007) Purification, characterization and application of laccase from Trametes versicolor for colour and phenolic removal of olive mill wastewater in the presence of 1-hydroxybenzotriazole. African J Biotechnol 6:1248–1254
Muñoz C, Guillén F, Martínez AT, Martínez MJ (1997) Induction and characterization of laccase in the ligninolytic fungus Pleurotus eryngii. Curr Microbiol 34:1–5
Petrov N, Budinova T, Razvigorova M, Parra J, Galiatsatou P (2008) Conversion of olive wastes to volatiles and carbon adsorbents. Biomass Bioenerg 32:1303–1310
Quaratino D, Federici F, Petruccioli M, Fenice M, D'Annibale A (2007) Production, purification and partial characterisation of a novel laccase from the white-rot fungus Panus tigrinus CBS 577.79. Antonie van Leeuwenhoek 91:57–69
Quaratino D, Ciaffi M, Federici E, D'Annibale A (2008) Response surface methodology study of laccase production in Panus tigrinus liquid cultures. Biochem Eng J 39:236–245
Robles A, Lucas R, Alvárez de Cienfuegos G, Gálvez A (2000) Phenol-oxidase (laccase) activity in strains of the hyphomycete Chalara paradoxa isolated from olive mill wastewater disposal ponds. Enzyme Microb Tech 26:484–490
Ruiz-Dueñas FJ, Martínez MJ, Martínez AT (1999) Molecular characterization of a novel peroxidase isolated from the ligninolytic fungus Pleurotus eryngii. Mol Microbiol 31:223–236
Sampedro I, Romero C, Ocampo JA, Brenes M, Garcia I (2004) Removal of monomeric phenols in dry mill olive residue by saprobic fungi. J Agr Food Chem 52:4487–4492
Sampedro I, D'Annibale A, Ocampo JA, Stazi SR, García-Romera I (2005) Bioconversion of olive-mill dry residue by Fusarium lateritium and subsequent impact on its phytotoxicity. Chemosphere 60:1393–1400
Sampedro I, Marinari S, D'Annibale A, Grego S, Ocampo JA, García-Romera I (2007) Organic matter evolution and partial detoxification in two-phase olive mill waste colonized by white-rot fungi. Int Biodeter Biodegr 60:116–125
Sánchez-López MI, Vanhulle SF, Mertens V, Guerra G, Figueroa SH, Decock C, Corbisier AM, Penninckx MJ (2008) Autochthonous white rot fungi from the tropical forest: potential of Cuban strains for dyes and textile industrial effluents decolourisation. Afr J Biotechnol 7:1983–1990
Saparrat MCN, Guillén F (2005) Ligninolytic ability and potential biotechnology applications of the South American fungus Pleurotus laciniatocrenatus. Folia Microbiol 50:155–160
Saparrat MCN, Guillén F, Arambarri AM, Martínez AT, Martínez MJ (2002) Induction, isolation, and characterization of two laccases from the white-rot basidiomycete Coriolopsis rigida. Appl Environ Microb 68:1534–1540
Saparrat MCN, Mocchiutti P, Liggieri CS, Aulicino MB, Caffini NO, Balatti PA, Martínez MJ (2008) Ligninolytic enzyme ability and potential biotechnology applications of the white-rot fungus Grammothele subargentea LPSC no. 436 strain. Process Biochem 43:368–375
Saparrat MCN, Jurado M, Díaz R, Romera IG, Martinez MJ (2010) Transformation of the water soluble fraction from “alpeorujo” by Coriolopsis rigida: the role of laccase in the process and its impact on Azospirillum brasiliense survival. Chemosphere 78:72–76
Sarkanen S, Teller DC, Abramowski E, Mccarthy JL (1982) Kraft lignin component conformation and associated complex configuration in aqueous alkaline-solution. Macromolecules 15:1098–1104
Schoemaker HE, Tuor U, Muheim A, Schmidt HWH, Leisola MSA (1991) White-rot degradation of lignin and xenobiotics. In: Betts WB (ed) Biodegradation: natural and synthetic materials. Springer-Verlag, London, pp 157–174
Singleton V, Rossi JA (1965) Colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagents. Am J Enol Viticult 16:144–158
Thurston CF (1994) The structure and function of fungal laccases. Microbiology 140:19–26
Tomati U, Galli E, Dilena G, Buffone R (1991) Induction of laccase in Pleurotus ostreatus mycelium grown in olive oil waste-waters. Agrochimica 35:275–279
Tsioulpas A, Dimou D, Iconomou D, Aggelis G (2002) Phenolic removal in olive oil mill wastewater by strains of Pleurotus spp. in respect to their phenol oxidase (laccase) activity. Bioresource Technol 84:251–257
Vlyssides AG, Loizidou M, Gimouhopoulos K, Zorpas A (1998) Olive oil processing wastes production and their characteristics in relation to olive oil extraction methods. Fresen Environ Bull 7:308–313
Acknowledgements
This work was supported by the Spanish projects S-0505/AMB0100, A/019945/08, AGL2008-00572/AGR and P06-AGR-01906. R. Díaz thanks the Spanish Ministry for mobility fellowship to the Centro de Investigaciones Biológicas (CSIC), Madrid, Spain.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Díaz, R., Saparrat, M.C.N., Jurado, M. et al. Biochemical and molecular characterization of Coriolopsis rigida laccases involved in transformation of the solid waste from olive oil production. Appl Microbiol Biotechnol 88, 133–142 (2010). https://doi.org/10.1007/s00253-010-2723-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2723-z