
Abstract
Two alkaline keratinases-I and II secreted by Bacillus halodurans PPKS-2 were purified and characterized. Both the keratinases were purified using ammonium sulfate, DEAE-Sephadex followed by Sephadex G-200 column chromatography. The purification was 21.5-fold and 11.17% yield for keratinase-I and 23.7-fold with yield 18.46 for keratinase-II and its molecular weights 30 and 66 kDa. Both purified enzymes were relatively stable over a broad pH range 7.0–13.0 and optimally active at pH 11.0 and 60–70 °C. Keratinase-II was found to be more stable at 70 °C for 3 h and retained 100% of its activity, whereas keratinase-I lost 10% activity. Keratinase-I had high keratin disulfide reductase activity with low keratinase activity whereas keratinase-II had high keratinase activity with low keratin disulfide reductase activity. Keratinase activities of both the enzymes were completely inhibited by PMSF at 1 mM, whereas keratin disulfide reductase activity of keratinase-I was not affected. Enzymes were active and stable in the presence of the surfactants, bleaching agents (20% H2O2), commercial detergents (1%), and SDS (20%). Both the enzymes were partially sequenced and found that keratinase-I and II had a homology with disulfide reductases and serine type of proteases, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Keratin, a structural protein found in feathers, wool, and hair is resistant to degradation by common proteases such as trypsin, pepsin, and papain (Riffel et al. 2007). The mechanical stability of keratin depends on the tight packing of the protein chains in α helix (α keratin) or β sheet (β keratin) structures. Feather waste represents a potential protein alternative to more expensive dietary ingredients for animal feed (Onifade et al. 1998; El-Refai et al. 2005). Worldwide commercial poultry processing units generate million tons of feather per year which are currently converted to feather meal through steam pressure and chemical treatment (Shih 1993). This process of chemical treatment makes keratin waste more digestible but it is expensive and destroys certain amino acids (Papdopoulos et al. 1986). Therefore the bioconversion of keratinous residues is attracting increasing biotechnological interest since it might represent an alternative way of waste management that could couple with the production of valuable products. Bacterial keratinases are of particular interest because of their action on insoluble keratin substrates and generally on a broad range of protein substrates (Brandelli 2008).
The purification and characterization studies of keratinases would be a main step towards a better understanding of the mechanism of keratin degradation, molecular identification of the genes involved, and the basis to develop further the production and industrial uses of these enzymes. Besides, keratinases are finding applications for the manufacturing of biodegradable plastics, nitrogenous fertilizers, glues, cosmetic, medicine, and foils (Onifade et al. 1998; Gupta and Ramnani 2006; Brandelli 2008). Alkaline keratinases produced by strains of Bacillus licheniformis like PWD-1 (Cheng et al. 1995; Lin et al. 1992), and NCINB (Evans et al. 2000) have been isolated and characterized.
While many keratinases act readily on soluble and native insoluble substrates, the mechanism of keratinolysis is complex and poorly understood. Although most purified keratinases cannot degrade keratin (Ignatova et al. 1999), some purified keratinases of B. licheniformis PWD-1 (Lin et al. 1992) and Kocuria rosea (Bernal et al. 2006) were able to hydrolyse feather keratin. The efficient hydrolysis of native keratinaceous substrates also requires the reduction of disulfide linkages; therefore keratinolysis needs the synergistic action of the protease with any of the various redox mechanisms such as reductases, sulfite production, or redox potential of cells (Yamamura et al. 2002; Ramnani et al. 2005). Most of the investigations focused on the action of keratinolytic proteases. However, the cleavage of the cystine bonds may have a significant influence on keratin degradation (Böckle and Müller 1997). A disulfide reductase-like protein was purified and characterized from Stenotrophomonas sp. and shown to degrade keratin with the cooperation of protease D-1 (Yamamura et al. 2002). However, there were no reports on the disulfide reductase-like proteins from alkaliphilic Bacillus sp. Alkaline keratinases from different microorganism vary in their properties and they have been purified and characterized (Gupta and Ramnani 2006). These keratinases have pH optima, in neutral to alkaline range, i.e., 7.0–9.0. However, there were few reports on keratinases possessed extreme alkaliphilic optima of pH 11.0 (Mitsuiki et al. 2004; Takami et al. 1999) and they were not compatible with bleach and detergents.
Earlier we have reported a keratin-degrading bacterium Bacillus sp. strain PPKS-2 with the aim of effective keratin degradation by solid state fermentation. We attempted to characterize this new strain and optimized the conditions for production of keratinase by free and immobilized cells (Prakash et al. 2009). In this study, we have purified and characterized two new enzymes, (a) alkaline keratinase and (b) keratin disulfide reductase responsible for keratin degradation from the new strain PPKS-2.
Materials and methods
Cultivation of bacteria: Isolation, characterization of the new strain Bacillus halodurans PPKS-2, and its cultivation for producing keratinolytic enzyme were described in the previous report (Prakash et al. 2009). This organism was deposited in National Center for Industrial Microorganisms, Pune, India with an accession number 5292.
Soluble keratin, casein, was purchased from Hi-media Mumbai, India. Azocoll was purchased from Sigma chemicals, USA. The soluble feather keratin was prepared from chicken feathers by the method of Khawar et al. (1997). Feathers were washed thoroughly with Triton ×100 (1% w/v), rinsed with distilled water and oven dried (70 °C for 15 h) and treated with 0.2 M mercaptoethanol in 8 M urea at pH 10.0–11.0 for 2 h at 45 °C. The feather fibers sonicated in a cell disruptor (Vibra-cell Ultrasonicator model 175 USA) continuous mode 30 s and incubated to digest at 45 °C for an additional 30 min, after this time most of the fibers were dissolved. The supernatant was subjected to centrifugation for 5 min at 12,000 g and dialyzed against distilled water with two changes for 12 h and stored at 10 °C for further used in keratinolytic activity assay. All other reagents used were of analytical grade.
Enzyme assay and substrate specificity
The caseinolytic activity of the enzyme was determined by using casein as a substrate (Vidyasagar et al. 2006). In brief, 2 ml of reaction mixture containing 1 ml of 1% casein (Hammerstein) and 0.5 ml of enzyme in the presence of 50 mM glycine–NaOH buffer of pH 11.0 and incubated for 15 min at 60 °C. One unit of caseinolytic activity is defined as the amount of enzyme that liberates 1 μg of tyrosine per minute under assay conditions. Keratinolytic activity was measured using 1% keratin or elastin-congo red as a substrate in place of casein as in the above. One unit of keratinolytic activity was expressed in keratin units, defined as an increase of 0.01 OD at 660 nm for keratin or 495 nm for elastin-congo red in 1 h. Collagenase activity was measured by incubating the enzyme with 4 mg of azocoll (Janssen et al. 1994) in 1 ml of 50 mM glycine–NaOH buffer (pH 11.0) at 60 °C for 1 h with constant agitation using a rotary shaker at 250 rpm. The samples were centrifuged at 12,000 rpm for 10 min and the absorbance of the supernatant was measured at 595 nm. One unit of collagenase activity was defined as the amount of the enzyme that gives rise to an increase in absorbance at 595 nm of 0.01 under the experimental conditions. Protein concentration was determined by the method of Lowry et al. (1951) using bovine serum albumin as standard.
Assay for disulfide bond-reducing activity: The disulfide bond-reducing activity was determined spectrophotometrically at 412 nm by measuring the yellow-colored sulfide formed upon reduction of 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB; Ellman 1959). The enzyme (50 μl) was incubated with 200 μl of 1% keratin or glutathione (oxidized) in presence or absence of 1 mM NADH and 50 mM glycine–NaOH buffer (pH 11.0) at 60 °C for 30 min. The reaction was terminated by addition of 200 μl 5% TCA, the reaction mixture was mixed with 10 μl of 100 mM DTNB. After 10 min, the reaction mixture was centrifuged at 1,000 g for 10 min, and the supernatant was used to measure disulfide bond-reducing activity at 412 nm. One unit of disulfide bond-reducing activity was defined as the amount of enzyme that catalyzes the formation of 1 μmole of sulfide per minute.
Enzyme purification
The cells were harvested by centrifugation at 6,700 × g for 10 min at 4 °C. To the cell free culture supernatant was added solid ammonium sulfate to achieve 60% saturation. The precipitate was centrifuged and resuspended in a minimal volume of 50 mM glycine–NaOH buffer pH 11.0. The clear supernatant was applied to DEAE-Sephadex column (2.5 × 15 cm) equilibrated with the above buffer. The column was washed with same buffer and eluted with step wise with the above buffer containing 0, 0.2, 0.5, and 1 M NaCl. The active fractions (5 ml each) were pooled and dialyzed against the same buffer without NaCl for 18 h. Gel filtration chromatography was performed using G-200 (1× 65 cm) column equilibrated with 50 mM glycine–NaOH buffer of pH 11.0. The above concentrated sample was loaded on to the column followed by elution with same buffer at a flow rate of 0.1 ml/min with fraction size of 1 ml. The eluted active fractions were pooled and concentrated with a VIVA Cell 250 (10 kDa) protein concentrator to 5 ml. These purified enzymes were used for further biochemical characterization.
Gel electrophoresis and molecular weight determination
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was done essentially as described by Laemmli (1970) with 10% acrylamide. For activity staining, ammonium sulfate fraction or purified keratinases were used. After the SDS-PAGE gel was soaked three times in 2.5% (v/v) Triton X-100 for 20 min at room temperature. Enzyme activity was visualized by incubating the gel for 2 h in a buffer (0.05 M glycine–NaOH buffer pH 11.0) containing 1 mM NADH or DTT and soluble keratin (5 mg/ml) at room temperature. The gel was stained with 0.5% of Coomassie brilliant blue R-250 and destained. Keratinase bands appeared as clear zones on blue back ground.
The effect of pH temperature and NaCl concentration
The effect of pH on the activity of purified keratinases was measured in the pH range 5.0–13.0, using the appropriate buffers at a concentration of 50 mM (5.0–6.0 sodium acetate, 7.0–8.0 Tris–HCl, and 9.0–13.0 glycine–NaOH) under the standard assay conditions. The effect of temperature on the purified enzymes was measured in the range of 20–100 °C. The effect of NaCl on the purified enzymes was measured in the range of 0–20% (w/v).
Determination of kinetic parameters
The V max and Km values of the purified enzymes were determined from a Lineweaver–Burk plot generated from increasing substrate concentrations of glutathione (1–10 mM) or keratin (1–10 mg).
Effect of surfactants, local detergents, and bleach on enzyme activity
The purified enzymes were preincubated for 30 min before with the following detergents like SDS (5%, 10%, and 20%) and Triton X-100 (1% and 5%), Tween 20 and 80 (1%), H2O2 (20%), and local detergents (1%). Keratinase and keratin disulfide reductase activities were determined as explained earlier.
Effect of protease inhibitors and chelators on enzyme activity
The effect of some known enzyme inhibitors, divalent metal chelators, and reducing agents (Table 5) was investigated by preincubating them with the purified enzymes for 30 min at room temperature and then measured the keratinase or keratin disulfide reductase activity under the standard experimental conditions.
N-terminal sequencing
The N-terminal amino acid sequence of keratinase-I and II was determined using a protein sequencer (PPSQ-21, Shimadzu). It was carried at the Department of Biosciences and Bioengineering, Indian Institute of Technology, Mumbai, India.
Results
Purification of keratinases
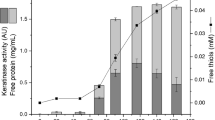
Strain PPKS2 was found to secrete two keratinases in to the extracellular medium as confirmed by activity staining in the gel (Fig. 1a).Total keratinase activity eluted as a single peak at 0.5 M NaCl on a DEAE-Sephadex chromatography. However, two forms of keratinase could be separated by size exclusion chromatography on Sephadex G-200. This procedure typically resulted in 23.7- and 21.5-fold purification with respective yield of 18.46 and 11.17 for keratinase-II and I (Table 1). Each preparation was tested for homogeneity by SDS-PAGE. Molecular masses of keratinase-I and II were estimated at 30 and 66 kDa, respectively, by comparison with molecular mass standard (Fig. 1b). Purified enzymes showed high activity in presence of 1 mM NADH or DTT in the gel as compared with the zymogrames in absence of reducing agents (Fig. 1c and d).

a Zymogram of the keratinases partially purified from Bacillus halodurans PPKS-2 in SDS-PAGE. b SDS-PAGE analysis of crude and purified keratinase-II and keratinase-I. Lane-1, molecular mass markers; phosphorylase (97 kDa), bovine serum albumin (66 kDa), ovalbumin (43 kDa), and carbonic anhydrase (29 kDa); Lane 2, crude extract; Lane 3, purified keratinase-II; Lane 4, DEAE-Sephadex column fraction; Lane 5, purified keratinase-I. c Zymogram of purified keratinases, lane 1, keratinase-I; lane 2, keratinase-II. d Zymogram of purified keratinaes in presence of reducing agents. Lane 1 and 2, in presence of 1 mM DTT; lane 3 and 4 in presence of.1 mM NADH
Substrate specificity
The substrate specificity of the keratinases was determined independently using casein, keratin, elastin-cong-red, azocasein, glutathione (oxidized), and azocoll. Both the enzymes showed very high activity toward casein, keratin, and elastin-congo. Azocoll was found to be a poor substrate (Table 2). On mixing these two enzymes, their activity increased by 49% and 83% compared with their individual activities, respectively. Kinetic studies of the two enzymes were performed under standard assay conditions using keratin or glutathione as the substrates. The Km and V max values were calculated from Lineweaver–Burk plot and are shown in (Table 3). Keratinase-I has high keratin disulfide reductase activity compared with keratinase activity whereas keratinase-II has low disulfide reductase activity compared with keratinolytic activity with respect to keratin as a substrate. However, glutathione was a good substrate for keratin disulfide reductase activity of keratinase-I, but keratinase-II has no action on glutathione (Table 3).
Effect of pH on the activities and stabilities of keratinase-I and II
The pH profiles of both the enzymes were determined using different buffers of varying pH values. Both the purified keratinases were found to be active between pH ranges of 9.0–13.0 with an optimum at pH 11.0 (Fig. 2). Enzyme stability was tested at pH values between 7.0 and 13.0 and the optimum was shown to be in the neutral to basic range (7.0–11.0) in which both keratinases retained 100% activity.

The effect of pH on the activity (solid line) and stability (dotted line) of purified keratinase-I (filled circle) and keratinase-II (circle) from Bacillus halodurans PPKS-2. The activity was determined according to the standard assay with different buffers. For stability studies, purified enzymes were incubated in various buffers (50 mM) from 7 to 13 at room temperature (28 °C) for 3 h. Each value represents the mean ± SE of three independent experiments
Effect of temperature and NaCl on keratinase-I and II activity and stability
The optimum temperature for keratinase-I and II were found to be 60–70 °C (Fig. 3). Both enzymes are stable at their optimum temperature for 3 h (Fig. 4). The enzymes were active over a broad range of NaCl (0–20%) by retaining 100% activity at 14% NaCl, 80% of activity at 16% NaCl under experimental conditions.

The effect of temperature on the activity of purified keratinase-I (filled circle) and II (circle). The activity was determined according to the standard assay temperatures from 20 °C to 100 °C at pH 11.0. Each value represents the mean ± SE of three independent experiments

Stability of the purified keratinase-I (filled circle) and II (circle) from Bacillus halodurans PPKS-2. Enzymes were incubated at 70 °C for different hours and residual activity was measured at 70 °C under standard assay conditions. Each value represents the mean ± SE of three independent experiments
Effect of protease inhibitors, metal ions, chelators, surfactants, local detergents, and bleach on enzyme activity
Keratinases are not affected by most metal ions tested. However, keratinase-I and II was inhibited by arsenite and cadmium ions. PMSF a potent inhibitor of serine proteases inhibited the keratinase activities of both the purified enzymes completely and instantaneously. However, keratinase-I retained its disulfide reductase activity in presence of 1 mM PMSF (Table 4). The EDTA and 1–10 phenanthroline did not inhibit the enzymes (Table 5). Reducing agents such as β-mercaptoethanol and had no significant effect but DTT enhance activity of purified keratinases by 50% to 65%. However the anionic detergent, SDS (20%) and nonionic Triton X-100 (1–5%) had no effect on the total activity, whereas commercial detergents had a positive effect except Nirma and Surf for both keratinases, enhanced the activity by 25% (Table 5).
Identification of keratinase by N-terminal sequence analysis
A total of ten residues of the N-terminal amino acid sequence of keratinase-I and 1I residues were determined successfully. The results showed that the keratinase-I had a significant homology to reductase enzymes whereas keratinase-II had significant homology with the serine proteases (Table 6).
Discussion
Keratinases from Bacillus sp. PPKS-2 were shown to possess high keratinolytic activity, especially at a high alkaline pH of 11.0 to 12.0. Strain PPKS-2 was shown to produce two forms of keratinases with molecular weights of 30 and 66 kDa for keratinase-I and II, respectively. The molecular weights of keratinase-I and II are obviously different from other microbial keratinases, such as, the keratinase of Fervidobacterium pennavorans 130 kDa (Friedrich and Antranikian 1996) and K. rosea 240 kDa (Bernal et al. 2006). The microbial keratinases that have the similar molecular weight as keratinase-II include the keratinase of Aspergillus oryzae, 60 kDa (Farag and Hassan 2004) and Chryseobacterium sp. kr6, 64 kDa (Riffel et al. 2007). However, the molecular weight of Ker-I was similar to the majority of keratinases, which vary from 20 to 50 kDa (Böckle et al. 1995; Cheng et al. 1995; Nilegaonkar et al. 2007). Chryseobacterium indologenes TKU014 was shown to produce three keratinolytic metalloproteases (Wang et al. 2007) with a molecular weight of 56, 40, and 40 kDa.
The pH of both enzymes was found to be a typical alkaline side displaying their activity for casein and keratin predominantly in the alkaline region of pH 7.0–12.0. There were only few similar reports from K. rosea keratinase, pH 10.0–11.0 (Bernal et al. 2006), Nocardiopsis sp. TOA-1 keratinase, pH 12.0 (Mitsuiki et al. 2004) and Bacillus sp. AH-101, pH 11.0–12.0 (Takami et al. 1999). Both keratinases of the strain PPKS-2 were active and stable over the temperature range of 60–70 °C. The optimum temperatures for both the keratinases are similar to keratinases isolated from F. pennavorans 70 °C (Friedrich and Antranikian 1996), Nocardiopsis sp TOA-1 60 °C (Mitsuiki et al. 2004).
Both the enzymes exhibited unusual stability in presence of SDS and H2O2, EDTA, and commercial detergents. In the detergent compatible enzymes in general are alkaline, thermostable in nature with high pH optima, because the pH of the laundry detergent is generally in the range of 9.0–11.0 and varying thermostability at laundry temperatures (50 °C to 60 °C; Gupta et al. 1999). Besides pH and temperature stability, bleach stability is important because bleach stable enzymes are not generally available except for a few reports (Bakhtiar et al. 2002; Gupta et al. 1999; Tuschiya et al. 1992). Very few published reports are available on the compatibility of the alkaline proteases with detergents (Gupta and Ramnani 2006). Thus the reported keratinases of Bacillus sp. PPKS-2 outstands with respect to pH, temperature, stability, detergent compatibility, and above all bleach stability for its future application in detergent formulation.
Purified keratinases were tested by using different protein substrates (casein, keratin, elastin, and azocoll in order to know the substrate specificity of the enzymes. Interestingly, both the keratinases degrade different protein substrates and exhibited a marked preference for casein, keratin, and elastin but no activity toward azocoll (Table 2). It is generally accepted that proteases to be applicable as depilatory enzymes, should have minimum action on collagen, the leather forming protein (Aravindhan et al. 2004), so as to break the collagen bundles without action on collagen fibers. Recently, we have demonstrated that the cell-free extract of B. halodurans PPKS-2 could be used to dehair the goat skin (Prakash et al. 2009).
The inhibition by PMSF may indicate that both the keratinases belong to the serine protease family with keratinolytic activity. Most of the keratinases described, particularly those from Bacillus sp. and Streptomyces sp. were also serine type proteases (Böckle et al. 1995; Lin et al. 1995). Keratinase-I was inhibited by arsenite and cadmium ions (Table 4) which are the known inhibitors of disulfide reductases of yeast (Ashahi et al. 1961) and pea seeds (Hatch and Turner 1960). Substrate specificity and homology of the N-terminal sequence with different reductases suggest that keratinase-I could be disulfide reductase, as it has high disulfide reductase activity with low Km for glutathione and keratin compared with keratinase activity (high Km for keratin). Further PMSF has no effect on disulfide reductase activity of keratinase-I (Table 4). These results indicate that keratinase-I has keratin disulfide reductase as well as protease activities. But it has low Km for disulfide bonds rather than peptide bonds. Keratinase-I was active in the reduction of oxidized glutathione in the presence or absence of electron donors. Further, its reductase activity was increased by fivefold (Table 2) in the presence of electron donor (1 mM NADH) and also evidenced by zymograms (Fig. 1d). The interesting feature of this enzyme was that it showing disulfide reductase activity in absence of any added reducing agents. At this time we have no clear explanation for the reduction disulfide bonds in the absence of electron donors. Keratin disulfide reductase-like protein (15 kDa) was purified and shown to reduce disulfide bonds of keratin in absence of any added electron donors (Yamamura et al. 2002). Takami et al. (1990) have characterized an alkaline protease from Bacillus sp. AH-101 which had high keratinase and elastage activity in the absence of any added reducing agents.
On the other hand, keratinase-II could be serine type of protease as it was also inhibited by PMSF and its N-terminal sequence was similar to most of the serine type of proteases. Further, comparison of N-terminal sequence of keratinases indicates that they share homology with subtilisin like serine proteases and serine proteases of Bacillus sp. (Table 6). Keratinase PWD1, which is the most well-studied keratinase to date (Lin et al. 1995), shows 99% homology with subtilisin Carlsberg, 65% with subtilisin E, 64% with B. pumilis dehairing protease, 62% Savinase, 30% with proteinase K, and 10% Nap A again indicating their relatedness to subtilisins. Further, it has high protease activity (low Km) and low disulfide reductase activity (high Km) towards keratin, but no action on glutathione–disulfide bonds (Table 3). This indicates that keratinase-II was more specific to disulfide bonds of keratin (with high Km) rather than glutathione. Earlier we have reported that the cell free extract or enzyme secreted by immobilized cells of strain PPKS-2 degraded feather keratin (Prakash et al. 2009). Now we interpret our results to support our earlier report that the disulfide bonds in keratin are attacked by keratinase-I first, yielding a partially cleaved protein which acts as a good substrate for keratinase-II which releases short peptides and amino acids. Reports on feather degradation propose that keratinase acts on disulfide bonds, responsible for rigid structure of keratin, thus making it easier for degradation. On the other hand, some reports suggest that the reduction of disulfide bonds by disulfide reductase (Böckle and Müller 1997; Yamamura et al. 2002) or the production of some reductants (Ramnani et al. 2005). Therefore it has been well documented that keratinolysis is often assisted by keratin disulfide reductase activity in extracellular medium.
Purified keratinases exhibited high stability towards nonionic detergents and reducing agents at a concentration as high as 10% is notable (Table 5). The keratinolytic protease from Streptomyces pactum was completely inhibited by 2% sodium thioglycolate (Böckle et al. 1995). For industrial purposes the resistance to reducing agents is highly preferable because the tight structure of keratin can weaken by disruption of the disulfide bonds. In addition, alkaline keratinases produced by strain PPKS-2 do not require Ca+2 for their stability could offer tremendous potential for detergent applications. In the process of detergent formulation where alkaline proteases are commonly added chelating agents are included to overcome the problem of hardness. In presence of such chelating agents, Ca+2 can be easily stripped off thus greatly affecting enzyme activity. Therefore enzymes which do not require Ca+2 for stability could offer tremendous potential for detergent application. These biochemical characteristics of the keratinases isolated, besides its extraordinary stability towards detergents bleaching agents, and divalent ions makes possible that these enzymes may be included in bating enzyme preparations for the leather industry and detergent industries.
The alkaline keratinases from B. halodurans PPKS-2 were significant for an industrial perspective because of their ability to function in broad pH and temperature ranges in addition to their tolerance and stability in the presence of bleaching and chelating agents. The enzymatic properties of the alkaline keratinases also suggest their use in detergent formulations, leather industry, and feather meal production. Since the strain grows on cheaper media can be exploited for enzyme production on an industrial scale.
References
Aravindhan R, Saravanabhavan S, Thanikaivelan P, Rao JR, Chandrasekaran B, Nair BU (2004) A bio-driven lime and pickle free tanning paves way for greener garment leather production. J Am Leather Chem Assoc 99:53–66
Ashahi RST, Bandurski LG, Wilson (1961) Yeast sulfate-reducing system. J Biol Chem 236:1830–1835
Bakhtiar S, Andersson MM, Gessesse A, Mattiasson B, Hatti-Kaul R (2002) Stability characteristics of a calcium-independent alkaline protease Nesterenkonia sp. Enzyme Microb Technol 32:525–531
Bernal C, Cairo J, Coello N (2006) Purification and characterization of a novel exocellular keratinase from Kocuria rosea. Enzyme Microbial Technol 38:49–54
Brandelli A (2008) Bacterial keratinases: useful enzymes for bio processing agro industrial waste and beyond. Food Bioproce Technol 1:105–116
Böckle B, Müller R (1997) Reduction of disulfide bonds by Streptomyces pactum during growth on chicken feathers. Appl Environ Microbiol 63:790–792
Böckle B, Galunsky B, Müller R (1995) Characterization of a keratinolytic serine proteinase from Streptomyces pactum DSM 40530. Appl Environ Microbiol 61:3705–3710
Cheng SW, Hu HM, Shen SW, Takagi H, Asano M, Tsai YC (1995) Production and characterization of keratinase of a feather degrading Bacillus licheniformis PWD-1. Biosci Biotechnol Biochem 59:2239–2243
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77
El-Refai HA, Naby MAA, Gaballa A, El-Araby MH, Fattah AAF (2005) Improvement of the newly isolated Bacillus pumilis FH9 keratinolytic activity. Process Biochem 40:2325–2332
Evans KL, Crowder J, Miller ES (2000) Subtilisins of Bacillus spp. Hydrolyse keratin and allow growth on feathers. Can J Microbiol 46:1004–1011
Farag AM, Hassan MA (2004) Purification, characterization and immobilization of a keratinase from Aspergillus orizae. Enzyme Microb Technol 34:85–93
Friedrich AB, Antranikian G (1996) Keratin Degradation by Fervidobacterium pennavorans, a novel thermophilic an aerobic species of the order thermotogales. Appl Environ Microbiol 62:2875–2882
Gupta R, Ramnani P (2006) Microbial keratinases and their applications: an overview. Appl Microbiol Biotechnol 70:21–33
Gupta R, Gupta K, Saxena RK, Khan S (1999) Bleach-stable, alkaline protease from Bacillus sp. Biotechnol Letts 21:135–138
Hatch MDJF, Turner D (1960) A protein disulphide reductase from pea seeds. Biochem J 76:556–562
Ignatova Z, Gousterova A, Spassov G, Nedkov P (1999) Isolation and partial characterization of extracellular keratinase from a wool degrading thermophilic actinomycete strain Thermoactinomyces candidus. Can J Microbiol 45:217–222
Janssen PH, Peck K, Morgan HW (1994) Effect of cultural conditions on the production of extra cellular proteinase by Thermmus sp. R141A. Appl Microbiol Biotechnol 43:400–406
Khawar SL, Watson K, Jones GL (1997) A comparative electrophortic analysis of mammalian hair and avian feather proteins. Int J Biochem Cell Biol 29:367–380
Laemmli UK (1970) Cleavage of structural proteins during assembly of head of bacteriophage T4. Nature 227:680–685
Lin X, Lee CG, Casale ES, Shih JCH (1992) Purification and characterization of a keratinase from a feather-degrading Bacillus licheniformis strain. Appl Environ Microbiol 58:3271–3275
Lin X, Kelemen DW, Miller ES, Shih JCH (1995) Nucleotide sequence and expression of ker A, the gene encoding keratinolytic protease of Bacillus licheniformis PWD-1. Appl Environ Microbiol 61:1469–1474
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Mitsuiki S, Ichikawa M, Oka T, Sakai M, Moriyama Y, Sameshma Y, Goto M, Furukawa K (2004) Molecular characterization of keratinolytic enzyme from an alkaliphilic Nocardiopsis sp. TOA-1. Enzyme Microb Technol 34:482–489
Nilegaonkar SS, Zambare VP, Kanekar PP, Dhakephalkar PK, Sarnaik SS (2007) Production and partial characterization of dehairing protease from Bacillus cereus MCM B-326. Biores Technol 98:1238–1245
Onifade AA, Al-Sane NA, Al-Musallam AA, Al-Zarban S (1998) A review: potentials for biotechnological applications of keratin degrading microorganisms and their enzymes for nutritional improvement of feathers and other keratins as livestock feed resources. Biores Technol 66:1–11
Ramnani P, Singh R, Gupta R (2005) Keratinolytic potential of Bacillus licheniformis RG 1: structural and biochemical mechanism of feather degradation. Can J Microbiol 51:191–196
Riffel A, Brandelli A, Bellato S, de M, Gustavo HMF, Eberlin MN, Tavares FCA (2007) Purification and characterization of a keratinolytic metalloprotease from Chryseobacterium sp. kr6. J Biotechnol 128:693–703
Papdopoulos MC, Boushy AR EI, Podbeen AE, Ketelaars EH (1986) Effect of processing time and moisture content on amino acid composition and nitrogen characteristics of feather meal. A Feed Sci Technol 14:279–290
Prakash P, Jayalakshmi SK, Sreeramulu K (2009) Production of keratinase by free and immobilized cells of Bacillus halodurans strain PPKS-2 partial characterization and its application in feather degradation and dehairing of the goat skin. Appl Biochem Biotechnol. doi:10.1007/s12010-009-8702-0
Shih JCH (1993) Recent development in poultry waste digestion and feather utilization a review. Poultry Sci 72:1617–1620
Takami H, Akiba T, Horikoshi K (1990) Characterization of an alkaline protease from Bacillus sp. no. AH-101. Appl Microbiol Biotechnol 3:519–523
Takami H, Nogi Y, Horikoshi K (1999) Reidentification of the keratinase-producing facultatively alkaliphilic Bacillus sp. AH-101 as Bacillus halodurans. Extremophiles 3:293–296
Tuschiya K, Nakamura Y, Sakashita H, Kimura T (1992) Purification and characterization of a thermostable alkaline protease from alkaliphilic Thermoactinomyces sp. HS682. Biosci Biotechnol Biochem 56:245–250
Wang SL, Hsu WT, Liang TW, Yen YH, Wang CL (2007) Purification and characterization of three novel keratinolytic metalloproteases produced by Chryseobacterium indlogenes TKU014 in a shrimp shell powder medium. Biores Technol 99:5679–5686
Yamamura S, Morita Y, Hasan Q, Yokoyama K, Tamiya E (2002) Keratin degradation: a cooperative action of two enzymes from Stenotrophomonas sp. Biochem Biophys Res Comm 294:1138–1143
Vidyasagar M, Prakash S, Jayalakshmi SK, Sreeramulu K (2006) Optimization of cultural conditions for the production of halothermophilic protease from halophilic bacterium Chromohalobacter sp. TVSP101.World. J Microbiol Biotechnol 23:655–662
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOC 32 kb)
Rights and permissions
About this article
Cite this article
Prakash, P., Jayalakshmi, S.K. & Sreeramulu, K. Purification and characterization of extreme alkaline, thermostable keratinase, and keratin disulfide reductase produced by Bacillus halodurans PPKS-2. Appl Microbiol Biotechnol 87, 625–633 (2010). https://doi.org/10.1007/s00253-010-2499-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2499-1