
Abstract
The bacterial population during malolactic fermentation of Tempranillo wine was studied using the polymerase chain reaction-denaturing gradient gel electrophoresis, a culture-independent method successfully used for identification and monitoring of bacterial population in different habitats included food fermentations. The results showed that Oenococcus oeni was the predominant species in the malolactic fermentation of Tempranillo wines, although the presence of Gluconobacter oxydans, Asaia siamensis, Serratia sp., and Enterobacter sp. was also observed. These results were partly coincidental with those obtained from a culture-dependent method, using a selective medium. Therefore, it may be concluded that for a more complete knowledge of the bacterial community present during malolactic fermentation of Tempranillo wine, an approach that combines a culture-independent method and a culture-dependent method would be advisable.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Malolactic fermentation (MLF), a process in which l-malate is converted into l-lactate and carbon dioxide, has been described as having a significant influence on wine quality (Henick-Kling 1993; Lonvaud-Funel 1999). Lactic acid bacteria (LAB) are responsible for this process, although other species of bacteria may also be present (Bae et al. 2006; Renouf et al. 2007).
Information regarding the composition and dynamics of microbial communities throughout the vinification process is always useful to control the process, which will contribute to improving wine quality. Both traditional and molecular methods have been used to study the microbial population dynamics during wine fermentation, mainly those of yeasts and LAB, which has allowed for a better understanding of the relations and interactions between the different species involved (Andorrà et al. 2008; Reguant and Bordons 2003; Renouf et al. 2007).
In recent years, randomly amplified polymorphic DNA-polymerase chain reaction (RAPD-PCR) has been frequently used in the genetic characterization of strains of bacteria participating in different food fermentations, including MLF (Coppola et al. 2006; Lechiancole et al. 2006; Rodas et al. 2005; Ruiz et al. 2010; Sánchez et al. 2004; Zapparoli et al. 2000). This culture-dependent method provides significant insight into specific isolates and microbial populations, but it is well known that only a small proportion of microorganisms are cultivable and, therefore, culture-dependent techniques often result in an incomplete representation of the true bacterial diversity present (Amann et al. 1995; Hugenholtz et al. 1998). Thus, recent microbial ecology studies of foods have employed novel culture-independent molecular approaches, such as those that use polymerase chain reaction amplification with different primers, in combination with denaturing gradient gel electrophoresis (DGGE) or temporal temperature gradient electrophoresis (Ampe et al. 2001; Ercolini 2004; Giannino et al. 2009; Meroth et al. 2003; Miambi et al. 2003). These methods allow for a rapid detection of individual species and offer a profile of the changes in community structure with time (Lopez et al. 2003; Pérez Pulido et al. 2005). They have revealed microbial constituents and microbial interactions not observed by previous plating analysis (Giraffa and Neviani 2001), although they also present some limitations (Prakitchaiwattana et al. 2004).
Some recent studies that use PCR-DGGE to examine the bacterial population during winemaking have been reported. Bae et al. (2006) used PCR-DGGE to examine bacteria growing in enrichment cultures from wine grapes cultivated in Australia and reported that the main malolactic bacterium, Oenococcus oeni, could not be found on grapes using this method, though its recovery could be obscured by overgrowth from other species. On the contrary, Renouf et al. (2006) reported that this technique made it possible to follow the evolution of the predominant species during laboratory microvinification and in several winemaking chateaux, although they proposed (Renouf et al. 2007) to use it in combination with population enumeration in selective media in order to monitor microbial changes at all stages of production since it does not provide quantitative data.
Spano et al. (2007) used PCR-DGGE to study bacterial populations in red wine and reported that it may be considered a reliable technique to monitor the bacterial starters extensively used in fermented beverages. On the other hand, Andorrà et al. (2008) reported that it is ideal for detecting species diversity in a mixed population with similar relative proportions, although the massive presence of a species did decrease the chances of detecting other minor species.
The aim of this research was to study the bacterial population during spontaneous MLF of Tempranillo wine produced in two vintages at cellars in Castilla-La Mancha (Spain), using PCR-DGGE, in order to complete and compare the results previously obtained from a culture-dependent method.
Materials and methods
Sampling
A total of 60 samples of Tempranillo wine were taken during the 2006 and 2007 vintages at five wineries (A–E) located in four provinces of the Castilla-La Mancha region (Spain). The wineries were selected because they had never used commercial starters for MLF. The winemaking process involves manual harvesting of grapes, followed by the vinification practices typical of this wine-producing area. Briefly, they consist of a controlled alcoholic fermentation at 25 ± 2°C in stainless steel tanks or jars, followed by spontaneous MLF.
Two batches (I and II) were sampled at each winery. The samples were aseptically collected at the end of alcoholic fermentation (stage 0) and at the middle and the end of MLF (stages 1 and 2, respectively). The criterion for defining the middle and the end of MLF was the content of l-malic acid and residual sugar (glucose + fructose) in wines, which were determined using the enzymatic tests purchased from Boehringer (Boehringer Mannheim, Mannheim, Germany). l-Malic acid content ranged between 1.27 and 2.31 g/L at stage 0 and between 0.00 and 0.34 g/L at stage 2. Residual sugar content ranged between 0.00 and 6.24 g/L at the end of alcoholic fermentation and reached values between 0.00 and 0.12 g/L at the end of MLF. The samples were kept refrigerated until analysis.
Bacterial strains and culture conditions
The reference strains Lactobacillus plantarum CECT 4645, Lactobacillus casei CECT 4045, Lactobacillus hilgardii CECT 4659, Leuconostoc mesenteroides CECT 394, O. oeni CECT 218, Gluconobacter oxydans CECT 4009, Serratia rubidaea CECT 868, and Enterobacter gergoviae CECT 857 from the Spanish Type Culture Collection (CECT) and Asaia siamensis JCM 10715T from the Japan Collection of Microorganisms (JCM) were used in this study.
Following CECT recommendations, the Lactobacillus and Leuconostoc species were grown in Man, Rogosa, and Sharpe medium (MRS) broth (Scharlab, Barcelona, Spain), O. oeni was grown in Leuconostoc oenos medium (MLO) broth (Claus et al. 1983; Scharlab), G. oxydans was grown in mannitol medium (Scharlab), and the Serratia and Enterobacter species were grown in trypticasein soy broth (Pronadisa, Madrid, Spain). A. siamensis was grown in AG medium composed of d-glucose (0.1%), glycerol (1.5%), peptone (0.5%), yeast extract (0.5%), malt extract (0.2%), and CaCO3 (0.7%) at pH 3.0. All cultures were incubated at 30°C.
PCR-DGGE analysis
DNA extraction
The DNA extraction kit (DNeasy Blood & Tissue) supplied by Izasa (Barcelona, Spain) was used. A total of 1.5 mL of wine were centrifuged (10 min, 5,000×g, 4°C), and the pellet was resuspended in 180 µL of a lysis buffer containing 24 g/L Tris, 7.4 g/L EDTA, 1.2% Triton, and 40 mg/mL lysozyme (Sigma-Aldrich, Madrid, Spain) and incubated at 37°C for 30 min. Subsequently, 25 µL of proteinase K and 200 µL of lysis buffer were added and, after mixing for 20 s, incubated at 56°C for 30 min. Two hundred microliters of ethanol (96–100%; Panreac, Barcelona, Spain) were added and mixed for 20 s. The mixture was transferred to a DNeasy Mini spin column placed in a 2-mL collection tube and centrifuged (1 min, 6,000×g, 25°C). The DNeasy Mini spin column was placed in a new 2-mL collection tube, and 500 µL of wash buffer (Izasa) was added. Following another centrifugation (3 min, 18,000×g, 25°C), the DNeasy Mini spin column was placed in a clean 1.5-mL microcentrifuge tube, and 50 µL of elution buffer (Izasa) was added. After centrifugation (1 min, 6,000×g, 25°C), the DNA was stored at −20°C.
PCR amplification of the microbial community 16S rRNA gene
The DGGE samples were prepared by two successive PCR amplifications (nested PCR), using the primer pairs described elsewhere (Ogier et al. 2002). First, a 700-bp fragment of the 16S rRNA gene that included the V3 region was amplified. The PCR was carried out in a total volume of 50 µL, containing 5 µL of 10× Taq reaction buffer, 2 mM MgCl2, each dNTP (Biotools) at a concentration of 200 µM, 1 µM primer W01 (5′-AGAGTTTGATC[AC]TGGCTC-3′), 1 µM primer W012 (5′-TACGCATTTCACC[GT]CTACA-3′), 2.0 U of Taq polymerase (Biotools), and 10 µL of template DNA. The amplification program was 96°C for 2 min; 30 cycles of 96°C for 1 min, 50°C for 30 s, and 72°C for 1 min; and, finally, 72°C for 2 min.
Secondly, the 700-bp fragment was used to amplify the V3 region as described by Ogier et al. (2002) using the primers HDA1-GC (5′-CGCCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACGGGGGGACTCCTACGGGAGGCAGCAGT-3′) and HDA2 (5′-GTATTACCGCGGCTGCTGGCA-3′). The PCR was carried out in a total volume of 50 µL, containing reaction buffer (10 mM Tris-HCl, 2 mM MgCl2, 50 mM KCl—final concentrations), each dNTP at a concentration of 200 µM, 1 µM of each primer, 2.0 U of Taq polymerase (Ecogen, Madrid, Spain), and 1 µL of the amplified 700-bp fragment. The amplification program was 94°C for 2 min; 30 cycles of 94°C for 1 min, 58°C for 30 s, and 72°C for 1 min; and, finally, 72°C for 7 min. The sizes and quantities of the PCR products were determined using 1.5% agarose gel electrophoresis.
PCR amplification of the rpoB gene
Primers rpoB1 (5′-ATTGACCACTTGGGTAACCGTCG-3′), rpoB10 (5′-ATCGATCACTTAGGCAATCGTCG-3′), and rpoB2 (5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGGGCACGATCACGGGTCAAACCACC-3), spanning the 336-bp region of the rpoB gene (Renouf et al. 2006), were also assayed in this study. Primer rpoB2 has a GC-rich clamp DNA sequence that improves DGGE separation (Scheffield et al. 1989). The reactions were carried out in a total volume of 50 µL, containing the same reaction buffer used above, each dNTP at a concentration of 200 µM, 1 µM of each primer, 2.0 U of Taq polymerase (Ecogen), and 10 µL of template DNA. The amplification program was 94°C for 5 min; 94°C for 1 min, 58°C for 1 min, and 72°C for 1 min for the first 15 cycles, followed by 15 cycles at 52°C as the annealing temperature; and, finally, 72°C for 10 min. The sizes of the PCR products were determined using 1.5% agarose gel electrophoresis.
Analysis of PCR products by DGGE
The amplification products obtained as described above were subjected to DGGE analysis using the DCode Universal Mutation Detection System (Bio-Rad Laboratories, Richmond, CA, USA) on 16 cm × 16 cm × 1 mm gels. Electrophoresis was performed at 60°C in 1× TAE buffer (40 mM Tris-acetate, 2 mM EDTA; pH 8.0) using 8% polyacrylamide gels containing 30–60% urea–formamide linear denaturing gradient (100% corresponded to 7 M urea and 40% (v/v) formamide) increasing in the direction of electrophoresis for 2 h at 180 V. Following electrophoresis, the gels were fixed for 5 min in fixation buffer (10% ethanol, 0.5% acetic acid), stained for 30 min in a SYBR Green solution (Sigma-Aldrich), and photographed with a KODAK DC290 Zoom Digital Camera.
DNA sequencing and data analysis
In order to identify the microbial populations, DGGE bands were excised from the gels immediately after staining. DNA from the selected bands was eluted in 50 µL of sterile water, overnight at 4°C, cloned into the pSTBlue-1 plasmid using the Blunt Cloning Kit (Novagen, USA), and sequenced with the ABI Prism 3700 DNA analyzer (Applied Biosystems).
Homology of the 16S rRNA gene was used to determine the closest known relative species; to this end, partial 16S rRNA gene sequences were compared to those available in the GenBank database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and the European Molecular Biology Library (EMBL; http://www.ebi.ac.uk/embl/Submission/webin.html).
Results
When the reference strains were analyzed in order to determine the discriminant capacity of primers rpoB (rpo B1 and rpo B2) and HDA (HDA1-GC and HDA2), a poorer discrimination was obtained with the rpoB primers since the species O. oeni CECT 218 and L. plantarum CECT 4645, both usually present in MLF, were not separated using these primers. The remanining reference species were adequately separated by both (rpo and HDA), and therefore, primers HDA were selected for the analysis of wine samples.
Samples taken at different stages during both vintages at all wineries showed profiles with a variable number of dominant bands, ranging from 3 to 5. As an example, Fig. 1 shows the electrophoretic profiles of samples taken at different stages during both vintages in winery A.

DGGE patterns of PCR products from the V3 region of the rRNA gene obtained from samples taken at different stages during both vintages in winery A. Band 1, O. oeni; band 2, nonidentified; band 3, G. oxydans; band 4, A. siamensis; band 5, Enterobacter sp.
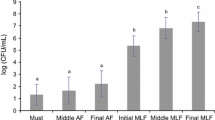
A higher number of bands were present in samples taken at the end of alcoholic fermentation (stage 0), decreasing throughout MLF. At the end of MLF (stage 2), samples from both vintages at all wineries exhibited three bands (bands 1, 3, and 5 in Fig. 1) corresponding to the dominant species.
In order to identify the bacterial species, PCR-DGGE bands were eluted from polyacrylamide gels and sequenced. All the sequences retrieved corresponded to portions of 16S rRNA genes. Comparison of sequences from the excised bands with those available in the GenBank and the EMBL databases revealed that all were ≥97% similar to 16S rRNA fragments already in the databases, except for band 2, which showed only a 90% homology with a fragment related to L. casei, and therefore, it could not be assigned to this species. Presence of common oenological bacteria, such as O. oeni and G. oxydans, and other less common ones, such as A. siamensis, Serratia sp., and Enterobacter sp., was displayed. In addition, one band present in some samples was identified as belonging to plant chloroplasts.
The EMBL accession numbers of the sequences and the percentages of homology obtained are listed in Table 1.
As regards the presence of these species at each winery (Table 2), it was observed that, while O. oeni, G. oxydans, A. siamensis, and Enterobacter sp. were present at all wineries, Serratia sp. was only present at wineries C, D, and E.
It is worth highlighting that G. oxydans and Enterobacter sp. were present in 57 and 59 samples, respectively, of a total of 60 samples, while O. oeni was absent in 14 of 20 samples taken at stage 0, appearing in all samples taken at later stages. Only the species O. oeni, G. oxydans, and Enterobacter sp. remained until the end of MLF (stage 2).
The band corresponding to O. oeni always showed the highest intensity, and it was much higher during the later stages of MLF. The intensities of the bands corresponding to G. oxydans and Enterobacter sp. were higher at the early stages of MLF, but always lower than that of the O. oeni band.
Discussion
In this study, the bacterial community from spontaneous MLF in Tempranillo wine was analyzed using a culture-independent method. PCR-DGGE has been used to study the structure and evolution of microbial communities from different habitats (Li et al. 2010; Liu et al. 2009; Lubbs et al. 2009; Petersson et al. 2009; Ponnusamy et al. 2008; Puglisi et al. 2009) included food fermentations (Endo and Okada 2005; Giannino et al. 2009; Meroth et al. 2003). All authors agreed that this method was well suited to study of microbial communities in each of the samples.
In contrast with the results obtained by Renouf et al. (2006), the discriminant capacity obtained with PCR-rpoB/DGGE was not sufficient for monitoring bacterial composition during winemaking because some of the species usually present during MLF in wines were not adequately separated. However, these authors advised that the detection of species present at low concentrations was difficult.
HDA primers have been successfully used by different authors for PCR-DGGE analysis (Giannino et al. 2009; Pérez Pulido et al. 2005). However, others (Lopez et al. 2003; Miambi et al. 2003) have reported, as occurred in our study, co-amplification of nonbacterial DNAs, including plant chloroplast, when using HDA and gc338f and 518r primers, to study bacterial population in food such as wine and fermented cassava. They affirm that it can be problematic since competition between bacterial and nontarget templates during PCR may mask lower bacterial populations. Therefore, in order to avoid this problem, it seems advisable to design more bacterium-specific PCR primers on future studies.
The band sequencing results from the profiles obtained by PCR-DGGE for the different samples were partially coincident with those reported by Renouf et al. (2006), who described the presence of O. oeni, L. casei, and G. oxydans during winemaking in different chateaux. In that study, in agreement with our results, O. oeni was the predominant species, which was represented by a highly intense band in most of the samples analyzed. Presence on the surface of grapes of wine-related acetic acid bacteria, such as members of Gluconobacter, has been reported and these bacteria may represent significant populations in musts (Lonvaud-Funel 1999).
On the contrary, the presence of species belonging to Serratia and Enterobacter has not been frequently reported in wines. They could have its origin in the grape surface, as mentioned for Gluconobacter, since both genera are widely distributed in nature, occurring in the soil, plant surfaces, and vegetables (Holt et al. 1994). Renouf et al. (2005) stated that these species play a significant role in the microbial consortium on grape surfaces, producing exopolysaccharides; years later, Renouf et al. (2007) described the presence of these species on grape surfaces from several vineyards in the Bordeaux area. On the other hand, Bae et al. (2006) identified A. siamensis in enrichment cultures from wine grapes cultivated in Australia.
Comparison of the PCR-DGGE results with those obtained from the identification of isolates obtained from MLOA plates inoculated with the same wine samples (Ruiz et al. 2010) revealed coincident results in terms of species diversity. However and as it was suspected, significant differences were observed in the species identified from both methods since only a selective medium for LABs was used in that study.
Thus, while L. plantarum, L. hilgardii, L. casei, and Leuconostoc mesenteroides were only identified by plating analysis, G. oxydans, A. siamensis, Serratia sp., and Enterobacter sp. were only detected by PCR-DGGE analysis. Only O. oeni was displayed by both methods. Results from other studies (Meroth et al. 2003; Miambi et al. 2003; Pérez Pulido et al. 2005) also revealed differences in the microbial composition of fermented foods depending on whether culture-dependent or culture-independent methods were used.
Enrichment cultures, such as those on MLO or MRS medium, favor the detection of a group of bacteria even at low concentrations but have the disadvantage of limiting the groups of bacteria that can be detected. In contrast, analysis of microbial populations by culture-independent methods allows for the identification of various groups of bacteria, although these must be present at higher concentrations. Renouf et al. (2007) reported that PCR-DGGE was only able to reveal the predominant species and that the detection of the numerous different species present at low concentrations was difficult using this technique. It could also be the reason why O. oeni was not detected at some samples taken at the beginning of MLF in this study.
Therefore, failure to detect certain species on PCR-DGGE gels does not necessarily mean that the species are absent, but only that they are less numerous than others. On this respect, Renouf et al. (2007) affirmed that depending on the environmental conditions, the best-adapted species constitute the overwhelming majority, and the population ratio of different species can exceed 1,000-fold, making detection of minor species difficult.
The limitations of PCR-DGGE analysis in ecological studies have been discussed (Prakitchaiwattana et al. 2004) and include different affinity of the primer DNA for template DNA in different species and competitive influences when template DNAs are present in different relative amounts. Miambi et al. (2003) also reported that the use of the16S rRNA gene may represent a limitation in the bacterial community analysis present at vegetable samples because interferences with plant chloroplasts are possible.
From the results obtained in this study, it may be concluded that, although PCR-DGGE analysis provides a broad picture of the different groups of bacteria present in malolactic fermentation in wine, it does not provide a complete picture either. The variations observed between the results obtained from culture-dependent and culture-independent methods suggest that a combined approach is needed to detect dominant and minor species and to better understand the bacterial ecosystem present in wine fermentation.
Moreover, it is worth highlighting that the use of a culture-dependent method with a high intraspecific discrimination capacity, such as RAPD-PCR, would make it possible to obtain additional information about the participating genotypes, which may be significant from a technological point of view.
This study has provided a more complete view of the composition of the bacterial community present during MLF of Tempranillo wine produced at cellars in Castilla-La Mancha. Our research has shown the presence of the species G. oxydans, A. siamensis, Serratia sp., and Enterobacter sp. in Tempranillo wine for the first time. Their significant presence in these wines suggests that it would be interesting to determine in future researches (1) their viability during MLF and (2) the influence on the process and on the organoleptic properties of the wines in case of survival (if they survive).
References
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Ampe F, Sirvent A, Zakhia N (2001) Dynamics of the microbial community responsible for traditional sour cassava starch fermentation studied by denaturing gradient gel electrophoresis and quantitative rRNA hybridization. Int J Food Microbiol 65:45–54
Andorrà I, Landi S, Mas A, Guillamón JM, Esteve-Zarzoso B (2008) Effect of oenological practices on microbiol populations using culture-independent techniques. Food Microbiol 25(7):849–856
Bae S, Fleet GH, Heard GM (2006) Lactic acid bacteria associated with wine grapes from several Australian vineyards. J Appl Microbiol 100(4):712–725
Claus D, Lack P, Neu P (1983) D.S.M. catalogue of strains. Deutsche Sammlung yon Mikroorganismen
Coppola S, Fusco V, Andolfi R, Aponte M, Blaiotta G, Ercolini D, Moschetti G (2006) Evaluation of microbial diversity during the manufacture of Fior di Latte di Agerola, a traditional raw milk pasta-filata cheese of the Naples area. J Dairy Res 29:1–9
Endo A, Okada S (2005) Monitoring the lactic acid bacterial diversity during shochu fermentation by PCR-Denaturing gradient gel electrophoresis. J Biosci Bioeng 99:216–221
Ercolini D (2004) PCR-DGGE fingerprinting: novel strategies for detection of microbes in food. J Microbiol Methods 56:297–314
Giannino ML, Marzotto M, Dellaglio F, Feligini M (2009) Study of microbial diversity in raw milk and fresh curd used for Fontina cheese production by culture-independent methods. Int J Food Microbiol 130:188–195
Giraffa G, Neviani E (2001) DNA-based, culture-independent strategies for evaluating microbial communities in food-associated ecosystems. Int J Food Microbiol 67:19–34
Henick-Kling T (1993) Malolactic fermentation. In: Fleet GH (ed) Wine microbiology and biotechnology. Harwood Academic, Switzerland, pp 289–326
Holt JH, Krieg NR, Sneath PHA, Staley JT, Williams ST (1994) Bergey’s manual of determinative bacteriology, 9th edn. Williams & Wilkins, Baltimore
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Lechiancole T, Blaiotta G, Messina D, Fusco V, Villani F, Salzano G (2006) Evaluation of intra-specific diversities in Oenococcus oeni through analysis of genomic and expressed DNA. Syst Appl Microbiol 29:375–381
Li P, Wang Y, Wang Y, Liu K, Tong L (2010) Bacterial community structure and diversity during establishment of an anaerobic bioreactor to treat swine wastewater. Water Sci Technol 61(1):243–252
Liu FH, Lin GH, Gao G, Qin BQ, Zhang JS, Zhao GP, Zhou ZH, Shen JH (2009) Bacterial and archaeal assemblages in sediments of a large shallow freshwater lake, Lake Taihu, as revealed by denaturing gradient gel electrophoresis. J Appl Microbiol 106(3):1022–1032
Lonvaud-Funel A (1999) Lactic acid bacteria in the quality improvement and depreciation of wine. Antonie Van Leeuwenhoek 76:317–331
Lopez I, Ruiz-Larrea F, Cocolin L, Orr E, Phister T, Marshall M, VanderGheynst J, Mills DA (2003) Design and evaluation of PCR primers for analysis of bacterial populations in wine by denaturing gradient gel electrophoresis. Appl Environ Microbiol 69:6801–6807
Lubbs DC, Vester BM, Fastinger ND, Swanson KS (2009) Dietary protein concentration affects intestinal microbiota of adult cats: a study using DGGE and qPCR to evaluate differences in microbial populations in the feline gastrointestinal tract. J Anim Physiol Anim Nutr 93(1):113–121
Meroth CB, Walter J, Hertel C, Brandt MJ, Hammes WP (2003) Monitoring the bacterial population dynamics in sourdough fermentation processes by using PCR-denaturing gradient gel electrophoresis. Appl Environ Microbiol 69:475–482
Miambi E, Guyot JP, Ampe F (2003) Identification, isolation and quantification of representative bacteria from fermented cassava dough using an integrated approach of culture-dependent and culture-independent methods. Int J Food Microbiol 82:111–120
Ogier JC, Son O, Gruss A, Tailliez P, Delacroix-Buchet A (2002) Identification of the bacterial microflora in dairy products by temporal temperature gradient gel electrophoresis. Appl Environ Microbiol 68:3691–3701
Pérez Pulido R, Ben Omar N, Abriouel H, Lucas R, Martínez M, Gálvez A (2005) Microbiological study of lactic acid fermentation of caper berries by molecular and culture-dependent methods. Appl Environ Microbiol 71:7872–7879
Petersson A, Domig KJ, Nagel P, Zollitsch W, Hagmüller W, Kneifeld W (2009) Denaturing gradient gel electrophoresis (DGGE)-based monitoring of intestinal lactobacilli and bifidobacteria of pigs during a feeding trial. Arch Anim Nutr 63(2):112–126
Ponnusamy L, Xu N, Stav G, Wesson DM, Schal C, Apperson CS (2008) Diversity of bacterial communities in container habitats of mosquitoes. Microb Ecol 56(4):593–603
Prakitchaiwattana CJ, Fleet GH, Heard GM (2004) Application and evaluation of denaturing gradient gel electrophoresis to analyse the yeast ecology of wine grapes. FEMS Yeast Res 4:865–877
Puglisi E, Fragoulis G, Ricciuti P, Cappa F, Spaccini R, Piccolo A, Trevisan M, Crecchio C (2009) Effects of a humic acid and its size-fractions on the bacterial community of soil rhizosphere under maize (Zea mays L.). Chemosphere 77(6):829–837
Reguant C, Bordons A (2003) Typification of Oenococcus oeni strains by multiplex RAPD-PCR and study of population dynamics during malolactic fermentation. J Appl Microbiol 95(2):344–353
Renouf V, Claisse O, Lonvaud-Funel A (2005) Numeration, identification and understanding of microbial biofilm on grape berry surface. Aust J Grape Wine Res 11:316–327
Renouf V, Claisse O, Miot-Sertier C, Lonvaud-Funel A (2006) Lactic acid bacteria evolution during winemaking: use of rpoB gene as a target for PCR-DGGE analysis. Food Microbiol 23:136–145
Renouf V, Claisse O, Lonvaud-Funel A (2007) Inventory and monitoring of wine microbial consortia. Appl Microbiol Biotechnol 75:149–164
Rodas AM, Ferrer S, Pardo I (2005) Polyphasic study of wine Lactobacillus strains: taxonomic implications. Int J Syst Evol Microbiol 55:197–207
Ruiz P, Izquierdo PM, Seseña S, MLl P (2010) Analysis of lactic acid bacteria populations during spontaneous malolactic fermentation of Tempranillo wines at five wineries during two consecutive vintages. Food Control 21:70–75
Sánchez I, Seseña S, MLl P (2004) Polyphasic study of the genetic diversity of lactobacilli associated with “Almagro” eggplants spontaneous fermentation based on combined numerical analysis of randomly amplified polymorphic DNA and pulsed field gel electrophoresis patterns. J Appl Microbiol 97:446–458
Scheffield VC, Cox DR, Lerman LS, Myers RM (1989) Attachment of a 40-base pair G + C rich sequence (GC-clamp) to genomic DNA fragments by the polymerase chain reaction results in improved detection of single-base changes. Proc Natl Acad Sci 86:232–235
Spano G, Lonvaud-Funel A, Claisse O, Massa S (2007) In vivo PCR-DGGE analysis of Lactobacillus plantarum and Oenococcus oeni populations in red wine. Curr Microbiol 54:9–13
Zapparoli G, Reguant C, Bordons A, Torriani S, Dellaglio F (2000) Genomic DNA fingerprinting of Oenococcus oeni strains by pulsed-field gel electrophoresis and randomly amplified polymorphic DNA-PCR. Curr Microbiol 40:351–355
Acknowledgments
The authors wish to thank the Ministry of Education and Science (INIA-MEC) for its financial support (project RM 2006-00011-C02-02). P. Ruiz is supported by a grant from the Council of Communities of Castilla-La Mancha (JCCM).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ruiz, P., Seseña, S., Izquierdo, P.M. et al. Bacterial biodiversity and dynamics during malolactic fermentation of Tempranillo wines as determined by a culture-independent method (PCR-DGGE). Appl Microbiol Biotechnol 86, 1555–1562 (2010). https://doi.org/10.1007/s00253-010-2492-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2492-8