
Abstract
A recombinant polyester-degrading hydrolase from Thermobifida sp. BCC23166 targeting on aliphatic-aromatic copolyester (rTfH) was produced in Streptomyces rimosus R7. rTfH was expressed by induction with thiostrepton as a C-terminal His6 fusion from the native gene sequence under the control of tipA promoter and purified from the culture supernatant to high homogeneity by a single step affinity purification on Ni-Sepharose matrix. The enzyme worked optimally at 50–55°C and showed esterase activity on C3-C16 p-nitrophenyl alkanoates with a specific activity of 76.5 U/mg on p-nitrophenyl palmitate. Study of rTfH catalysis on surface degradation of polyester films using surface plasmon resonance analysis revealed that the degradation rates were in the order of poly-ε-caprolactone > Ecoflex® > polyhydroxybutyrate. Efficient hydrolysis of Ecoflex® by rTfH was observed in mild alkaline conditions, with the highest activity at pH 8.0 and ionic strength at 250 mM sodium chloride, with the maximal specific activity of 0.79 mg−1min−1mg−1 protein. Under the optimal conditions, rTfH showed a remarkable 110-time higher specific activity on Ecoflex® in comparison to a lipase from Thermomyces lanuginosus, while less difference in degradation efficiency of the two enzymes was observed on the aliphatic polyesters, suggesting greater specificities of rTfH to the aliphatic-aromatic copolyester. This study demonstrated the use of streptomycetes as an alternative expression system for production of the multi-polyester-degrading enzyme of actinomycete origin and provided insights on its catalytic properties on surface degradation contributing to further biotechnological application of this enzyme.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biodegradable plastics are promising alternative environmentally friendly materials which play increasing roles in a wide range of conventional and specialized plastic utilization. Continual development in this field has resulted in a diverse range of biodegradable plastics with properties comparable to those from petroleum origins. Polyester is one of the largest groups of biodegradable plastic with diversified physical and chemical properties, which can be either produced from biological or petrochemical origins (Luengo et al. 2003). Aliphatic polyesters, e.g., polyhydroxylakanoates (PHAs), poly-ε-caprolactone (PCL), and polybutylene succinate, are among the most extensively studied commercially promising aliphatic polyesters. These polyesters are completely degradable by microbial processes (Tokiwa and Calabia 2004). However, aliphatic polyesters exhibit disadvantages concerning their material properties, e.g., low tensile strength and melting points, which limit their commercial applications. In contrast, aromatic polyesters, e.g., poly(ethylene-terephthalate) (PET) and poly(butylene-terephthalate) have excellent material properties but are not degradable under natural conditions (Müller et al. 2001). Aliphatic-aromatic copolyesters have been developed with the combined biodegradable nature of aliphatic polyesters and practical material properties of the aromatic ones. Copolyesters based on a combination of 1,4-butanediol, terephthalic acid, and adipic acid (BTA) are potential biodegradable plastics based on their desirable material properties (Witt et al. 1997). A number of biodegradable plastics based on the BTA structure are now commercially available, e.g., Ecoflex® (BASF AG, Germany) and Eastar™ (Eastman, US).
Thermophilic actinomycetes are important organisms in the degradation of polyesters under high-temperature composting conditions. Diverse polyester degrading thermophilic actomonycetes have been isolated (Kalabia and Tokiwa 2004; Hoang et al. 2007; Phithakrotchanakoon et al. 2009a). Thermobifida fusca was isolated from compost and showed remarkable activities towards degradation of BTA and aliphatic polyesters e.g. PHAs (Kleeberg et al. 1998). Extracellular polyester degrading enzymes from this bacterium including BTA hydrolase and PHA depolymerase have been isolated and characterized (Gouda et al. 2002; Phithakrotchanakoon et al. 2009b). The BTA hydrolase (TfH) is a serine hydrolase which is active on degradation of BTA, PCL and even PET (Müller et al. 2005a). The enzyme is classified as a member of the lipase/esterase family with the catalytic behavior between a lipase and esterase, and has been proposed as a cutinase (Müller et al. 2005b). The genes for this enzyme are located on an operon containing two nearly identical genes (bta1 and bta2) encoding two enzyme homologues with 92% identity. In T. fusca, the enzyme is expressed only in the presence of its target polyesters and its level also depends on other phenomena, including enzyme adsorption and inhibition (Gouda et al. 2002). In order to avoid the complicated induction behavior in the wild type, recombinant expression systems have been developed for the production of BTA hydrolase in heterologous hosts, including as an OmpA fusion for secretion in Escherichia coli system (Dresler et al. 2006) and in Bacillus megaterium using a codon-optimized gene (Yang et al. 2006). These systems allow large-scale production of BTA hydrolase for biochemical characterization and further biotechnological applications, including polyester degradation and textile fiber pretreatment and modifications (Deckwer et al. 2001; Heumann et al. 2006).
Conventional catalysis activity analyses on bioplastic degradation have been based on a variety of mostly insensitive crude methods (reviewed in Jendrossek 2007). Currently, most studies on BTA hydrolase have been based on indirect activity analysis e.g. on PCL or synthetic small substrates while the catalytic study on BTA has been based on the assay of released degradation products from BTA nanoparticle and films in solution (Gouda et al. 2002; Kleeberg et al. 2005). Direct mass change determination of BTA hydrolase activity on surface degradation phenomenon of BTA thin films is of interest to provide complementary catalytic and biochemical characteristics of the enzyme. In this study, an alternative recombinant expression system has been developed in an actinomycete to avoid the problems of enzyme secretion and codon usage. This included the expression of the full-length bta1 gene with no codon optimization in Streptomyces rimosus. The catalytic activity and specificities of the recombinant hydrolase (rTfH) on surface degradation of a commercial BTA films (Ecoflex®) and aliphatic polyesters have been characterized in comparison to the lipase from Thermomyces lanuginosus, previously grouped together as “polyesterase” capable on aromatic polyester degradation (Eberl et al. 2008), using the recently introduced technique based on surface plasmon resonance analysis (Phithakrotchanakoon et al. 2009b). The effects of reaction parameters on rTfH catalysis have also been investigated. The study gives insights towards the understanding of the catalytic properties of this potent enzyme and its further biotechnological application.
Materials and methods
Materials
Poly-[(R)-3-hydroxybutyrate] (PHB) powder (natural origin; Mw 554,503 Da) and poly(ε-caprolactone) (PCL; Mw 175,693 Da) were purchased from Sigma-Aldrich, Germany. Ecoflex® (Mw 867,885 Da) was from BASF (Ludwidshafen, Germany). Lipase from T. lanuginosus was from Sigma-Aldrich. All reagents were analytical or molecular biology grade and purchased from major chemical suppliers.
Strains, plasmids, and culturing conditions
E. coli DH5α was used as a host for plasmid propagation. E. coli ET12567 (dam-, dcm-) containing pUZ8002 (MacNeil et al. 1992) was used as a donor strain for conjugation. E. coli strains were cultured in Luria–Burtani medium (1% tryptone, 0.5% yeast extract, 1% NaCl). S. rimosus R7 (ATCC10970; Hranueli et al. 1979) was maintained on mannitol soya agar (MS: 2% mannitol, 2% soya flour, 2% agar; Hobbs et al. 1989) and was incubated for 3–5 d at 30°C. For submerged culture, S. rimosus was grown by inoculating the spore suspension (109–1010 spores/ml) at 1:100 dilution in tryptone soya broth (TSB; HiMedia Laboratories, Mumbai, India). The culture was incubated at 30°C with rotary shaking at 200 rpm. A thermophilic actinomycete, Thermobifida sp. BCC23166 was from the BIOTEC Culture Collection (www.biotec.or.th/bcc). This bacterium was isolated from a landfill site in Suphanburi province, Thailand based on the ability to form a clear zone on PHB suspended agar containing basal medium and identified as previously described (Phithakrotchanakoon et al. 2009b) The isolate was maintained on PHB suspended agar plate (Calabia and Tokiwa 2004). The conjugative vector pIJ8600 (Sun et al. 1999) was used for recombinant expression of the target enzyme in S. rimosus.
Construction of expression plasmid
Genomic DNA of Thermobifida sp. BCC23166 was extracted from cells grown on MS agar using phenol/chloroform extraction, followed by precipitation with isopropanol (Kieser et al. 2000). The purified DNA was used as a template for polymerase chain reaction for amplification of the complete bta gene, including the signal peptide encoding sequence using the primers designed based on the bta1 gene from T. fusca strain YX (GenBank accession number AJ810119). A His6 encoding sequence was included in the reverse primer for expression of the enzyme as a C-terminal His6 fusion protein. The amplification reaction (50 μl) contained 20 ng genomic DNA, 0.2 mM dNTPs, 0.5 μM BTA-F primer (5′-gcgccatatggctgtgatgaccccccg-3′) and BTA-R primer (5′-atatggatcctcagtggtggtggtgg tggtgtgctgcgaacgggcaggtggagcgg-3′) (the restriction sites for cloning and the His6 tag sequence are underlined and shown in italics, respectively), and 0.5 U DyNAzyme DNA polymerase (Finnzymes, Espoo, Finland) in ×1 DyNAzyme buffer. The PCR conditions were as follows: pre-denaturation at 94°C, 4 min; 30 cycles of 94°C for 1 min, 55°C for 1 min, 74°C for 2 min; and 74°C for 10 min. Amplicons were gel-purified using a QIAquick Gel Extraction kit (QIAGEN, Hilden, Germany) and cloned into a TA-cloning vector, pTZ57R/T (Fermentas, Vilnius, Lithuania). Recombinant plasmids were DNA sequenced at Macrogen (Seoul, South Korea). The gene was then subcloned by digesting with NdeI and BamHI and ligated with pIJ8600 digested with the same restriction enzymes. The ligation mixture was transformed into E. coli DH5α and screened for recombinant plasmid on LB agar containing apramycin (50 μg/ml). The resultant plasmid pIJ-BTA contained the bta1 gene fused in-frame to the downstream His6 tag encoding sequence.
Plasmid transfer by intergeneric conjugation
The recombinant plasmid was transferred into S. rimosus R7 using intergeneric conjugation. For donor cell preparation, pIJ-BTA was transformed into a donor strain, E. coli ET12567 containing pUZ8002 by the conventional heat-shock method (Sambrook and Russell 1989). A single colony of E. coli strain containing pIJ-BTA was grown in 5 ml LB containing apramycin (50 μg/ml), chloramphenicol (25 μg/ml), and kanamycin (25 μg/ml) and incubated at 37°C with rotary shaking at 200 rpm overnight. The overnight culture was inoculated at 1:100 dilution into 100 ml of fresh LB medium containing the same antibiotics and incubated at 37°C until the absorbance at 600 nm reached 0.4–0.6; 1.5 ml of the cell culture was then collected and the cells washed twice with 0.5 ml LB before resuspension in 0.5 ml of fresh LB medium. For preparation of recipient cells, a spore suspension of S. rimosus R7 (109–1010 spores/ml) was inoculated at 1:100 dilution in 5 ml TSB and incubated at 28°C with rotary shaking at 200 rpm for 24 h. The culture was then diluted at 10−3–10−4 in 0.5 ml of TSB medium. For integeneric conjugation, 0.5 ml of the donor and recipient cells were mixed together and incubated at room temperature for 10 min. The cells were then harvested and resuspended in 0.2 ml TSB before spreading on tryptone soya agar (Oxoid, Hampshire, UK) supplemented with 10 mM MgCl2. The culture was incubated at 30°C for 16–20 h before flooding with 1 ml of solution containing 1 mg/ml thiostrepton and 0.5 mg/ml nalidixic acid in water and further incubated for 7 days under the same conditions. The transconjugant grown on the plate was then subcultivated thrice in MS agar containing antibiotics as described above to obtain the stable recombinant S. rimosus strain.
Expression of rTfH
The S. rimosus transconjugant containing pIJ-BTA was grown in 50 ml TSB containing 25 μg/ml thiostrepton at 28°C with rotary shaking at 200 rpm. Thiostrepton (5 μl of 50 mg/ml stock solution) was sequentially added everyday during the incubation period for 7 days. The cells were then separated by centrifugation at 10,000×g for 10 min, and the supernatant was collected for further analysis of the recombinant protein.
Purification of rTfH
The recombinant enzyme with a C-terminal His6 tag was purified from the culture supernatant (500 ml total volume) using a Ni-Sepharose Fast Flow column (GE-Healthcare Biosciences, Uppsala, Sweden) according to the manufacturer’s protocol. The purified enzyme was desalted and concentrated by ultrafiltration using an Amicon centrifugal unit, MWCO 10 kDa (Millipore, Billerica, MA) in 50 mM sodium phosphate buffer, pH 7.0. Protein expression and purification profiles were analyzed on SDS-PAGE and stained with Coomassie Brilliant Blue (Sambrook and Russell 1989). Western blot analysis was based on detection of the His6 tag using an anti-His6 antibody-linked with alkaline phosphatase (Invitrogen, Carlsbad, CA) as described previously (Phithakrotchanakoon et al. 2009b). Protein concentration was analyzed with Bio-Rad Protein Assay Reagent based on Bradford’s method (Bio-Rad, Hercules, CA) using bovine serum albumin as the standard. Identification of the target protein was performed by analyzing the tryptic peptides using LC/MS/MS on a Finnigan LTQ Linear Ion Trap Mass Spectrometer (Thermo Electron, San Jose, CA). MS/MS spectra were searched using Biowork™ 3.3 software (Sequest algorithm) against the NCBI-nr database.
Esterase activity assay
The esterase activity of BTA hydrolase was analyzed from the initial rate of p-nitrophenolate formation based on the method modified from Schmidt-Dannert et al. 1994. The standard reaction (1 ml) contained 50 mM sodium phosphate buffer, pH 8.0, 2.5 mM of p-nitrophenyl palmitate (pNPP; or otherwise indicated) and an appropriate dilution of the purified enzyme. The reaction was incubated at 50°C in a temperature controlled spectrophotometer (Citra 404 equipped with a GBC Thermocell, GBC Scientific Equipment, Dandenong, Australia). The formation of p-nitrophenolate was determined by measuring the absorbance at 405 nm over a 5-min time course. Control reactions with no enzyme were included in all experiments to correct for non-enzymatic hydrolysis of substrates. The initial rate was calculated by least square analysis. One unit of the enzyme activity was defined as the amount of enzyme catalyzing the release of 1 µmole p-nitrophenolate per min. The reactions were performed in triplicate and the averages of the results were reported (SD < 5%).
Surface plasmon resonance analysis
Surface plasmon resonance configuration
A Surface plasmon resonance spectroscope (SPR) was constructed by the Electro-optics laboratory, National Electronics and Computer Technology Center, Thailand. Polymer coated on gold substrate (gold thickness of 50 on 10 nm of chromium) was placed on the prism with index matching fluid. The flow cell contains seven channels; channels 1–6 were used as reaction chambers for enzyme degradation while one channel was used as the control channel. A flow rate of 2.0 µl/min pumped with a syringe pump over the sensor chip surface was used throughout this study.
Sensor chip preparation
Solutions of Ecoflex® and PCL were prepared by dissolving in chloroform. The dissolution of PHB was carried out in boiling chloroform. The polymer solutions were stirred overnight at room temperature. The gold substrates were cleaned with Piranha solution (H2SO4: H2O2 = 70:30) for 15 min, sonicated in water and followed by methanol immersion for 15 min, and then dried with nitrogen gas. Ecoflex®, PCL, and PHB were then coated on gold substrates with a spin coater (Model P6700D, Specialty Coating Systems, Indianapolis, IN). The polymer solution was deposited onto the substrate by centrifugation at 1,500 rpm for 60 s.
SPR data analysis
The running buffer (50 mM MOPS or sodium phosphate buffer, pH 8.0, or otherwise indicated) was injected over the multi-channel sensor chip at 2 μl/min in order to establish the baseline. BTA hydrolase at different dilutions, ranging from 4.5–0.0045 μg/ml in the same buffer was injected into the sample channel until the enzyme completely filled the reaction chamber. The flow was then stopped and the enzyme was incubated on the sensor surface for the time indicated. The degradation rate was determined from the sensogram based on the initial slope, assuming from system calibration that an SPR angle shift of 1 millidegree corresponds to a mass change of 1.05 ng/cm2 (Phithakrotchanakoon et al. 2009b). The specific activity was calculated based on the system configuration of which the mass reduction of 1 ng/cm2/min is equal to the specific activity of 44.4 μg−1min−1mg−1 protein (using initial enzyme concentration of 0.45 μg/ml). The reactions with lipase from T. lanuginosus were performed using the same method as described above. All of the reactions were carried out at room temperature (25°C). A minimum of two replicates were performed for each experiment, with SD < 5%.
Result
Construction of transconjugant S. rimosus containing pIJ-BTA
Thermobifida sp. BCC23166, was isolated initially from landfill for its capability on degradation of emulsified PHB agar. The isolate was found to efficiently degrade Ecoflex® films in liquid basal salt medium within 2 days at 50°C under aerobic conditions, suggesting the presence of BTA hydrolase homologous to the previously reported TfH (Gouda et al. 2002). Amplification of the bta gene from the isolate using the primers designed based on the homologous bta gene from T. fusca strain YX resulted in amplification of a 1.0 kb DNA fragment. Sequencing of the amplicon showed 100% homology to the bta1 gene of the reference strain, covering the full-length gene including the native signal peptide. The gene was then ligated to pIJ8600 (Sun et al. 1999), which is an E. coli–Streptomyces shuttle vector, at the NdeI and BamHI sites. The resultant plasmid, pIJ-BTA was transferred into the expression host S. rimosus using intergeneric conjugation. Ten transconjugants were selected after subcultivation of the transconjugants to eliminate non-transconjugant and E. coli background. As pIJ8600 is a non-replicative plasmid, incorporation of the recombinant gene is based on site-specific recombination between attP on the vector and attB sites on the bacterial chromosome (Sun et al. 1999). The presence of bta gene in all transconjugants was confirmed using PCR amplication of the target bta gene (1.0 kb) and the vector-encoded tsr gene (0.8 kb; data not shown).
Recombinant expression of rTfH
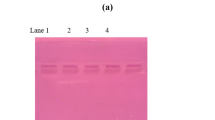
In this study, expression of the bta gene in the S. rimosus transconjugants was regulated under the control of tipA promoter, derived from a thiostrepton resistant gene. Heterologous expression of the target enzyme was studied after induction by thiostrepton for 7 days. The secretion of BTA hydrolase in the culture supernatant was analyzed by SDS-PAGE and western blot analysis using an antiHis6 antibody, revealing an induction of the target enzyme with the corresponding size (29.38 kDa, without the signal peptide) in comparison to the non-induced transconjugant and the wild-type (non-conjugated) S. rimosus (Fig. 1a, b). The peptide was recognized by an antibody against His6, indicating that it was in a C-terminally intact form. The bands of low molecular weight peptides with increased intensity in the induced transconjugants observed on the SDS-PAGE were not reacted with the antibody, suggesting that they were not the C-terminally truncated forms of the target enzyme; however, the identities of these peptides were not further investigated. The target enzyme was then purified to high homogeneity (>95% purity as determined from Coomassie Brilliant Blue staining) with a single step Ni-Sepharose affinity chromatography from the starting culture volume of 500 ml with the purification yield of 82.5%. Its identity was confirmed using LC/MS/MS analysis in which the peptide sequence showed 100% match to a triacyl glycerol lipase (BTA hydrolase) from T. fusca YX (P-value = 7.54E-12; GenBank accession YP_288944.1). The purified recombinant BTA hydrolase rTfH was then used for subsequent study.

Expression and purification of rTfH in S. rimosus transconjugant containing pIJ-BTA. a Protein profiles on SDS-PAGE. Lane M, protein molecular weight marker; lane 1, culture supernatant of S. rimosus transconjugant induced with thiostrepton; lane 2, non-induced S. rimosus transconjugant; lane 3, wild-type (non-conjugated) S. rimosus; lane 4, purified rTfH. b Western blot analysis using antiHis6 antibody. The lane labels are corresponded to A
Biochemical characterization of rTfH
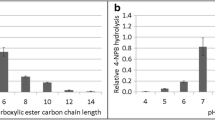
The purified exhibited an optimal temperature range of 50–55°C and optimal pH in the mild basidic range based on the assay on pNPP (Fig. 2a, b). Substrate specificity of the enzyme was assayed on p-nitrophenyl alkanoates with different alkyl chain length (C4–C16). The enzyme showed esterase activity with preference towards short side chain p-nitrophenyl alkanoates with the highest activity on p-nitrophenyl butyrate (5.5-fold of that for pNPP), while significant activity on long chain alkanoates (myristate and palmitate) was also observed. The rTfH specific activity on pNPP was 76.5 U/mg under the assay conditions.

Optimal working conditions of rTfH. The reaction contained an appropriate dilution of rTfH with 2.5 mM p-nitrophenyl palmitate. a Temperature dependence in 50 mM sodium phosphate buffer, pH 8.0; b pH dependence at 50 °C. The activity at 50 °C and pH 8 (76.5 U/mg) represents 100% relative activity
Surface plasmon resonance analysis
Degradation kinetics on different polyesters
To demonstrate the measurement of rTfH activity on different polyesters, different concentrations of rTfH were incubated over the sensor surface containing Ecoflex®, PCL, and PHB and the change in the reflectivity was measured at different angles. The angle shift corresponds to the reduction of the adsorbed polyester mass on the chip surface upon enzymatic degradation, which results in the alteration of the refractive index. The kinetics of polyester degradation by rTfH was monitored by recording the change in the reflectivity at the linear range of the SPR curve as a function of time. Different concentrations of rTfH ranging from 4.5 to 0.0045 μg/ml were used under the initial conditions in 50 mM MOPS buffer, pH 8.0 based on Phithakrotchanakoon et al. (2009b) in order to determine the comparative catalytic activity and the limit of sensitivity on different polyesters (Fig. 3). After injection, the enzyme was incubated on the sensor surface for up to 120 min while the change of the SPR angle was recorded as a function of time. The rate of change of SPR angle was proportional to rTfH concentration, and no change was seen in the control indicating that autohydrolysis of the polyester films was negligible under the experimental conditions tested.

SPR sensograms on degradation of polyesters at different rTfH concentration. The polymer thin films were incubated with different concentrations of rTfH in 50 mM MOPS buffer, pH 8.0. a Ecoflex®; b PCL; c PHB. rTfH concentration: a 4.5 μg/ml; b 0.45 μg/ml; c 0.22 μg/ml; d 0.045 μg/ml; e 0.0045 μg/ml
At the lowest enzyme concentration (0.0045 μg/ml), the SPR sensogram was poorly defined, whereas exponential decay was observed at higher concentrations (≥0.045 μg/ml). The degradation of polyesters can be quantified from the SPR sensograms. The degradation of polyesters was linear in the early phase (0–10 min) and the degradation rates were estimated at 2.0, 15.7, 1.8 ng−1cm−2min−1 equivalent to 88.3, 698, and 80 μg−1min−1mg protein for Ecoflex®, PCL, and PHB, respectively, using rTfH at 0.45 μg/ml. The degradation rates declined thereafter for all polyesters. At the maximal enzyme concentration, continual PCL and PHB degradation was seen over the 120 min incubation period although the rate tended to decrease along the incubation. In contrast, degradation of Ecoflex® almost ceased after 60 min of incubation under identical conditions (data not shown). The differences in degradation rate and kinetics would reflect the differences in the relative crystalline/amorphous phases of different polyesters and the enzyme adsorption kinetics and catalysis on the polyester surface.
Effects of reaction parameters on Ecoflex® degradation
Further investigation was focused on the study of reaction conditions on degradation of Ecoflex®. The effect of pH on rTfH catalysis was studied in the pH range from 5–8 in sodium phosphate buffer while fixing the enzyme concentration at 0.45 μg/ml. Degradation kinetics were pH dependent and the enzyme worked optimally in the mild basidic pH range with the highest degradation rate at pH 8.0, equivalent to 2.49 ng−1cm−2min−1, which was higher than the degradation rate in MOPS, pH 8.0 (Fig. 4). At pH 8.0, the catalytic activity was strongly affected by the buffer component, in which the highest activity was observed in sodium phosphate, which was 1.25 and 1.44 times higher than that in MOPS and Tris-HCl buffer, respectively. This suggested the effect of buffer components on rTfH catalysis or enzyme interaction with the polyester surface.

Effects of pH on degradation of Ecoflex®. The polymer film was incubated with 0.45 μg/ml rTfH in 50 mM sodium phosphate (pH 5.0–8.0); MOPS (pH 8.0); and Tris-HCl (pH 8.0) for 10 min
The effects of ionic strength on rTfH catalysis were also studied in different NaCl concentrations ranging from 0–1 M in 50 mM sodium phosphate buffer, pH 8.0. The degradation rate increased at low ionic strength with the optimal NaCl concentration at 250 mM and then decreased at higher NaCl concentration (Fig. 5). The highest degradation rate at the optimal conditions (pH 8.0 and 250 mM NaCl) was estimated at 17.9 ng−1cm−2min−1, equivalent to the specific activity of 0.79 mg−1min−1mg−1 protein.

Effects of ionic strength on degradation of Ecoflex®. The polymer film was incubated with 0.45 μg/ml rTfH in 50 mM sodium phosphate buffer, pH 8.0 containing 0–1 M sodium chloride for 10 min
Comparison of polyester degradation by rTfH and lipase from T. lanuginosus
The comparison of rTfH and the lipase from T. lanuginosus (Lipase LT) activities on degradation of different aliphatic and aliphatic-aromatic polyesters is shown in Fig. 6. Lipase LT was relatively active on degradation of aliphatic polyesters with the specific activities of 0.80 and 0.21 mg−1min−1mg−1 protein for PHB and PCL, respectively. However, the enzyme showed rather low specific activity on degradation of Ecoflex® (7.1 μg−1min−1mg−1 protein). Obviously, a remarkable higher specific activity was shown for rTfH on Ecoflex® (0.79 mg−1min−1mg−1 protein), which corresponded to 110 times higher degradation activity in comparison to that of Lipase LT while comparable activities was observed on PHB and only 17 times higher activity was observed on PCL. It can be clearly seen that rTfH displayed a marked higher catalytic and reaction specificities towards the aliphatic-aromatic copolyester Ecoflex® when compared to the lipase counterpart. The result thus implied significant differences on substrate specificities between these two enzymes.

Specific activity of rTfH and lipase from T. lanuginosus (Lipase LT) on different polyesters. The polymer thin films were incubated with 0.45 μg/ml enzyme in 50 mM sodium phosphate buffer, pH 8.0 and 250 mM NaCl for 10 min
Discussion
Study on microbial and enzymatic degradation of promising biodegradable polyesters is a key issue for their commercial application as well as their recycling. The Thermobifida BTA hydrolase has been reported as the first enzyme efficiently attacking the commercial aliphatic-aromatic copolyester. The enzyme has been considered very potent for application on degradation of BTA and related polyesters, together with an interest on investigation on its biochemical mechanism and molecular characteristics for further engineering of the enzyme. Our initial trial on recombinant expression of BTA hydrolase as an intracellular mature form in E. coli and as a secreted form in Pichia pastoris resulted in no significant expression of the target enzyme (data not shown). In order to develop a system for production of BTA hydrolase for further study, development of a heterologous expression system in a phylogenetically related streptomycete was considered an attractive approach owing to compatible codon usage in actinomycetes and the nature of the strain on high level protein secretion (Vrancken and Anné 2009).
Streptomyces species are potential hosts for heterologous protein production (Beki et al. 2003; Díaz et al. 2008). This genus has been described for general usefulness and versatility as a host for the expression of bioactive proteins (Brawner et al. 1991; Gilbert et al. 1995). The application of streptomycetes system for heterologous expression has been increasingly reported with the development on more efficient gene transfer systems via intergeneric conjugation (Mazodier et al. 1989; Flett et al. 1997). Among streptomycetes expression systems, S. rimosus R7 was previously reported for expression of green fluorescent protein and was described as a reliable host for expression under the tipA promoter from plasmid pIJ8600 (Phornphisutthimas et al, unpublished data). In this work, we used S. rimosus R7 as an expression host for the target enzyme in order to take the advantage of its highly protein secretion nature and the similarity in codon usage, with C or G preferentially. Our results showed that the expression of the bta1 gene with a codon usage similar to Streptomyces clearly improved the probability of success. Compared to previous studies, our system allows production of rTFH from the full-length unmodified gene with no prior codon optimization (as for B. megaterium system; Yang et al. 2006) or gene modification by fusion to surface protein (as for E. coli system; Dresler et al. 2006). However, at this stage the initial yield of expression was rather low (0.058 mg/L) in comparison to those from the E. coli or B. megaterium systems. There have been several reports on yield improvement such as using alternative strong promoter (Díaz et al. 2008) for expression or morphological engineering to reduce viscosity and pellet formation, resulting in enhanced growth rates in batch fermentations (van Wezel et al. 2006). To our knowledge, this is the first report on heterologous expression of a bacterial bioplastic degrading enzyme in a streptomycete system and provides a promising alternative system for expression of heterologous bio-plastic degrading enzymes originated from actinomycetes, which might be difficult for expression in commonly used recombinant systems.
Specific activity of rTfH in this study was in the same range as those from previous reports based on the activity analysis on p-nitrophenyl alkanoate substrates (Dresler et al. 2006; Yang et al. 2006). However, the optimal working temperature of rTfH (50–55°C) differs significantly from that previously reported (65–70°C) (Gouda et al. 2002; Kleeberg et al. 2005), which may be due to the use of different substrates and assay techniques. The higher optimal temperatures were observed for the enzymes assayed with polymers in comparison to short chain p-nitrophenyl substrates. The apparent lower optimal temperature observed in this study would be due to the influence of temperature on the mobility of macromolecular polymer substrate, which resulted in increased temperature maximum (Marten et al. 2003). In addition, it would also be of interest to further explore the effect of polymer substrate on stabilization of the enzyme at high temperature, analogous to the protection effect of carbohydrate polymers on hydrolytic enzymes (Champreda et al. 2007). The pH dependence of rTfH based on SPR analysis agreed well with the optimal pH from spectroscopic assay using the p-nitrophenyl substrate. The pH optimum of rTfH in this study was similar to that previously reported by Gouda et al. 2002 in which a pH-stat titration technique on analysis of BTA nanoparticle degradation was used but slightly differed to the optimal pH reported for the purified enzyme based on UV test (pH 6–7) (Kleeberg et al. 2005). This would be due to the effect of buffer concentration, reaction components, and ionic strength on the optimal enzyme working conditions as previously reported (Gouda et al. 2002; Kleeberg et al. 2005).
Application of the surface plasmon resonance technique to study hydrolysis of biodegradable plastic has been recently introduced for catalytic activity characterization of the PHB depolymerase from this Thermobifida strain based on direct substrate mass determination via optical property analysis (Phithakrotchanakoon et al. 2009b). In this work, using the SPR-based analytical technique, we demonstrated the substrate preference of rTfH on different aliphatic and aliphatic-aromatic polyesters in the order of PCL > Ecoflex® > PHB. This would be due to (1) the specificities inherent in the enzyme structure which results in variation in binding (enzyme adsorption) kinetics and catalytic specificity on different polyesters; and (2) differences in the physicochemical property of substrates e.g. relative crystalline/amorphous content. The specific activity on degradation of Ecoflex® with rTfH obtained in our study under the initial conditions (88.3 μg/min/mg) was comparable to that of the purified wild-type BTA hydrolase on polybutyleneadipate SP4/6 nanoparticle determined by titration method (Eberl et al. 2008). Optimization of the reaction conditions led to a marked increase (9 times) on the enzyme’s catalysis on Ecoflex®, resulting in the specific activity of 0.79 mg/min/mg protein under the optimal conditions. The reactivity of rTfH on PHB was different to that previously reported for the purified BTA hydrolase which was inactive on PHB degradation (Kleeberg et al. 2005). This could be due to differences on substrate film preparation methods and assay techniques. The catalytic activities of rTfH on polyesters tested in this study were higher compared to the previously reported activity of the recombinant PHB depolymerase from the Thermobifida strain on polyhydroxyalkanoates, which was in the range of 1.02 μg/min/mg protein at its optimal condition for polyhydroxybutyrate-co-valerate (Phithakrotchanakoon et al. 2009b).
Lipases are generally active on degradation of aliphatic polyester substrates and the polymer-related factors controlling its catalysis have been systematically investigated (Marten et al. 2003). BTA hydrolase has been shown to possess a unique catalysis behavior between a lipase and esterase based on its substrate specificities and activation mechanism, and previously proposed as a cutinase. However, this classification has been still questionable as mentioned by Kleeberg et al. (2005). Recent study has suggested that the BTA hydrolase and the lipase from T. lanuginosus together with few other serine hydrolases capable of aromatic polymer degradation constitute a so-called “polyesterases” group and cannot be classified into any distinct EC class of enzymes (Eberl et al. 2008). In comparison to the lipase from T. lanuginosus, rTfH showed efficient degradation efficiency on various polyester substrates with different structures and physicochemical properties and displayed remarkably higher reaction specificities towards degradation of the aliphatic-aromatic copolyester. The higher reaction specificities of rTfH on Ecoflex® was corresponded to the previous work showing higher efficiency of this enzyme on degradation of a linear aromatic polyester poly(trimethylene terephthalate) (PTT) in comparison to the lipase from T. lanuginosus (Eberl et al. 2008). Our recent analysis using sequence homology search revealed that BTA hydrolase showed high similarity (62% identities and 76% similarity) to a predicted lipase with putative diene lactone hydrolase function from S. albus J1074 (ZP_04702335), in addition to several lipases of acinomycete origins. Together with its specificities towards degradation of aromatic containing polyester substrates, this would suggest classification of the enzyme into a new subgroup in lipase/esterase family. However, further systematic analysis is needed.
In conclusion, an alternative recombinant expression system for the multi-polyester-degrading hydrolase from Thermobifida sp. has been reported in this study, which would be applicable on heterologous expression of bio-plastic degrading enzymes of actinomycete origins. The catalytic activity, reaction kinetics, and substrate specificities of rTfH have been characterized using the sensitive SPR analytical technique focusing on the surface degradation phenomenon on polyester films providing complementary information on catalytic characteristics of this biotechnologically potent enzyme. Further application of the SPR technique on the study of rTfH catalysis and kinetics is of interest for elucidation of the enzyme’s catalytic and mechanistic properties.
References
Beki E, Nagy I, Vanderleyden J, Jager S, Kiss L, Fulop L, Hornok L, Kukolya J (2003) Cloning and heterologous expression of a beta-d-mannosidase (EC 3.2.1.25)-encoding gene from Thermobifida fusca TM51. Appl Environ Microbiol 69:1944–1952
Brawner M, Poste G, Rosenberg M, Westpheling J (1991) Streptomyces: a host for heterologous gene expression. Curr Opin Biotechnol 2:674–681
Calabia BP, Tokiwa Y (2004) Microbial degradation of poly(d-3-hydroxybutyrate) by a new thermophilic streptomyces isolate. Biotechnol Lett 26:15–19
Champreda V, Kanokratana P, Sriprang R, Tanapongpipat S, Eurwilaichitr L (2007) Purification, biochemical characterization, and gene cloning of a new extracellular thermotolerant and glucose tolerant malto-oligosaccharide forming α-amylase from an endophytic ascomycete Fusicoccum sp. BCC4124. Biosci Biotechnol Biochem 71:2010–2020
Deckwer W-D, Müller RJ, Van den Heuvel J, Kleeberg I (2001) Enzyme which cleaves ester groups and which is derived from Thermomonospora fusca. GBF patent W001123581A1
Díaz M, Ferreras E, Moreno R, Yepes A, Berenguer J, Santamaría R (2008) High-level overproduction of Thermus enzymes in Streptomyces lividans. Appl Microbiol Biotechnol 79:1001–1008
Dresler K, van den Heuvel J, Müller R-J, Deckwer W-D (2006) Production of a recombinant polyester-cleaving hydrolase from Thermobifida fusca in Escherchia coli. Bioprocess Biosys Eng 29:169–183
Eberl A, Heumann S, Kotek R, Kaufmann F, Mitsche S, Cavaco-Paulo A, Gübitz GM (2008) Enzymatic hydrolysis of polymers and oligomers. J Biotechnol 135:45–51
Flett F, Mersinias V, Smith CP (1997) High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol Lett 135:223–229
Gilbert M, Morosoli R, Shareck F, Kluepfel D (1995) Production and secretion of proteins by streptomycetes. Crit Rev Biotechnol 15:13–39
Gouda MK, Kleeberg I, van den Heuvel J, Müller R-J, Deckwer W-D (2002) Production of a polyester degrading extracellular hydrolase from Thermomonospora fusca. Biotechnol Prog 18:927–934
Heumann S, Eberl A, Pobeheim H, Liebminger S, Fischer-Colbrie G, Almansa E, Cavaco-Paulo A, Gübitz GM (2006) New model substrates for enzyme hydrolyzing polyethyleneterepthalate and polyamide fibers. J Biochem Biophys Methods 69:89–99
Hoang K-C, Tseng M, Shu W-J (2007) Degradation of polyethylene succinate (PES) by a new thermophilic Microbispora strain. Biodegradation 18:333–342
Hobbs G, Frazer CM, Gardner DCJ, Cullum JA, Oliver SG (1989) Dispersed growth of Streptomyces in liquid culture. Appl Microbiol Biotechnol 31:272–277
Hranueli D, Pigac J, Vesligaj M (1979) Characterization and persistence if actinophage RP2 isolated from Streptomyces rimosus ATCC10970. J Gen Microbiol 114:295–303
Jendrossek D (2007) Peculiarities of PHA granules preparation and PHA depolymerase activity determination. Appl Microbiol Biotechnol 74:1186–1196
Kalabia BP, Tokiwa Y (2004) Microbial degradation of poly(d-3-hydroxybutyrate) by a new thermophilic Streptomyces isolate. Biotechnol Lett 26:15–19
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces Genetics. Society for General Microbiology, Reading UK
Kleeberg I, Hetz C, Kroppenstedt RM, Müller R-J, Deckwer W-D (1998) Biodegradation of aliphatic-aromatic copolyesters by Thermomonospora fusca and other thermophilic compost isolates. Appl Environ Microbiol 64:1731–1735
Kleeberg I, Welzel K, VandenHeuvel J, Müller R-J, Deckwer W-D (2005) Characterization of a new extracellular hydrolase from Thermobifida fusca degrading aliphatic-aromatic copolyesters. Biomacromolecules 6:262–270
Luengo JM, Garsia B, Sandoval A, Naharro G, Olivera ER (2003) Bioplastics from microorganisms. Curr Opin Microbiol 6:251–260
MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T (1992) Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111:61–68
Marten E, Müller R-J, Deckwer W-D (2003) Studies on the enzymatic hydrolysis of polyesters I. Low molecular mass model esters and aliphatic polyesters. Polym Degrad Stab 80:485–501
Mazodier P, Petter R, Thompson C (1989) Intergeneric conjugation between Escherichia coli and Streptomyces species. J Bacteriol 171:3583–3585
Müller RJ, Kleeberg I, Deckwer W-D (2001) Biodegradation of polyesters containing aromatic constituents. J Biotechnol 86:87–95
Müller RJ, Schrader H, Profe J, Dresler K, Deckwer W-D (2005a) Enzymatic degradation of poly(ethylene terephthalate): rapid hydrolyse using a hydrolase from T. fusca. Macromol Rapid Commun 26:1400–1405
Müller I, Welzel K, VandenHeuvel J, Müller R-J, Deckwer WD (2005b) Characterization of a new extracellular hydrolase from Thermobifida fusca degrading aliphatic-aromatic copolyesters. Biomacromolecules 6:262–270
Phithakrotchanakoon C, Rudeekit Y, Tanapongpipat S, Leejakpai T, Aiba S, Noda I, Champreda V (2009a) Microbial degradation and physico-chemical alteration of polyhydroxyalkanoates by a thermophilic Streptomyces sp. Biologia 64:246–251
Phithakrotchanakoon C, Daduang R, Thamchaipenet A, Wangkam T, Srikhirin T, Eurwilaichitr L, Champreda V (2009b) Heterologous expression of polyhydroxyalkanoate depolymerase from Thermobifida sp. in Pichia pastoris and catalytic analysis by surface plasmon resonance. Appl Microbiol Biotechnol 82:131–140
Sambrook J, Russell DW (1989) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, New York
Schmidt-Dannert C, Sztajer H, Stocklein W, Menge U, Schmid RD (1994) Screening, purification and properties of a thermophilic lipase from Bacillus thermocatenulatus. Biochim Biophys Acta 1214:43–53
Sun J, Kelemen GH, Fernandez-Abalos JM, Bibb MJ (1999) Green fluorescent protein as a receptor for spatial and temporal gene expression in Streptomyces coelicolor A3(2). Microbiology 145:2221–2227
Tokiwa Y, Calabia BP (2004) Degradation of microbial polyesters. Biotechnol Lett 26:1181–1189
van Wezel GP, Krabben P, Traag BA, Keijser BJF, Kerste R, Vijgenboom E, Heijnen JJ, Kraal B (2006) Unlocking Streptomyces spp. for use as sustainable industrial production platforms by morphological engineering. Appl Environ Microbiol 72:5283–5288
Vrancken K, Anné J (2009) Secretory production of recombinant proteins by Streptomyces. Future Microbiol 4:181–188
Witt U, Müller RJ, Deckwer WD (1997) Biodegradation behavior and material properties of aliphatic-aromatic polyesters of commercial importance. J Environ Polym Degrad 5:81–89
Yang Y, Malten M, Grote A, Jahn D, Deckwer W-D (2006) Codon optimized Thermobifida fusca hydrolase secreted by Bacillus megaterium. Biotechnol Bioeng 96:780–794
Acknowledgements
This project was supported by the Thailand Research Fund (TRF). The authors would like to thank Dr. Phillip James Shaw for manuscript proofreading. N.S. was granted YSTP senior project studentship from NSTDA. T.W. was granted the Development and Promotion of Science and Technology Talents Project (DPST) Fund.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sinsereekul, N., Wangkam, T., Thamchaipenet, A. et al. Recombinant expression of BTA hydrolase in Streptomyces rimosus and catalytic analysis on polyesters by surface plasmon resonance. Appl Microbiol Biotechnol 86, 1775–1784 (2010). https://doi.org/10.1007/s00253-010-2465-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2465-y