
Abstract
Biotechnology needs to explore the capacity of different organisms to overproduce proteins of interest at low cost. In this paper, we show that Streptomyces lividans is a suitable host for the expression of Thermus thermophilus genes and report the overproduction of the corresponding proteins. This capacity was corroborated after cloning the genes corresponding to an alkaline phosphatase (a periplasmic enzyme in T. thermophilus) and that corresponding to a beta-glycosidase (an intracellular enzyme) in Escherichia coli and in S. lividans. Comparison of the production in both hosts revealed that the expression of active protein achieved in S. lividans was much higher than in E. coli, especially in the case of the periplasmic enzyme. In fact, the native signal peptide of the T. thermophilus phosphatase was functional in S. lividans, being processed at the same peptide bond in both organisms, allowing the overproduction and secretion of this protein to the S. lividans culture supernatant. As in E. coli, the thermostability of the expressed proteins allowed a huge purification factor upon thermal denaturation and precipitation of the host proteins. We conclude that S. lividans is a very efficient and industry-friendly host for the expression of thermophilic proteins from Thermus spp.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thermostable enzymes (thermozymes) from several sources are useful tools for different biotechnological process (Pantazaki et al. 2002). Frequent sources of these enzymes are strains of the genus Thermus, and the most common way to overproduce them involves use of a surrogate mesophilic host as cell factory, Escherichia coli being the one most widely used (Pessela et al. 2004). When production of the active form of a thermophilic enzyme in this mesophile is possible, the use of a purification step involving thermal denaturalization of the host proteins strongly facilitates its purification.
Alternative methods for the overexpression of these proteins in engineered expression hosts of the same species have been successfully applied to the overproduction of different enzymes (Hidalgo et al. 2004; Kayser and Kilbane 2001; Moreno et al. 2003, 2005). However, the scaling up of these thermophilic expression systems is still poorly known in comparison to the long experience already available for classical industrial microorganisms such as Streptomyces.
We have developed an expression system for Streptomyces based on the promoter of a xylanase gene: the xysAp promoter (Santamaría et al. 2005). This is a strong promoter that is repressed by glucose and induced by xylose or xylan (Rodríguez et al. 2005). The system has proved useful for the expression of genes from different Streptomyces species, up to several grams per liter being achieved for some enzymes, such as cellulases or amylases (Díaz M and Santamaría RI, unpublished). In addition, a 100-fold overexpression of the xylanase X22 from Aspergillus nidulans has been obtained, representing a 20-fold improvement with respect to production in Saccharomyces cerevisiae (Díaz et al. 2004), despite relevant differences between Aspergillus and Streptomyces in codon usage.
Owing to similarities in their codon usage, with C or G preferentially as the third base of most codons, we speculated that Streptomyces could be an ideal host for the expression of genes from Thermus spp. Two genes from Thermus thermophilus encoding two thermostable enzymes were selected to study this capacity of Streptomyces and to compare it with the production obtained in E. coli. One of them encodes an extracellular alkaline phosphatase (Castán et al. 2002) and the other encodes an intracellular ß-glycosidase (TTP0042) (Henne et al. 2004). The reason for selecting these genes lies in the potential biotechnological usefulness of these enzymes and their different locations in the cell. Thus, alkaline phosphatases are usually used to eliminate phosphate from DNA in different molecular biology techniques and as a biochemical marker in ELISA and in non-radioactive techniques such as blotting and sequencing systems (Gong et al. 2005). The ß-D glycosidase can be used as a tool in organic chemistry and for the hydrolysis of lactose and dairy products (Pessela et al. 2003).
Materials and methods
Strains and culture conditions
The T. thermophilus strains HB8 (ATCC 27634) and HB27 (ATCC BAA-163) were the source of the DNA encoding a hyperalkaline phosphatase PhoA, (Castán et al. 2002) (Accession number AJ309568; EC number: 3.1.3.1) and a putative beta-glycosidase (TTP0042, Accession number: AAS82372.1; EC number: 3.2.1.21), respectively. E. coli DH5α (Hanahan 1983) was used for the cloning and isolation of plasmids, following standard procedures, and E. coli BL21(DE3) (Stratagene) was used for the overexpression of genes under the control of a promoter dependent on the T7-RNA polymerase. Streptomyces lividans JI66 (DSM 46482) (Kieser et al. 2000) was used for cloning and protein expression.
Growth and manipulation of E. coli and S. lividans were accomplished as described previously (Hanahan 1983; Kieser et al. 2000).
Plasmid construction and protein expression
The phoA gene was PCR amplified from plasmid pAPA (Castán et al. 2002) with the primers OpaNdeI (GGCATATGAAGCGAAGG) and OpaSalI (GGGGTCGACGGCCCAGAC), which included NdeI and SalI restrictions sites (underlined), respectively. The gene encoding the TTP-0042 protein was PCR-amplified from total DNA of T. thermophilus HB27 with the primers oTTP0042NdeI (AAACATATGACCGAGAACGCCGA) and oTTP0042HindIII (AAAAGCTTAGGTCTGGGCCCG), which included NdeI and HindIII restriction sites (underlined).
The phoA gene was cloned into plasmid pET22b (Novagen), which provides a C-terminal (6×)Histidine tag to the produced protein, whereas the putative beta-glycosidase gene was cloned in pET28b (Novagen), from which the corresponding protein is also produced with a N-terminal (6×)Histidine tag. The final plasmids were designated pET22PA and pTTP0042.
For protein overproduction, the E. coli BL21DE3 strains transformed with each of these plasmids were grown at 37°C in Luria-Bertani (LB) up to an OD550 of 0.5, after which 1 mM isopropyl-beta-d-thiogalactopyranoside (IPTG) was added to the cultures, which were kept under the same growth conditions for 4 more hours before cell harvesting.
The Streptomyces expression plasmids were obtained in two steps. In the first step, the phoA and TTP0042 genes were cloned under the control of the xylanase xysA promoter (xysAp) of Streptomyces halstedii between the mmrt (T1) and fdt (T2) transcriptional terminators. In the second step, these expression cassettes were transferred to the Streptomyces multicopy vector pIJ702 (Katz et al. 1983). To accomplish this, the phoA gene DNA was isolated from plasmid pET22PA by digestion with the SalI and NdeI restriction enzymes. The TTP0042 gene was obtained by digestion with NdeI and HindIII from the plasmid pTTP0042. Both ORFs were inserted into the corresponding sites of the plasmid pXHis1 (Adham et al. 2001), yielding the E. coli multicopy plasmids pXFOS1 and pXGAL1, respectively.
The expression cassettes of both plasmids were obtained by digestion with BglII, and after agarose gel purification they were ligated to the plasmid pIJ702 digested with the same enzyme and treated with CIAP (calf intestine alkaline phosphatase). After transformation of S. lividans protoplasts, the plasmids pTXF1 (carrying the phoA gene) and pTXGal1 (carrying the TTP0042 gene) were obtained.
The production of both enzymes with the C-terminal 6×His tag by S. lividans transformed with the above plasmids was carried out in YES medium supplemented with 1% xylose as described (Díaz et al. 2004).
Protein analysis and enzymatic assays
Protein profiles were analyzed with denaturing polyacrylamide gel electrophoresis (SDS-PAGE) (Ruiz-Arribas et al. 1995). Proteins were detected by 0.5% Coomassie brilliant blue R staining. Protein contents were determined using the Bio-Rad D C protein assay, with bovine serum albumin as standard. The amino terminus of the secreted phosphatase was determined with an Applied Biosystems 470A Protein Sequenator.
β-glycosidase activity was determined as described by Miller (Miller 1972) by mixing samples (2–5 µl) with buffer Z 1× (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, pH 7) up to final volume 500 µl. After incubation at 70°C for 5 min, 100 µl of an ONPG (ortho-nitrophenyl-beta-d-galactopyranoside) solution (4 mg/ml in 100 mM sodium phosphate buffer, pH 7.5) was added as substrate. The samples were incubated for 5 min at 70°C and the incubation was finally stopped by adding 250 µl of 1 M sodium carbonate and, cooling on ice. Activity was measured at 420 nm DO and is referred to as DO420 ml−1 min−1.
Alkaline phosphatase activity was assayed by mixing 100 µl of the sample, 800 µl of 0.2 M sodium carbonate buffer pH 11.3 and, as substrate, 50 µl of PNPP 0.4% (para-nitrophenylphosphate/Sigma104 phosphatase substrate) in the same buffer. The reaction time was 10 min at 60°C. Reactions were stopped by adding 50 µl of 1 M K2HPO4. The samples were centrifuged at 12,000×g and measured at 420 nm (modified from Brickman and Beckwith 1975). One enzyme unit was defined as µmoles PNP ml−1 min−1 produced.
Purification of His-tagged proteins
His-tagged phosphatase and beta-glycosidase were purified in a first step of thermal denaturation of the host proteins (70°C, 30 min) followed by affinity chromatography of the soluble thermostable fraction on Ni-NTA-agarose, according to the standard procedures of the manufacturer (Qiagen). Briefly, samples were adjusted to 50 mM sodium phosphate, 300 mM NaCl, pH 8.0, for binding to the resin, washed twice with 5 mM imidazole in the same buffer and eluted with 250 mM imidazole. Fractions containing highly purified protein were pooled and dialyzed against 50 mM sodium phosphate buffer, pH 7.5, plus 50 mM NaCl. Purified fractions containing PhoA were incubated with 40 mM CoSO4 for 15 min at 65°C to reactivate the enzyme.
Results
Cloning and expression of TTP0042 in S. lividans JI66 versus E. coli.
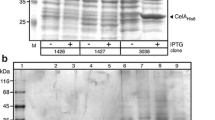
The gene encoding the beta-glycosidase from T. thermophilus was cloned into the Streptomyces multicopy plasmid pIJ702 (“Materials and methods”). The expression plasmid, pTXGAL1, containing the sequence encoding TTP0042 transcribed under the control of the Streptomyces promoter xysA (xysAp) (Fig. 1a, left), was transformed in S. lividans JI66 protoplasts and the plasmid vector pIJ702 was used as a negative control. Transformants were cultured in liquid media (YES medium supplemented with 1% xylose) for 4 and 6 days, and the cells were harvested and mechanically disrupted (fast prep), obtaining a cell extract (CE). A good production of the beta-glycosidase was observed with SDS-PAGE stained with Coomassie Blue (Fig. 1b). A clear extra protein band of 48.6 kDa with respect to the negative control cultures was detected in the cell extracts of the pTXGAL1 cultures (Fig. 1b, lane 2).

a Expression plasmids for Streptomyces (pTXGal1, left) and E. coli (pTTP0042, right) schemes: tsr: thiostrepton resistance; melC2: tyrosinase gene; T1: mmrt transcriptional terminator; xysAp: xylanase promoter; Tthgal1: T. thermophilus glycosidase; T2: fdt transcriptional terminator; kan: kanamycin resistance; lacI: galactosidase gene; T7p: T7-polymerase promoter; T: T7 transcriptional terminator; bThermus ß-glycosidase production in Streptomyces versus E. coli transformed with pTXGal1 or pTTP0042. Coomassie-stained gel protein of 10 µl of total cell extract protein culture (CE); S. lividans: Sp: supernatant; E. coli: P: particulate fraction after cell lysis; S: soluble proteins after cell lysis; empty plasmids pIJ702 or pET28b(+) were used as negative controls (C−), lanes 1 and 4, respectively; c Soluble proteins after treatment of the same fractions at 70°C for 30 min; d Glycosidase activity of the different fractions (activity unit: DO420 nm ml−1 min−1). Error bars correspond to the standard deviations of four different assays. Numbers between c and d correspond to the different lanes or reactions
Part of the enzyme could also be observed in the supernatant fraction (Fig. 1b, lane 3) in spite of being an intracellular enzyme, and hence without a signal peptide sequence. The specific activity of this enzyme was confirmed by colorimetric assays with ONPG, as described in “Materials and methods,” in both fractions after 4 days of culture (Fig. 1d). These pTXGAL1 cultures had about 260 enzyme units per milliliter (U/ml) in the cell extract fraction whereas 28 U/ml were detected in the supernatant. The amino terminal sequence of the secreted protein was determined as “TENAEKFL”, corresponding to the second amino acid (only lacking the methionine), and hence the protein was not processed by any known secretion pathway. Production was not enhanced in the cell extract fraction (229 U/ml) at longer incubation time (6 days) and was even slightly more abundant in the supernatant fraction (55 U/ml). The presence of the protein in the supernatant could be explained in terms of partial cell lysis.
Simultaneously, E. coli BL21 (DE3) was transformed with plasmids pTTP0042 (Fig. 1a, right) and pET28b+ as negative controls (C−) and the production of beta-glycosidase under the T7-promoter was induced with 1 mM IPTG, as described in “Materials and methods.” The cells were harvested and disrupted by sonication (total cell extract: CE), after which the cell extract was centrifuged to separate the soluble fraction (S) from the particulate fraction (P), and their activities were measured. As shown in Fig. 1b lane 5, a large amount of beta-glycosidase (about half of that produced by Streptomyces) was produced in the cells (CE) as seen in Coomassie-stained SDS-PAGE protein gels, but the ß-gal activity of the protein overexpressed in E. coli was quite low (3.6 U/ml) in comparison with that expressed in Streptomyces (about 260 U/ml) (Fig. 1d). In fact, all the activity present in whole cells extracts of E. coli corresponded to the small amount of the protein that remained soluble after disruption of the cells (S, Fig. 1b, d lane 7) (3.4 U/ml). This result indicates that most of the protein produced in E. coli under these conditions does not fold properly and precipitates as inclusion bodies (P fraction, Fig. 1c lane 6). Thus, despite the good production of the protein in E. coli its specific activity was very poor in contrast to the high specific activity of the enzyme produced in Streptomyces.
As TTP0042 is a thermozyme, we subjected the different fractions containing the protein to a heat treatment (70°C during 30 to 60 min). The results with the Streptomyces fractions revealed a clear enrichment in the thermophilic protein (Fig. 1c, lane 2), which remained active in both fractions (205 U/ml in CE and 30 U/ml in Sp) even after 60 min at 70°C. However, it is worth noting that different host proteins remained soluble after the treatment. As expected, the heat treatment applied did not result in an increase in the amount of soluble and active forms of the enzyme in the CE and S fractions from E. coli, the same activity (3.5 U/ml in CE and 3.2 U/ml in S) being detected as that measured before the treatment. This shows that a single heat treatment is not sufficient to refold the enzyme from the inclusion bodies produced during its expression in E. coli.
Cloning and expression of PhoA in S. lividans JI66
The phoA gene encoding the periplasmic hyperalkaline phosphatase from Thermus thermophilus was also cloned under the control of the S. halstedii xysA promoter (see “Materials and methods”). The final plasmid—pTXF1 (Fig. 2a, left)—was obtained in S. lividans JI66 and its map was corroborated by digestion with restriction enzymes. Cultures of S. lividans JI66 transformed with pTXF1 or with the control plasmid pIJ702, were grown at 30°C for several days in liquid YES media supplemented with 1% xylose. The protein pattern was analyzed by SDS-PAGE and the phosphatase activity (AP) was measured in culture supernatants and in whole cell extracts. An accumulation of a protein band corresponding to the expected size of the PhoA protein (54.7 kDa) was observed along the culture time in the supernatant of the cultures carrying pTXF1 (Fig. 2b, lanes 3 and 4) but not in the 4- and 6-day cultures of S. lividans JI66 transformed with pIJ702 (Fig. 2b, lanes 1 and 2).

a Expression plasmids for Streptomyces (pTXF1, left) and E. coli (pET22PA, right) schemes: tsr: thiostrepton resistance; melC2: tyrosinase gene; T1: mmrt transcriptional terminator; xysAp: xylanase promoter; TthphoA: T. thermophilus phosphatase; T2: fdt transcriptional terminator; amp: ampicillin resistance; lacI: galactosidase gene; T7p: T7-polymerase promoter; T: T7 transcriptional terminator; bThermus phosphatase production in Streptomyces versus E. coli transformed with pTXF1 or pET22PA. Coomassie-stained protein gel showing production of 10 µl liquid culture. S. lividans: Sp(4): 4-day supernatant; Sp(6): 6-day supernatant; E. coli: CE: total cell extract protein; P: particulate fraction after cell lysis; S: soluble proteins after cell lysis; empty plasmids pIJ702 or pET22b(+) were used as negative controls (C−) lanes 1 and 5 respectively; c Soluble proteins after treatment of the same fractions at 70°C for 30 min; d Phosphatase activity of the different fractions (activity unit: µmoles PNP ml−1 min−1). Error bars correspond to the standard deviations of four different assays. Numbers between c and d correspond to the different lanes or reactions
In the latter fractions, a protein slightly larger than PhoA was observed, but it did not accumulate as the growth time advanced. In fact, these fractions showed a basal level of alkaline phosphatase activity (about 13 U/ml). In contrast, an activity of about 267 U/ml of phosphatase was detected in the supernatant of 6-day cultures carrying pTXF1 (Fig. 2d); this supports the notion that the accumulating protein detected by Coomassie blue staining in fact corresponded to the T. thermophilus PhoA. Confirmation of this was obtained by N-terminal sequencing of this secreted protein, which revealed the existence of two consecutive processing sites (+29 and +34: Q↓NQPSL↓GRRYNL...) that were identical to those detected for the PhoA protein purified directly from the periplasmic fraction of T. thermophilus (Castán 2004).
It is interesting to note only a small fraction of phosphatase activity remained bound to the cells (data not shown), showing that almost all the protein was efficiently secreted to the growth medium in Streptomyces, thus avoiding, on one hand, the limitations that the volume of the periplasmic space imposes on the amount of overexpressed proteins in E. coli (Castán et al. 2002) and, on the other, reducing the requirements for subsequent purification steps.
A clear example of the limitations to the expression of secreted proteins in E. coli was observed when the PhoA protein was expressed in E. coli BL21 (DE3) from plasmid pET22PA (Fig. 2a, right). After induction with IPTG, the cells were disrupted and the soluble and particulate fractions were analyzed. As shown in Fig. 2b, the enzyme could not be easily detected by Coomassie blue staining in any of the fractions. However, a low activity was detected in the whole-cell (CE, 15 U/ml) and soluble (S, 11 U/ml) fractions (Fig. 2d).
Owing to its thermophilic nature, the fractions from both organisms containing PhoA were also subjected to thermal treatment at 70°C for 30 min, as above. As shown in Fig. 2c, most of the Streptomyces-secreted proteins disappeared after this treatment, PhoA remaining as the major component of the soluble fraction from 4- and 6-day cultures (Sp(4), lane3 and Sp(6), lane 4). The AP activity recovered from the culture supernatant after this treatment was more than 85% of that detected before it.
Biochemical properties of the thermophilic proteins
After thermal denaturalization of the host proteins we performed a His-tag affinity chromatography of the thermostable soluble fraction to purify and characterize both proteins, as described in “Materials and methods.” Aliquots of the purified proteins were then used to analyze the optimal pH and temperature of both enzymes. As shown in Fig. 3a, the TTP0042 enzyme functions optimally between pH 6.5 and 7.5, being inhibited in the presence of Tris–HCl buffer, as described for other beta-glycosidases (Dion et al. 1999). At the optimal pH, the activity at 75°C was the maximum (Fig. 3b). A further stability assay was carried out, and it was observed that the enzyme was stable for more than 2 h at 70 and 80°C, but that it was completely inactivated after 40 min at 90°C (Fig. 3c). No activity was detected when PNPG (para-nitrophenyl-alpha-d-galactopyranoside) was used as substrate instead of ONPG (ortho-nitrophenyl-beta-d-galactopyranoside) (not shown), supporting the idea that the enzyme has no alpha-glycosidase activity.

Biochemical properties of TTP0042. a optimal pH determination; b optimal temperature determination; c thermal inactivation pattern at different temperatures
However, the PhoA enzyme thus purified was completely inactive. As the activity was readily detected upon overproduction, we speculated that such absence of activity would be due to a loss of the necessary required cations during its purification through the chelating column. Accordingly, we incubated the purified protein with different salts (40 mM) at 60°C, observing activation in the following order: Co2+ > Zn2+ >Mg2+ > Cu2+ (data not shown). The optimal pH and temperature for the Co+ -reactivated enzyme was measured as above with appropriate buffers, values of pH 11.3 and 80°C, respectively, being obtained (Fig. 4).

Biochemical properties of PhoA. a optimal pH determination The buffers used were: Tris–HCl (7–10) and Carbonate (11–13); b optimal temperature determination
Discussion
Microorganisms included within the genus Streptomyces are used for the industrial production of a broad number of active compounds, such as antibiotics, antitumorals, and several enzymes, such as proteases, transglutaminase, or xylose isomerase (Challis and Hopwood 2003; Van Mellaert and Anne 2001). In this work, we observed that Streptomyces also has great potential for producing large amounts of thermophilic proteins. In particular, we used S. lividans for the production of enzymes from the thermophile Thermus thermophilus. Our results show that the expression of genes with a codon usage similar to Streptomyces (C or G in the third base), as is the case in Thermus, clearly improves the probabilities of success when expressing heterologous genes in this microorganism, as has also been demonstrated for Mycobacterium proteins (Vallín et al. 2006).
The interest of the present work derives from the fact that mesophilic organisms with a high industrial background can be used for the production of thermostable enzymes isolated from a thermophilic organism that is difficult to scale up. We used two different organisms (a Gram-negative E. coli and a Gram-positive Streptomyces) to produce a periplasmic alkaline phosphatase (PhoA) and an intracellular beta-glycosidase (TTP0042). We obtained a good expression level of the beta-glycosidase in both microorganisms, but whereas most of the protein was produced in an active and soluble form in Streptomyces, the protein produced in E. coli was improperly folded and was accumulated in the particulate fraction, most likely as inclusion bodies. Therefore, at least for this particular protein, the use of Streptomyces as an expression host represents a dramatic improvement in comparison with the use of E. coli.
The presence of relevant amounts of the beta-glycosidase in the supernatants of the overproducing S. lividans cultures (55 U/ml in a 6-day culture) was unexpected owing to the absence of any putative signal peptide or processing of the enzyme (only lacking the first methionine). The most likely first-hand explanation we considered was cell lysis, although such a phenomenon could not be detected microscopically. Moreover, no other major intracellular proteins were detected accompanying the beta-glycosidase in the culture supernatants, suggesting that if there were indeed some microscopically undetectable lysis, the Streptomyces-secreted proteases would be degrading such intracellular proteins without affecting the thermozyme. Whichever the case, it seems clear from the beta-glycosidase sequence that none of the main bacterial protein secretion systems described to date (Sec/SRP and Tat) is likely to be responsible for a putative active secretion of this protein. Nevertheless, the possibility still remains that any of the broad variety of new protein-targeting secretion systems described for Gram-positive bacteria, such as WXG100/ESAT-6, sortase, etc. (some of which do not require the presence of a typical signal peptide), could be involved in the putative secretion of this intracellular enzyme (Pallen et al. 2003).
An even more dramatic difference was found when the expression of the PhoA protein in both organisms was compared. In this case, the secreted nature of this protein, which is secreted to the periplasm of T. thermophilus through the TAT system (Cava and Berenguer 2006; Cava et al. 2008), was a problem when E. coli was used as overexpression host. In this bacterium, the amount of protein produced was basically detected only by its enzymatic activity, as there were insufficient amounts available for detection with Coomassie blue staining. This was probably the consequence of both a limitation in the number of TAT secretion systems in E. coli, the pathway by which this protein is secreted in this host (Angelini et al. 2001), and also to the volume limitations imposed by the periplasmic space.
In contrast to such limitations, the secretion machinery of Streptomyces does recognize the PhoA signal peptide and secretes it efficiently to the growth medium. Moreover, processing of the signal peptide of this protein was identical in both organisms, as revealed by comparison of the N-terminal sequence of the protein secreted by Streptomyces with that isolated and purified from the periplasmic space of T. thermophilus (Castán et al. 2002). This is a very relevant result, not only because it shows that no protein engineering would be required at signal peptide level for the expression of Thermus periplasmic proteins in this industrial microorganism, but also because its direct secretion to the growth medium bypasses the above-commented limitations imposed by the periplasm volume on the amount of expressed proteins in Gram-negative hosts.
In addition, the fact that active forms of the PhoA and the beta-glycosidase accumulated in S. lividans supernatants after several days of culture indicates that the proteins used here are stable and insensitive to S. lividans-secreted proteases. Similar results concerning protease resistance have been described for other heterologous secreted proteins produced by Streptomyces (Sianidis et al. 2006)). Nevertheless, there are other Streptomyces protease-defective strains that could be used as hosts in the event of the protein to be expressed not being so protease-resistant.
Another advantage of using a mesophilic organism as a host (Streptomyces), compared to recently developed Thermus homologous expression systems (Moreno et al. 2005), is the use of elevated temperatures to denature and precipitate most host cell proteins, allowing a very efficient first purification step. In the case of the secreted proteins, this results in an essentially pure product that would not require further purification for most applications, making the yield of the process highly profitable from an industrial point of view.
The most remarkable conclusion derived from this work is the higher capacity of Streptomyces to produce active Thermus heterologous proteins compared to E. coli. In particular, the 260 U/ml vs 3.5 U/ml of the ß-glycosidase intracellular enzyme and the 267 U/ml vs 15 U/ml of the periplasmic phosphatase enzyme are good examples of the appropriate choice of Streptomyces for protein expression in biotechnology.
References
Adham SA, Honrubia P, Díaz M, Fernández-Ábalos JM, Santamaría RI, Gil JA (2001) Expression of the genes coding for the xylanase Xys1 and the cellulase Cel1 from the straw-decomposing Streptomyces halstedii JM8 cloned into the amino-acid producer Brevibacterium lactofermentum ATCC13869. Arch Microbiol 177:91–97
Angelini S, Moreno R, Gouffi K, Santini C, Yamagishi A, Berenguer J, Wu L (2001) Export of Thermus thermophilus alkaline phosphatase via the twin-arginine translocation pathway in Escherichia coli. FEBS Lett 506:103–107
Brickman E, Beckwith J (1975) Analysis of the regulation of Escherichia coli alkaline phosphatase synthesis using deletions and phi80 transducing phages. J Mol Biol 96:307–316
Castán P (2004) Desarrollo de cepas de interés biotecnológico mediante la manipulación de los genes recA y slpA de Thermus thermophilus. PhD thesis. Departamento de Biología Molecular. Universidad Autónoma de Madrid. Madrid
Castán P, Zafra O, Moreno R, de Pedro MA, Vallés C, Cava F, Caro E, Schwarz H, Berenguer J (2002) The periplasmic space in Thermus thermophilus: evidence from a regulation-defective S-layer mutant overexpressing an alkaline phosphatase. Extremophiles 6:225–232
Cava F, Berenguer J (2006) Biochemical and regulatory properties of a respiratory island encoded by a conjugative plasmid in the extreme thermophile Thermus thermophilus. Biochem Soc Trans 34:97–100
Cava F, de Pedro MA, Blas-Galindo E, Waldo GS, Westblade LF, Berenguer J (2008) Expression and use of superfolder green fluorescent protein at high temperatures in vivo: a tool to study extreme thermophile biology. Environ Microbiol 10:605–613
Challis GL, Hopwood DA (2003) Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci U S A 100(Suppl 2):14555–14561
Dion M, Fourage L, Hallet JN, Colas B (1999) Cloning and expression of a beta-glycosidase gene from Thermus thermophilus. Sequence and biochemical characterization of the encoded enzyme. Glycoconj J 16:27–37
Díaz M, Adham SA, Ramón D, Gil JA, Santamaría RI (2004) Streptomyces lividans and Brevibacterium lactofermentum as heterologous hosts for the production of X22 xylanase from Aspergillus nidulans. Appl Microbiol Biotechnol 65:401–406
Gong N, Chen C, Xie L, Chen H, Lin X, Zhang R (2005) Characterization of a thermostable alkaline phosphatase from a novel species Thermus yunnanensis sp. nov. and investigation of its cobalt activation at high temperature. Biochim Biophys Acta 1750:103–111
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Henne A, Bruggemann H, Raasch C, Wiezer A, Hartsch T, Liesegang H, Johann A, Lienard T, Gohl O, Martinez-Arias R, Jacobi C, Starkuviene V, Schlenczeck S, Dencker S, Huber R, Klenk HP, Kramer W, Merkl R, Gottschalk G, Fritz HJ (2004) The genome sequence of the extreme thermophile Thermus thermophilus. Nat Biotechnol 22:547–553
Hidalgo A, Betancor L, Moreno R, Zafra O, Cava F, Fernández-Lafuente R, Guisán JM, Berenguer J (2004) Thermus thermophilus as a cell factory for the production of a thermophilic Mn-dependent catalase which fails to be synthesized in an active form in Escherichia coli. Appl Environ Microbiol 70:3839–3844
Katz E, Thompson CJ, Hopwood DA (1983) Cloning and expression of the tyrosinase gene from Streptomyces antibioticus in Streptomyces lividans. J Gen Microbiol 129:2703–2714
Kayser KJ, Kilbane JJ 2nd (2001) New host-vector system for Thermus spp. based on the malate dehydrogenase gene. J Bacteriol 183:1792–1795
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical streptomyces genetics. John Innes Centre, Norwich
Miller J (1972) Experiments in molecular genetics. Cold Spring Harbor, New York
Moreno R, Zafra O, Cava F, Berenguer J (2003) Development of a gene expression vector for Thermus thermophilus based on the promoter of the respiratory nitrate reductase. Plasmid 49:2–8
Moreno R, Haro A, Castellanos A, Berenguer J (2005) High-level overproduction of His-tagged Tth DNA polymerase in Thermus thermophilus. Appl Environ Microbiol 71:591–593
Pallen MJ, Chaudhuri RR, Henderson IR (2003) Genomic analysis of secretion systems. Curr Opin Microbiol 6:519–527
Pantazaki AA, Pritsa AA, Kyriakidis DA (2002) Biotechnologically relevant enzymes from Thermus thermophilus. Appl Microbiol Biotechnol 58:1–12
Pessela BC, Vian A, Mateo C, Fernández-Lafuente R, García JL, Guisán JM, Carrascosa AV (2003) Overproduction of Thermus sp. Strain T2 beta-galactosidase in Escherichia coli and preparation by using tailor-made metal chelate supports. Appl Environ Microbiol 69:1967–1972
Pessela BC, Torres R, Fuentes M, Mateo C, Filho M, Carrascosa AV, Vian A, García JL, Guisán JM, Fernández-Lafuente R (2004) A simple strategy for the purification of large thermophilic proteins overexpressed in mesophilic microorganisms: application to multimeric enzymes from Thermus sp. strain T2 expressed in Escherichia coli. Biotechnol Prog 20:1507–1511
Rodríguez S, Santamaría RI, Fernández-Ábalos JM, Díaz M (2005) Identification of the sequences involved in the glucose-repressed transcription of the Streptomyces halstedii JM8 xysA promoter. Gene 351:1–9
Ruiz-Arribas A, Fernández-Ábalos JM, Sánchez P, Garda AL, Santamaría RI (1995) Overproduction, purification, and biochemical characterization of a xylanase (Xys1) from Streptomyces halstedii JM8. Appl Environ Microbiol 61:2414–2419
Santamaría R, Ruiz-Arribas A, González Holgado G, Rodríguez S, Díaz M, Fernández-Ábalos JM (2005) Secuencia de nucleótidos promotora de la expresión génica derivada de la región promotora del gen xysA. Consejo Superior de Investigaciones Científicas (CSIC) España. ES 2 235–562
Sianidis G, Pozidis C, Becker F, Vrancken K, Sjoeholm C, Karamanou S, Takamiya-Wik M, van Mellaert L, Schaefer T, Anne J, Economou A (2006) Functional large-scale production of a novel Jonesia sp. xyloglucanase by heterologous secretion from Streptomyces lividans. J Biotechnol 121:498–507
Vallín C, Ramos A, Pimienta E, Rodríguez C, Hernández T, Hernández I, Del Sol R, Rosabal G, Van Mellaert L, Anne J (2006) Streptomyces as host for recombinant production of Mycobacterium tuberculosis proteins. Tuberculosis (Edinb) 86:198–202
Van Mellaert L, Anne J (2001) Gram-positive bacteria for the heterologous production of biopharmaceutical compounds. In: Van Broekhoven A, Shapiro F, Anne J (eds) Novel frontiers in the production of compounds for biomedical use. vol. 1. Kluwer Academic, New York, pp 277–300
Acknowledgments
This work has been supported by grants CSI02A05 from the Junta de Castilla y León to R. Santamaría and BIO2007-60245 and S0505/PPQ/0344 from the Ministry of Education and Science and the Comunidad Autónoma de Madrid, respectively, to J. Berenguer. Institutional grants from Fundación Ramón Areces to CBMSO and from Junta de Castilla y León are also acknowledged. We thank MJ Jimenez Rufo for her excellent technical work. Thanks are also due to N. Skinner for supervising the English version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Díaz, M., Ferreras, E., Moreno, R. et al. High-level overproduction of Thermus enzymes in Streptomyces lividans . Appl Microbiol Biotechnol 79, 1001–1008 (2008). https://doi.org/10.1007/s00253-008-1495-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-008-1495-1