
Abstract
Escherichia coli cells expressing Rhodococcus DK17 o-xylene dioxygenase genes were used for bioconversion of m-xylene. Gas chromatography–mass spectrometry analysis of the oxidation products detected 3-methylbenzylalcohol and 2,4-dimethylphenol in the ratio 9:1. Molecular modeling suggests that o-xylene dioxygenase can hold xylene isomers at a kink region between α6 and α7 helices of the active site and α9 helix covers the substrates. m-Xylene is unlikely to locate at the active site with a methyl group facing the kink region because this configuration would not fit within the substrate-binding pocket. The m-xylene molecule can flip horizontally to expose the meta-position methyl group to the catalytic motif. In this configuration, 3-methylbenzylalcohol could be formed, presumably due to the meta effect. Alternatively, the m-xylene molecule can rotate counterclockwise, allowing the catalytic motif to hydroxylate at C-4 yielding 2,4-dimethylphenol. Site-directed mutagenesis combined with structural and functional analyses suggests that the alanine-218 and the aspartic acid-262 in the α7 and the α9 helices play an important role in positioning m-xylene, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aromatic dioxygenases, which catalyze the initial reaction in many aerobic degradative pathways for aromatic compounds, are typically three-component enzyme systems consisting of a flavoprotein reductase, a ferredoxin containing a [2Fe-2S] or [3Fe-4S] center, and an oxygenase component containing a Rieske [2Fe-2S] center and nonheme iron (Fe2+). The reductase and ferredoxin components form a short electron transport chain that supplies electrons from NAD(P)H to the oxygenase component, which adds both atoms of molecular oxygen to the aromatic ring. The oxygenase component consists of a small subunit and a large subunit that contains the catalytic and substrate-binding domains (Ferraro et al. 2005; Kweon et al. 2008). During the past decade, aromatic dioxygenases have increasingly attracted interest, primarily due to their potential application as biocatalysts for regioselective and enantioselective synthesis of vicinal cis-dihydrodiols (Boyd and Sheldrake 1998; Hudlicky et al. 1999; Zhang et al. 2000; Boyd et al. 2001; Gakhar et al. 2005) and other oxygenated products such as catechols, epoxides, and phenolics (Nolan and O’Connor 2008).
The metabolically versatile Rhodococcus sp. strain DK17 was originally isolated for the ability to grow on o-xylene and has the capability to utilize aromatic compounds such as benzene, alkylbenzenes (toluene, ethylbenzene, isopropylbenzene, and n-propyl to n-hexylbenzenes), phenol, and phthalates as sole carbon and energy sources (Kim et al. 2002). The degradation of alkylbenzenes in DK17 is initiated by an o-xylene dioxygenase, which consists of a reductase (AkbA4), a ferredoxin (AkbA3) component, and a Rieske oxygenase component containing large and small subunits (AkbA1A2; Kim et al. 2004). The cloned akbA1A2A3 genes have been expressed in Escherichia coli and used in functional studies on the initial dioxygenation of various aromatic compounds. In many cases of heterologous expression of genes for dioxygenases in E. coli, the native reductase component may be left out as E. coli reductases may substitute for them (Haddad et al. 2001), and this is also the case for the DK17 o-xylene dioxygenase. The AkbA1A2A3 enzyme catalyzes dioxygenation on the aromatic ring of o-xylene, producing one form of dihydrodiol (o-xylene cis-3,4-dihydrodiol), or ethylbenzene, producing two different dihydrodiols (ethylbenzene cis-2,3- and cis-3,4-dihydrodiol; Kim et al. 2004). The same enzyme was also reported to oxidize the aromatic rings of p-xylene, biphenyl, and naphthalene, none of which can be used as a carbon and energy source by DK17, resulting in the formation of each corresponding cis-dihydrodiol (Kim et al. 2007). Here, we report benzylic and aryl hydroxylations of m-xylene by the DK17 o-xylene dioxygenase and present a functional modeling study of such reactions.
Materials and methods
Bioconversion experiments using o-xylene dioxygenase
Rhodococcus sp. strain DK17 (Korea patent no. 0469087) and the o-xylene dioxygenase gene sequences have been deposited in the Korean Culture Collection of Microorganisms (KCCM) and in the GenBank database under the accession numbers KCCM-10332 and AY502075, respectively. Chemically competent E. coli BL21(DE3) that had previously been transformed with recombinant pKEB051 containing the akbA1aA2aA3 genes (Kim et al. 2004) was cotransformed with the pKJE7 chaperone plasmid (Takara). A negative control strain was also developed by transformation of E. coli BL21(DE3) with pCR T7/CT-TOPO vector which was used to construct pKEB051. The resulting transformants were precultured by inoculating one colony into 20 ml LB medium supplemented with carbenicillin (100 μg/ml) and chloramphenicol (34 μg/ml) and incubating overnight at 37 °C. Four milliliters of the overnight culture was transferred to 200 ml LB plus carbenicillin/chloramphenicol and incubated under the same conditions. The culture was induced by addition of IPTG (1.0 mM) and arabinose (0.002%) when the cells reached an OD600 of approximately 0.6 and further incubated for 2 h at 37 °C. Cells were harvested by centrifugation at 10,000 × g for 15 min, washed twice with 50 mM potassium phosphate buffer (pH 7.4), and resuspended in 40 ml of the same buffer supplemented with 20 mM glucose plus carbenicillin and chloramphenicol. m-Xylene was provided in the vapor phase, and the bioconversion was carried out at 30 °C for 12 h.
Structural identification of m-xylene metabolites
E. coli cells were removed by centrifugation at 10,000 × g for 30 min, and the supernatant was extracted twice with an equal volume of ethyl acetate and dried by a rotary evaporator. Gas chromatography–mass spectrometry (GC-MS) analysis of m-xylene metabolites was performed using Perkin Elmer Clarus 500 MS (electron impact ionization, 70 eV) connected to Clarus 500 GC with an Elite-5 capillary column (0.25 mm × 30 m, 0.25 μm film thickness), under the following GC conditions: 1 ml He/min, on-column injection mode; oven temperature, 100 °C for 1 min; thermal gradient, 10 °C/min to 300 °C and then held at 300 °C for 10 min. For the detection of cis-dihydrodiol metabolites, the dried residue was derivatized with N-methyl-N-trimethylsilyltrifluoroacetamide as described previously (Kim et al. 2002). GC-MS analysis of trimethylsilyl ether derivatives was performed by a HP 5973 mass selective detector (70 eV) connected to a HP 6890 gas chromatograph fitted with a fused silica capillary column (HP-5, 0.25 mm × 30 m, 0.25 μm film thickness). The following conditions were used for the GC: 1 ml He/min, on-column injection mode; oven temperature, 60 °C for 2 min; thermal gradient 5 °C/min to 220–230 °C and then held at 220–230 °C for 10 min.
Molecular modeling and site-directed mutagenesis
The molecular structure of the Rieske oxygenase component large subunit of the DK17 o-xylene dioxygenase was predicted using the Swiss-Model Server (Schwede et al. 2003) based on the crystal structure of biphenyl oxygenase from Rhodococcus sp. strain RHA1 (PDB ID 1ULI). Site-directed mutagenesis was performed at A218, D262, L266, and V297 via overlap extension PCR as principally described by Sakamoto et al. (2001). The first round PCR reaction was performed: (1) using the AkbA1-forward primer (5′-ATGGAGTGGAGCATGTTG-3′) and A218-MID reverse (5′-GGTCCAGCCGAGGTGATAGGC-3′), D262-MID reverse (5′-CAAGCCGATAAGGCCCACACC-3′), L266-MID reverse (5′-CGGCGACCCGAACAAGCCGAT-3′) or V297-MID reverse (5′-GCACGAAGCGAGCGACCCGAA-3′) primers to amplify the 5′ portion of the akbA1A2A3 genes and (2) using the AkbA3- reverse primer (5′-TCATTGAGACTCGGCGCC-3′) and A218-MID forward (5′-GCCTATCACCTCGGCTGGACC-3′), D262-MID forward (5′-GGTGTGGGCCTTATCGGCTTG-3′), L266-MID forward (5′-ATCGGCTTGTTCGGGTCGCCG-3′), V297-MID forward (5′-TTCGGGTCGCTCGCTTCGTGC-3′) primers to amplify the 3′ portion of the genes. One microliter of each PCR reaction was combined for a second PCR reaction using only the AkbA1-forward and AkbA3-reverse primers. The final PCR products were cloned using the pCRT7/CT-TOPO TA expression kit (Invitrogen, USA) according to the manufacturer’s instructions. The thermal cycling program was a 10 min hot start (95 °C), 30 cycles of 30 s of denaturation (95 °C), 30 s of annealing (55 °C), and 1 min of extension (72 °C) and a final 10 min of extension (72 °C). The introduced mutations were confirmed by DNA sequencing.
Chemicals
Aromatic compounds used in this study were obtained from Sigma-Aldrich Korea (Korea). Solvents were purchased from Mallinckrodt Baker, Inc. (USA). All chemicals were analytical grade purity or above.
Results
Identification of m-xylene metabolites formed by the DK17 o-xylene dioxygenase
E. coli cells harboring the recombinant plasmid containing the DK17 o-xylene dioxygenase genes were cotransformed with a chaperone plasmid for higher expression and stable maintenance of o-xylene dioxygenase. Biotransformation experiments were performed as described in the “Materials and methods” section. GC-MS analysis of the original extract allowed detection of one major peak at 3.28 min (m-xylene metabolite I) and one minor peak at 3.38 min (m-xylene metabolite II) on the total ion chromatogram. Although both metabolites have the same molecular ions at m/z 122, they demonstrate different mass fragmentation patterns (Table 1). A subsequent GC-MS library search (NIST MS Search 2.0) tentatively identified metabolites I and II as 3-methylbenzylalcohol and 2,4-dimethylphenol, respectively. Indeed, the retention times and mass spectra of metabolites I and II were identical to those of authentic 3-methylbenzylalcohol and 2,4-dimethylphenol (Table 1). The relative amount of 3-methylbenzylalcohol and 2,4-dimethylphenol was determined to be a ratio of 9:1. This indicates that DK17 o-xylene dioxygenase has the ability to oxidize m-xylene through two different oxidative pathways, benzylic monohydroxylation and aryl monooxygenation, but favors the former. It should be noted that neither 3-methylbenzylalcohol nor 2,4-dimethylphenol were detectable from the culture of the negative control strain and the cell-free medium, which were incubated with m-xylene under the same conditions, respectively. The above results clearly show that o-xylene dioxygenase, to date known as an enzyme that dihydroxylates the aryl rings of aromatic compounds to cis-dihydrodiols, is also able to carry out a monohydroxylation reaction on the methyl substituent or aryl ring on m-xylene.
Molecular modeling of the DK17 o-xylene dioxygenase
An NCBI BLAST search revealed that the Rieske oxygenase component large subunit of the DK17 o-xylene dioxygenase (AkbA1) has significant sequence identity with iron sulfur protein large subunits (ISPLs) of several aromatic oxygenases, including a biphenyl dioxygenase from Rhodococcus sp. strain RHA1 (Furusawa et al. 2004), a toluene dioxygenase from Pseudomonas putida (Mosqueda et al. 1999), and a cumene dioxygenase from Pseudomonas fluorescens (Dong et al. 2005). The amino acid sequence alignment of these ISPLs clearly shows conservation of important cysteines and histidines in the [2Fe-2S] Rieske cluster and the mononuclear iron center (Fig. 1). Despite the relatively low identity (36%) in the primary amino acid sequences, it is still possible to build a structural model for the DK17 AkbA1 protein based on the crystal structure of the ISPL of a biphenyl dioxygenase from Rhodococcus sp. strain RHA1 (PDB ID 1ULI) using the Swiss-Model server. The residues surrounding a substrate at the active site are well conserved between the two proteins. This explains why the DK17 o-xylene dioxygenase can mediate dioxygenations of not only o-xylene, but also biphenyl or naphthalene (Kim et al. 2007).

Sequence alignment of an iron sulfur protein large subunit (ISPL) of o-xylene dioxygenase from Rhodococcus sp. strain DK17 and the most closely related proteins. BphA1 an ISPL of biphenyl dioxygenase from Rhodococcus sp. strain RHA1, TodC1 an ISPL of toluene dioxygenase from Pseudomonas putida, CumA1 an ISPL of cumene dioxygenase from P. fluorescens IP01, AkbA1 an ISPL of o-xylene dioxygenase from Rhodococcus sp. strain DK17. Numbering is according to the sequence of BphA1. The arrows and coils above the aligned sequences indicate secondary structural elements of BphA1. Circles indicate two cysteine–histidine pairs coordinated with the [2Fe-2S] Rieske center. Diamonds indicate residues associated with the mononuclear iron center. Triangles indicate the substrate-binding residues. Multiple alignment was visualized using ESPript (http://esprit.ibcp.fr/ESPript/ESPript/)
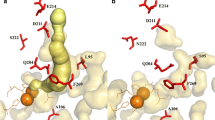
As shown in Fig. 2a, the RHA1 biphenyl dioxygenase holds a biphenyl molecule at a kink region between the α6 and α7 helices of its active site in a position where one side of the aromatic ring to be attacked is bordered by conserved D221 and H224 from α7 helix and Q217 and F218 from α6 helix. In addition, the side chain of H313, L323, and F368 surround the other side of the same aromatic ring. In contrast, the DK17 AkbA1 employs asparagine (N220) and serine (S299) at the positions corresponding to Q217 and H313, respectively. Since the substrate-binding pocket of AkbA1 is modeled to be large enough to accommodate a biphenyl, it would be spacious when a xylene isomer binds as a substrate. It is unlikely, however, that m-xylene locates at the active site with a methyl group facing the kink region because such a configuration would not fit within the substrate-binding pocket (Fig. 2b). The distance between biphenyl and the carbonyl group of D221 in the kink region is measured to be approximately 3.2 Å in the biphenyl-bound dioxygenase structure (PDB ID IULJ). In theory, m-xylene can be positioned in two other ways. In the first position, the m-xylene molecule flips horizontally to expose the meta-position methyl group to the catalytic motif (Fig. 2c). In this configuration, 3-methylbenzylalcohol could be formed as a product, presumably due to the meta effect. Alternatively, the m-xylene molecule can rotate counterclockwise from the position illustrated in Fig. 2c. This would allow the catalytic motif to hydroxylate at C-4, yielding 2,4-dimethylphenol as a product (Fig. 2d).

Molecular modeling of substrate binding in the DK17 AkbA1. a Biphenyl interaction at the active site of the RHA1 BphA1. The mononuclear iron and a water molecule in the active site are shown as spheres. Panels b, c, and d illustrate positioning of substrates within the active site of the DK17 AkbA1. Curved arrows indicate hydroxylation sites
Investigation for m-xylene-positioning residues
In order to test the hypothesis that the different positioning of m-xylene in the active site might cause the observed generation of two different products, the molecular model of the DK17 AkbA1 was examined for the potential residues affecting the positioning of the substrate. Subsequently, a total of four amino acid residues were selected for mutagenesis based on the proximity to the m-xylene molecule in the active site. Alanine, aspartic acid, leucine, and valine at positions 218, 262, 266, and 297 were changed to leucine (A218L), leucine (D262L), phenylalanine (L266F), and leucine (V297L) by site-directed mutagenesis.
The total amount of m-xylene metabolites produced by each mutant enzyme was almost equivalent to that of the wild-type enzyme. The mutant enzymes A218L and D262L were found to produce almost exclusively 3-methylbenzylalcohol (>99%) and 3-methylbenzylalcohol and 2,4-dimethylphenol in the ratio 7:3, respectively, while the mutant enzymes L266F and V297L produced them in the similar ratio as to the native enzyme (Fig. 3b). Based on the model shown in Fig. 2c, the methyl side chain of A218 in α7 helix locates closely to the ring structure of m-xylene. Thus, the substitution of the alanine at position 218 to leucine containing the larger isopropyl side chain is likely to hinder the approach of either of the methyl groups in m-xylene and result in increasing the preference for the m-xylene binding like in Fig. 2c. In the case of the mutant enzyme D262L, the hydrophobic side chain of the leucine allows a closer approach of a methyl group of m-xylene and promotes the production of 2,4-dimethylphenol. In contrast, a bulkier hydrophobic side chain in the mutant enzyme L266F seems not to affect m-xylene positioning although it was modeled to be in close proximity to either methyl group of m-xylene (Fig. 3a). Also, the fact that no change in the product ratio was observed with the mutant enzyme V297L suggests that the valine residue parallel to the ring plane hardly influence to the positioning of the methyl groups.

Generation and characterization of four site-directed mutants of the DK17 AkbA1. a A model for positioning m-xylene at the active site of wild-type AkbA1. A218, D262, L266, and V297 are the target residues for site-directed mutagenesis. b Comparison of relative amounts of m-xylene metabolites produced by the wild-type and mutant enzymes. Numbers above each bar represent the relative percentage of each metabolite. Relative metabolite amounts are the averages from at least three independent experiments
Discussion
2,4-Dimethylresorcinol was previously reported as a dead-end product formed when o-xylene induced cells of Rhodococcus sp. strain DK17 are incubated in the presence of m-xylene, although DK17 is unable to grow on m-xylene (Kim et al. 2003). The production of 2,4-dimethylresorcinol from m-xylene is somewhat surprising because an oxygen atom must be added to the aromatic ring of m-xylene between the two methyl groups. We previously proposed that this is achieved either through the action of a dioxygenase, forming a dihydrodiol that subsequently dehydrates to a phenolic compound, or through the action of a monooxygenase that directly hydroxylates or a combination of the two types of monooxygenase. No m-xylene dihydrodiols were detected despite thorough examination of all the significant peaks in the total ion chromatogram of both underivatized and derivatized m-xylene metabolites. This earlier observation, combined with the detection of 2,4-dimethylphenol in the present experiments, favors the hypothesis of two successive monohydroxylations.
Recently, Boyd and coworkers reported that toluene dioxygenase (TDO) from P. putida UV4 transforms m-xylene solely into 3-methylbenzylalcohol (Boyd et al. 2006). They explained this result in terms of the meta effect, which states that a substituent at a meta-position on a benzene ring does not allow binding and catalysis of arene cis-dihydroxylation of TDO. In contrast, the o-xylene dioxygenase from DK17 was found to have the ability to perform both benzylic and aryl hydroxylations of m-xylene. Furthermore, the detection of 3-methylbenzylalcohol and 2,4-dimethylphenol as m-xylene metabolites of DK17 o-xylene dioxygenase complements and reinforces the theory that arene cis-dihydroxylation by aromatic oxygenase enzymes is inhibited when the target compound contains a meta-substituent.
Prior to the present work, the DK17 o-xylene dioxygenase was known only to perform aryl dihydroxylation reactions. However, the rigorous structural identification of 3-methylbenzylalcohol and 2,4-dimethylphenol as oxidation products of m-xylene clearly shows that this enzyme is also able to perform benzylic and aryl hydroxylations. To our knowledge, 2,4-dimethylphenol has not previously been detected in the oxidation of m-xylene.
References
Boyd DR, Sheldrake GN (1998) The dioxygenase-catalysed formation of vicinal cis-diols. Nat Prod Rep 15:309–324
Boyd DR, Sharma ND, Allen CC (2001) Aromatic dioxygenases: molecular biocatalysis and applications. Curr Opin Biotechnol 12:564–573
Boyd DR, Sharma ND, Bowers NI, Dalton H, Garrett MD, Harrison JS, Sheldrake GN (2006) Dioxygenase-catalysed oxidation of disubstituted benzene substrates: benzylic monohydroxylation versus aryl cis-dihydroxylation and the meta effect. Org Biomol Chem 4:3343–3349
Dong X, Fushinobu S, Fukuda E, Terada T, Nakamura S, Shimizu K, Nojiri H, Omori T, Shoun H, Wakagi T (2005) Crystal structure of the terminal oxygenase component of cumene dioxygenase from Pseudomonas fluorescens IP01. J Bacteriol 187:2483–2490
Ferraro DJ, Gakhar L, Ramaswamy S (2005) Rieske business: structure-function of Rieske non-heme oxygenases. Biochem Biophys Res Commun 338:175–190
Furusawa Y, Nagarajan V, Tanokura M, Masai E, Fukuda M, Senda T (2004) Crystal structure of the terminal oxygenase component of biphenyl dioxygenase derived from Rhodococcus sp. strain RHA1. J Mol Biol 342:1041–1052
Gakhar L, Malik ZA, Allen CC, Lipscomb DA, Larkin MJ, Ramaswamy S (2005) Structure and increased thermostability of Rhodococcus sp. naphthalene 1, 2-dioxygenase. J Bacteriol 187:7222–7231
Haddad S, Eby DM, Neidle EL (2001) Cloning and expression of the benzoate dioxygenase genes from Rhodococcus sp. strain 19070. Appl Environ Microbiol 67:2507–2514
Hudlicky T, Gonzalez D, Gibson DT (1999) Enzymatic dihydroxylation of aromatics in enantioselective synthesis: expanding asymmetric methodology. Aldrichimica Acta 32:35–62
Kim D, Kim YS, Kim SK, Kim SW, Zylstra GJ, Kim YM, Kim E (2002) Monocyclic aromatic hydrocarbon degradation by Rhodococcus sp. strain DK17. Appl Environ Microbiol 68:3270–3278
Kim D, Kim YS, Jung JW, Zylstra GJ, Kim YM, Kim SK, Kim E (2003) Regioselective oxidation of xylene isomers by Rhodococcus sp. strain DK17. FEMS Microbiol Lett 223:211–214
Kim D, Chae JC, Zylstra GJ, Kim YS, Kim SK, Nam MH, Kim YM, Kim E (2004) Identification of a novel dioxygenase involved in metabolism of o-xylene, toluene, and ethylbenzene by Rhodococcus sp. strain DK17. Appl Environ Microbiol 70:7086–7092
Kim D, Lee JS, Choi KY, Kim YS, Choi JN, Kim SK, Chae JC, Zylstra GJ, Lee CH, Kim E (2007) Effect of functional groups on the regioselectivity of a novel o-xylene dioxygenase from Rhodococcus sp. strain DK17. Enzyme Microb Technol 41:221–225
Kweon O, Kim SJ, Baek S, Chae JC, Adjei MD, Baek DH, Kim YC, Cerniglia CE (2008) A new classification system for bacterial Rieske non-heme iron aromatic ring-hydroxylating oxygenases. BMC Biochem 9:11
Mosqueda G, Ramos-González MI, Ramos JL (1999) Toluene metabolism by the solvent-tolerant Pseudomonas putida DOT-T1 strain, and its role in solvent impermeabilization. Gene 232:69–76
Nolan LC, O'Connor KE (2008) Dioxygenase- and monooxygenase-catalysed synthesis of cis-dihydrodiols, catechols, epoxides and other oxygenated products. Biotechnol Lett 30:1879–1891
Sakamoto T, Joern JM, Arisawa A, Arnold FH (2001) Laboratory evolution of toluene dioxygenase to accept 4-picoline as a substrate. Appl Environ Microbiol 67:3882–3887
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385
Zhang N, Stewart BG, Moore JC, Greasham RL, Robinson DK, Buckland BC, Lee C (2000) Directed evolution of toluene dioxygenase from Pseudomonas putida for improved selectivity toward cis-indandiol during indene bioconversion. Metab Eng 2:339–348
Acknowledgment
This work was supported by a grant from the Ministry of Education, Science and Technology, of the Republic of Korea through the 21C Frontier Microbial Genomics and Applications Center Program and also by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2009-0079296). DK acknowledges the support of the Korea Polar Research Institute under project PE09050. KC is a recipient of the Brain Korea 21 scholarship.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Kim, D., Choi, K.Y., Yoo, M. et al. Benzylic and aryl hydroxylations of m-xylene by o-xylene dioxygenase from Rhodococcus sp. strain DK17. Appl Microbiol Biotechnol 86, 1841–1847 (2010). https://doi.org/10.1007/s00253-009-2418-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2418-5