
Abstract
An effective cell lysis method for extraction of bacterial genomic DNA from compost was developed in this study. Enzymatic disruption method, physical–chemical combination method, and commercial kit method were used to extract DNA from compost samples and were compared by analyzing DNA yield and efficient cell lysis. The results showed that all the three methods can be used to extract high-quality DNA from compost, but the enzymatic method had better cell lysis efficiency and DNA yields than others without the use of special equipment and expensive spending. Comparison of different methods for lysing gram-positive bacteria Bacillus subtilis indicated that the enzymatic cell lysis is superior for destroying the gram-positive cell wall. Spin-bind DNA column was used for DNA purification, and the purity of the purified sample was checked by polymerase chain reaction to amplify a region of the 16S rRNA. Results indicated that the part of 16S rRNA were amplified from all the purified DNA samples, and all the amplification products could be digested by the restriction enzyme HhaI.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Composting is an effective approach to degrade organic wastes from livestock farm and some other organic wastes like municipal refuses. It converts organic matter into useful products, like fertilizers which can be applied to the land without adversely affecting the environment (Goyal et al. 2005). A typical composting process goes through a series of stages, including rapid temperature increase, sustaining high temperature and gradual cooling of the composting mass. Gradients of oxygen, nutrients, and temperatures in compost support diverse microbial populations, which degrade organic matters and some complex polymers (Guo et al. 2007; Ishii et al. 2000). So there is an increased interest to the study of bacterial community in composting. But traditional cultivation-dependent approaches are based largely on isolation and plate count studies to analyze the microbial diversity. These classical culture-dependent methods are strongly biased because only a very small fraction (0.01–10%) of the microorganisms in natural environments can be identified (Amann et al. 1995; Torsvik et al. 1996). Molecular biology introduces new techniques to study microbial communities, especially unculturable microbes in various environments (Blanc et al. 1999; Cahyani et al. 2004; Franke-Whittle et al. 2005; Shan et al. 2008; Kowalchuk et al. 1999; Tiquia et al. 2005). They have been proven to be an advantageous technique and be applied widely. But, this molecular biological analysis of microbial community in compost requires efficient and unbiased DNA extraction methods which suffer from compounded inhibitory factors in the individual steps, such as incomplete cell lysis (Miller et al. 1999). Because the composting has specific conditions like high temperature, the live bacterial should be high-temperature-resistant. So it seems to be much more difficult to extract high-quality microbial DNA from compost than from other environmental samples.
The direct in situ lysis extraction method has been published for the extraction and purification of total community DNA from environmental samples such as soils and sediments (Bürgmann et al. 2001; Jiang et al. 2005; Luna et al. 2006; Martin-Laurent et al. 2001; Roose-Amsaleg et al. 2001). This method, which assumes complete in situ lysis of all microorganisms, generally provides the highest DNA yields within acceptable processing times.
Currently, three types of cell lysis (or membrane disruption) are used alone or in combination: (1) physical, (2) chemical, and (3) enzymatic disruption (Patrick et al. 2003). The physical methods have shown efficiencies for disruption of cells, but they often result in significant DNA shearing (Liesack et al. 1991; More et al. 1994). Many protocols including enzymatic lysis have been developed from soil and sediments (Niemi et al. 2001; Rochelle et al. 1992; Tsai and Olson 1991)
In compost, gram-positive bacteria which are more resistant than gram-negative bacteria are the predominant bacteria (Guo et al. 2007; Wakase et al. 2008). Here, we compared enzymatic-disruption method, physical–chemical combination method, and commercial kit method by analyzing total DNA yield, DNA purity, DNA recovery, and polymerase chain reaction (PCR) amplification. And a more effective lysis method for direct extraction of DNA from compost was developed.
Materials and methods
Sample collection and processing
The composting experiment was conducted on the Huazhong Agricultural University Swine Farm, which is subordinate to the National Engineering Research Center-Animal Science. A 1.2 × 1.0 × 1.0 m (length × width × height) composting bin was designed and built, which was covered to shield from sunshine and rain. The swine manure was collected from the swine farm, and sawdust was obtained from a local farm. About 0.5 kg of fresh sample was collected from surface of compost (20 cm to surface) containing swine manure and sawdust on day 16 when the temperature was 58°C. The collected samples were put into Ziploc and immediately placed into an adiabatic box, which was filled with ice for transportation to the laboratory, and then stored at −20°C.
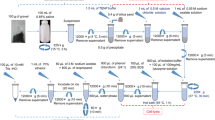
Cell lysis and DNA extraction
Enzymatic method (A)
The samples (0.2 g) were mixed with 1.0 ml extraction buffer (0.1 mol l−1 Tris-HCl, 0.1 mol l−1 ethylenediamine tetraacetic acid, 1.5 mol l−1 NaCl, pH 8.0) and 0.01 ml snailase solution (50 mg ml−1). After vortexing for 5 min, the mixtures were incubated in a 37°C water bath for 1 h. Then, 0.1 ml of 10% sodium dodecyl sulfate (SDS) and 0.01 ml proteinase K (10 mg ml−1) were added into the tubes and incubated in 65°C water bath for 1 h with agitation at a 15- to 20-min interval. Afterwards, the mixtures were centrifuged at 6,000×g for 5 min at room temperature. The supernatant was transferred to fresh tubes, and 0.5 ml sterile deionized water was added into the former tubes to wash the deposition with centrifugation (6,000×g, 1 min). Both supernatants were put together. All supernatants were mixed with 1× volume of chloroform/isoamyl alcohol (24:1) with gentle shaking. The aqueous layers were transferred to clean tubes after centrifugation (3,000 × g, 5 min) and were treated with 0.6 volume of isopropanol for 10 min at 4°C and centrifuged 10 min with 12,000×g. The pellets of crude DNA were washed twice with 700 μl ice-cold 70% ethanol and dried at room temperature. The crude DNA was dissolved in 100 μl deionized water and stored at −20°C for future research.
Physical–chemical method (B)
We modified the DNA extraction procedures based on the protocol described by Dees and Ghiorse (2001).The 0.2-g sample of subsamples were added to 1 ml of 0.1% sodium phosphoric acid (NaPP) and shaken for 30 min at room temperature on an orbital shaker at 220 rpm. The mixture was vibrated for 5 min then centrifuged for 10 min at 6,000×g. The precipitates were added to sterilized centrifugal vial containing 1.0 g of 0.1-mm diameter sterilized silica and zirconia beads. The vials received 1 ml each of: sodium phosphate buffer (100 mmol l−1NaH2PO4, pH 8.0); lysis buffer (100 mmol l−1 of NaCl, 500 mmol l−1 of Tris, pH 8.0, 10% SDS); and chloroform-isoamyl alcohol (24: 1). The mixture was vibrated for 10 min and centrifuged at 3,000×g for 5 min. The aqueous supernatant containing the community DNA was transferred to another sterile micro-centrifuge vial and were treated with 0.6 volume of isopropanol for 10 min at 4°C and centrifuged at 12,000×g for 10 min. The pellets of crude DNA were washed twice with 0.7 ml ice-cold 70% ethanol and dried at room temperature. The crude DNA was dissolved in 100 μl deionized water and stored at −20°C for future use.
Commercial kit method (C)
The Fast soil genome DNA isolation kit (Bioteke, Beijing) was used to extract DNA from compost according to the manufacturer's instructions.
Control extraction experiment was performed without compost following each protocol described above.
DNA extraction from gram-positive bacteria
The gram-positive bacteria Bacillus subtilis (China Center for Type Culture Collection no. M208122) was used as the control for comparing the efficiency of cell lysis methods. The B. subtilis was grown on beef extract peptone liquid medium. After collecting cell pellets by centrifuging at 8,000×g in 30 s from 10 ml pure cultures which contained 1010 cells ml−1 counted by plate count technique, the pellets were suspended with the DNA extraction buffer described above. Then, the DNA was extracted by the protocol described previously. DNA yield was employed to evaluate the efficiency of different cell lysis method. The DNA concentration was determined by the UV spectrophotometer.
DNA purification and recovery
The crude DNA of compost was precipitated by adding two volumes of anhydrous ethyl alcohol and 0.1 volume of 5 M NaCl. The samples were mixed by inverting gently and incubated for 10 min at 4°C. Then, the crude DNA was added into a SpinBind DNA column (Axygen Scientific, USA) for purification. The column was centrifuged for 1 min at 12,000×g. The pellet was washed twice with ice-cold 70% ethanol and centrifuged at 12,000×g for 1 min. And then, 200 μl Tris-Cl-Ethylenediaminetetraaceticacid buffer was added to the column, settled 1 min in room temperature, and centrifuged 1 min at 12,000×g. The DNA pellets were stored in −20°C.
The A 260/280 ratios and the concentrations of purified DNA were quantified on UV spectrophotometer. The DNA yields were estimated on the basis of at least three replicated determinations. The F test was carried out to analyze the variance of DNA yields that were extracted by the different methods.
Microscopic evaluation of cell lysis
Intact compost samples subjected to the lysis procedures were examined with a Zeiss Standard 18 microscope under phase contrast and epifluorescence viewing or a Zeiss laser scanning microscope (model LSM-10) equipped for fluorescence, phase, and differential interference contrast imaging under 488-nm light from an argon laser . The LSM-10 was configured so that a single field of view can be examined by conventional transmitted and epifluorescence illumination or by comparable laser-scanning illumination. Both microscopes were fitted with ×100 oil immersion objective lenses with numerical apertures of 1.3 or 1.4. An acridine orange direct count agar-smear procedure was used to assess the extent of lysis of the endospores and enumerate the total number of cells in the intact compost samples (More et al. 1994). Before DNA extraction, 0.1 g compost was added into 1 ml sterile water and was vortexed for 5 min. The coarse particles were allowed to settle for 1 min, and then a 0.1-ml subsample removed from the upper portion was added into 8.9 ml sterile water. One microliter of a freshly prepared 1% agar solution, cooled down to 50°C, was added, and the suspension was then vigorously shaken. Of the suspension, 0.01 ml was transferred onto a slide and quickly dried at 40°C. Dried smears were stained with 0.01% acridine orange and examined with a Zeiss Standard 18 microscope (Trolldenier 1973). The computerized imaging and analysis systems of the LSM-10 were used to document the size distribution of microbial cells surviving the various lytic procedures. The average count and standard deviation were computed from five duplicate smears prepared from three independent subsamples of the compost as described previously.
The cells per gram wet weight were calculated by the following formula:
Where E is the cells per gram wet compost; X is the cells per microscopic view field; S 1 is the area of smear (mm2); S 2 is the area of microscopic view field (mm2); V 1 is the volume of subsample of suspension which was transferred for smear (ml); n is the dilution factor; V is the volume of sterile water which the compost sample was added (ml); W is weight of compost sample (g).
After compost samples had suffered the lysis procedures, smears were prepared again, and the same general procedure was followed.
PCR amplification and restriction enzyme digestion
The purity of the purified sample was able to be checked by the ability of PCR to amplify a region of the 16S rRNA. Part of 16S rRNA gene was amplified by PCR using the universal primer pair 1492r (5′-TAC CTT GTT ACG ACT T-3′) and 27f (5′-AGA GTT TGA TCC TGG CTC AG-3′) (Dojka et al. 1998). The PCR reaction system was: 1.0 × 10−5 mmol of both primers, 1 μl genome DNA, 1.5 μl of 2 mmol l−1 dNTPs, 2.5 μl of 10× PCR buffer, 2.0 μl of 25 mmol l−1 MgCl2, 2.0 U of Taq DNA polymerase (1 U μl−1; Jingmei Biotech, Shenzhen), and 13 μl of sterile-filtered water. The PCR was done as follows: 94°C for 40 s, 55°C for 45 s, 72°C for 1 min 30 s, 30 cycles, then 72°C extension for 10 min, finally 25°C to terminate the reaction. The amplified products were electrophoretically separated and visualized in 1.0% agarose gels stained with ethidium bromide.
The PCR products were ligated into the pMD18-T Vector (Takara), and the ligation product was transformed into Escherichia coli DH5a competent cells. White colonies (Sambrook et al. 1989) were selected randomly, and the vector insert size was detected using M13⁄pUC universal primers P47 and P48.
About 10 μl of PCR product was digested with 1 U with HhaI restriction endonucleases (Fermentas) in 20 μl of the appropriate buffer for 12 to 16 h at 37°C, and the digested DNA fragments were electrophoresed in 1.5% agarose gels. After staining with ethidium bromide, the gels were photographed, and scanning image analyses were performed manually.
Result
DNA yield and recovery efficiency
Figure 1 shows the crude DNA extracted from compost after electrophoresis and staining with ethidium bromide. The F test was carried out using the data of the DNA yield. Because of the different cell lysis protocol, the DNA yields by the three methods were significantly different (P = 0.01). Of the three methods, method A yielded the most amount of DNA. The crude DNA was purified with the SpinBind column. Table 1 indicates that the DNA recovery efficiency of method B was obviously lower than A and C. The A 260/280 ratios of purified DNA were 1.7–1.8. This indicates that the DNA was of good quality. In control experiment, no DNA was detected.

Crude DNA extracted from compost. Lane 1 crude DNA extracted by method C, lane 2 crude DNA extracted by method B, lane 3 crude DNA extracted by method A, lane 4 DNA ladder
Comparison of different methods for lysing gram-positive bacteria
Because of the differences of the cell wall structure between gram-positive and gram-negative bacteria, the gram-positive bacteria are more difficult to destroy than gram-negative bacteria. So here, the gram-positive bacteria B. subtilis was used as the control organism to test the efficiency of cell lysis method. According to the DNA yields, the results indicate that method A was most efficient (Table 1).
Cell lysis efficiency
Cell count with the method of fluorescence microscopical enumeration was an effective approach for evaluating the effectiveness of cell lysis protocol. So the change of the cell counts before and after lysis was measured. After severe treatment with different method, there were still some small cells that survived. The data in Table 2 showed that after cell lysis, direct microscopic counts of three methods were considerably lower than before cell lysis. After treatment with snailase and proteinase K, the cells dropped to 1.8 ± 0.12 × 108 from 7.33 ± 0.75 × 109, and the efficiency of method A was 97.5%.
PCR amplification and restriction enzyme digestion
The amplification of the 16S rRNA genes was successful when DNA purified by all extraction methods was used as the PCR template. The results are shown in Fig. 2. No PCR product was observed with the negative control. Figure 3 indicates that the PCR products, which were amplified from the different 16S rRNA clone libraries, can be digested by HhaI. The different restriction fragment length polymorphisms (RFLP) are shown in Fig. 3.

16s rNA fragment amplified by primer pair 1492r and 27 f. Lane 1 (−) control no template DNA, lane 2 amplified with the DNA extracted by method A, lane 3 amplified with the DNA extracted by method B, lane 4 amplified with the DNA extracted by method C, lane 5 DNA ladder

Restriction enzymes digest of PCR-amplified products. Lane 1 HhaI digested with pMD 18-T Vector as a control, lane 2 16S rRNA PCR products from compost DNA extracted by method A digested with HhaI, lane 3 16S rRNA PCR products from compost DNA extracted by method B digested with HhaI, lane 4 16S rRNA PCR products from compost DNA extracted by method C digested with HhaI, lane 5 DNA ladder
Discussion
Although many studies have evaluated some DNA extraction protocols from environment samples (LaMontagne et al. 2002; Howeler et al. 2003; Yang et al. 2007; Zhou et al. 1996) and the cell lysis and DNA extraction efficiency is quite high, there are still some cells which have not been lysed especially some small, round cells (Yang et al. 2007). So an efficient cell lysis is a very important procedure for DNA extraction in compost. In this paper, we used snailase to destroy the cell wall of fungus (Jinxian et al. 1998; Shao and Sun 2007) to improve the lysis of recalcitrant bacteria.
DNA yield and cell lysis were employed by this study for the evaluation the effectiveness of DNA extraction and purification procedure. Among the three methods, method A has the highest cell lysis, which achieved 97.5%, compared with other studies (LaMontagne et al. 2002; Howeler et al. 2003; Yang et al. 2007), and obviously it is a better method. These results proved that the microorganism in compost were susceptible to snailase and proteinase K lysis. Method B is a chemical–physical method. It also shows an effective disruption (94.4%) percent of the bacterial cell wall, but it is limited to smaller cells and spores (Yang et al. 2007).
We have compared the DNA yields of gram-positive B. subtilis produced by the different extraction methods. Our findings show that higher DNA yields may be achieved by method A.
We found that using different extraction method will lead to different DNA yields. The quantity of DNA recovered using enzymatic cell lysis (method A) was much higher compared to physical–chemical disruption (method B). Because of the bead beating, method B was likely to cause DNA shearing, resulting in DNA decrease. Method A yielded about 66 ± 3.2 μg g−1, a little lower than the report of 83 ± 4.1 μg g−1 (Yang et al. 2007) but higher than other DNA yields that had been reported by 10–35 μg g−1 (Blanc et al. 1999) and 18.2 μg g−1 (Howeler et al. 2003). Assuming that bacteria were the predominant source of DNA in the compost and that each bacterium had a single stationary-phase genome weighing 5 × 10−15 g (based on data for E. coli) (Howeler et al. 2003), then theoretically, 1 g of compost might have DNA yields of about 35–39 μg in the samples. The result that the DNA yields obtained in the crude fraction were higher than the value may probably be due to greater genome DNA existence in the actively growing cells and eukaryotic cells.
In purification step, we used spin-bind cartridge to remove the humic acids and other contaminants. Because of the humic acids and other substances that may remain associated with the extracted DNA, it was difficult for molecular analysis, such as PCR amplification (Hilger and Myrold 1991; Tebbe and Vahjen 1993). So we performed the test including PCR amplification for 16S rRNA and restriction enzyme digestion to determine DNA purity. Figures 2 and 3 show that obtaining DNA from compost can be successfully amplified using eubacterial 16S rRNA primer pair of 27F and 1495R and digested by HhaI. Different RFLP types had been obtained indicating that the DNA extracted by different methods could be used for RFLP analysis.
The results of this study suggest that these three methods were suitable for DNA extraction from compost. Enzymatic lysis showed the best results with respect to cell lysis and DNA purity. Compared with enzymatic method, method B was effective in disrupting cell but shearing the DNA badly during the course of extraction led to a significant DNA loss. Method C was very expensive and not suitable for large-scale DNA extraction. So method A was the best choice to extract total DNA from compost without using any special equipment and expensive spending.
References
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Bürgmann H, Pesaro M, Widmer F, Zeyer J (2001) A strategy for optimizing quality and quantity of DNA extracted from soil. J Microbiol Methods 45:7–20
Blanc M, Marilley L, Beffa T, Aragno M (1999) Thermophilic bacterial communities in hot composts as revealed by most probable number counts and molecular (16S rDNA) methods. FEMS Microbiol Ecol 28:141–149
Cahyani V, Matsuya K, Asakawa S, Kimura M (2004) Succession and phylogenetic profile of eukaryotic communities in the composting process of rice straw estimated by PCR-DGGE analysis. Biol Fertil Soils 40:334–344
Dees PM, Ghiorse WC (2001) Microbial diversity in hot synthetic compost as revealed by PCR-amplified rRNA sequences from cultivated isolates and extracted DNA. FEMS Microbiol Ecol 64:3869–3877
Dojka MA, Hugenholtz P, Haack SK, Pace NR (1998) Microbial diversity in a hydrocarbon- and chlorinated-solvent contaminated aquifer undergoing intrinsic bioremediation. Appl Environ Microbiol 64:3869–3877
Franke-Whittle IH, Klammer SH, Insam H (2005) Design and application of an oligonucleotide microarray for the investigation of compost microbial communities. J Microbiol Methods 62:37–56
Goyal S, Dhull S, Kapoor K (2005) Chemical and biological changes during composting of different organic wastes and assessment of compost maturity. Bioresour Technol 96:1584–1591
Guo Y, Zhu N, Zhu S, Deng C (2007) Molecular phylogenetic diversity of bacteria and its spatial distribution in compost. J Appl Microbiol 103:1344–1354
Hilger AB, Myrold DD (1991) Method for extraction of Frankia DNA from soil. Agric Ecosyst Environ 34:107–113
Ishii K, Fukui M, Takii S (2000) Microbial succession during a composting process as evaluated by denaturing gradient gel electrophoresis analysis. J Appl Microbiol 89:768–777
Jiang J, Alderisio KA, Singh A, Xiao L (2005) Development of procedures for direct extraction of Cryptosporidium DNA from water concentrates and for relief of PCR inhibitors. Appl Environ Microbiol 71:1135–1141
Jinxian Luo, Li Z, Li W (1998) Expression and secretion of barley α-amylase and A. niger glucoamylase in Saccharomyces cerevisiae Science in China Series C. Life Sci 41:113–118
Kowalchuk GA, Naoumenko ZS, Derikx PJL, Felske A, Stephen JL, Arkhipchenko IA (1999) Molecular analysis of ammonia-oxidizing bacteria of the L subdivision of the class Proteobacteria in compost and composted materials. Appl Environ Microbiol 65:396–403
LaMontagne MG, Michael FC Jr, Holden PA, Reddy CA (2002) Evaluation of extraction and purification methods for obtaining PCR-amplifiable DNA from compost for microbial community analysis. J Microbiol Methods 49:255–264
Liesack W, Weyland H, Stackebrandt E (1991) Potential risks of gene amplification by PCR as determined by 16S rDNAanalysis of a mixed-culture of strict barophilic bacteria. Microb Ecol 21:191–198
Luna GM, Dell Anno A, Danovaro R (2006) DNA extraction procedure: a critical issue for bacterial assessment in marine sediments. Environ Microbiol 8:308–320
Martin-Laurent FPL, Hallet S, Chaussod R, Germon JC, Soulas G (2001) DNA extraction from soils: old bias for new microbial diversity analysie methods. Appl Environ Microbiol 67:2354–2359
Howeler M, Ghiorse WC, Walker LP (2003) A quantitative analysis of DNA extraction and purification from compost. J Microbiol Methods 54:37–45
Miller DN, Bryant JE, Madsen EL, Ghiorse WC (1999) Evaluation and optimization of DNA purification procedures for soil and sediment samples. Appl Environ Microbiol 65:4715–424
More MI, Herrick JB, Silva MC, Ghiorse WC, Madsen EL (1994) Quantitative cell lysis of indigenous microorganisms and rapid extraction of microbial DNA from sediment. Appl Environ Microbiol 60:1572–1580
Niemi RM, Heiskanen I, Wallenius K, Lindstrom K (2001) Extraction and purification of DNA in rhizosphere soil samples for PCR-DGGE analysis of bacterial consortia. J Microbiol Methods 45:155–165
Patrick Robe, Renaud Nalin, Carmela Capellano, Vogel TM, Simonet P (2003) Extraction of DNA from soil. European Journal of Soil Biology 39:183–190
Rochelle PA, Fry JC, Parkes RJ, Weightman AJ (1992) DNA extraction for 16S rRNA gene analysis to determine genetic diversity in deep sediment communities. Fems Microbiol Lett 79:59–65
Roose-Amsaleg CL, Garnier-Sillam E, Harry M (2001) Extraction and purification of microbial DNA from soil and sediment samples. Appl Soil Ecol 18:47–60
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: a Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, New York
Shan G, Jin W, Lam EKH, Xing X (2008) Purification of total DNA extracted from activated sludge. J Environ Sci 20:80–87
Shao Z, Sun F (2007) Intracellular sequestration of manganese and phosphorus in a metal-resistant fungus Cladosporium cladosporioides from deep-sea sediment. Extremophiles 11:435–443
Tebbe CC, Vahjen W (1993) Interference of humic acids and DNA extracted directly from soil in detection and tr. Appl Environ Microbiol 59:2657–2665
Tiquia SM, Ichida JM, Keener HM, Elwell DL, Burtt EH Jr, Michel F Jr (2005) Bacterial community profiles on feathers during composting as determined by terminal restriction fragment length polymorphism analysis of 16S rDNA genes. Appl Microbiol Biotechnol 67:412–419
Torsvik V, Sorheim R, Goksoyr J (1996) Total diversity in soil and sediment communities: a review. J Ind Microbiol 96:1584–1591
Trolldenier G (1973) The use of fluorescence microscopy for counting soil microorganisms Modern Methods in the Study of Microbial Ecology. Bull Ecol Res Comm 17:53–59
Tsai YL, Olson BH (1991) Rapid method for direct extraction of DNA from soil and sediments. Appl Environ Microbiol 57:1070–1074
Wakase S, Sasaki H, Itoh K, Otawa K, Kitazume O, Nonaka J, Satoh M, Sasaki T, Nakai Y (2008) Investigation of the microbial community in a microbiological additive used in a manure composting process. Bioresour Technol 99:2687–2693
Yang ZH, Xiao Y, Zeng GM, Xu ZY, Liu YS (2007) Comparison of methods for total community DNA extraction and purification from compost. Appl Microbiol Biotechnol 74:918–925
Zhou J, Burns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62:316–322
Acknowledgments
This study was financially supported by the National Key Technology R&D Program of China (2006BAD14A05-02) and (2008BADB2B00); National 863 Projects (2007AA10Z166); National Natural Science Foundation (30800797)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, L., Li, F., Deng, C. et al. A method for obtaining DNA from compost. Appl Microbiol Biotechnol 84, 389–395 (2009). https://doi.org/10.1007/s00253-009-2103-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2103-8