
Abstract
The prokaryotic diversity associated with organic household waste (OHW), leachate (start-up inoculum), and mesophilic anaerobic digestion processes in the degradation of OHW for 44 and 90 days was investigated using a culture-independent approach. Bacterial and archaeal 16S rRNA and mcrA gene clone libraries were constructed from community DNA preparations. Bacterial clones were affiliated with 13 phyla, of which Firmicutes, Proteobacteria, and Bacteroidetes were represented in all libraries, whereas Actinobacteria, Thermotogae, Lentisphaerae, Acidobacteria, Chloroflexi, Cyanobacteria, Synergistetes, Spirochaetes, Deferribacteres, and Deinococcus-Thermus were exclusively identified in a single library. Within the Archaea domain, the Euryarchaeota phylum was the only one represented. Corresponding sequences were associated with the following orders of hydrogenotrophic methanogens: Methanomicrobiales (Methanoculleus genus) and Methanobacteriales (Methanosphaera and Methanobacterium genera). One archaeal clone was not affiliated with any order and may represent a novel taxon. Diversity indices showed greater diversity of Bacteria when compared to methanogenic Archaea.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Millions of tons of household solid waste (HSW) are produced daily worldwide, and this has become a serious environmental, social, and public health problem for urban communities due to a lack of available areas for its disposal. Thus, one of the greatest challenges of contemporary society is to reduce and treat HSW.
Currently, HSW treatment has gained relevance due to an increased interest in renewable energy sources, as it combines the removal of organic pollutants and energy conservation in the form of biogas production (Lastella et al. 2002). Anaerobic digestion has been extensively and successfully applied to organic waste treatment over the past decades (Abegglen et al. 2008; Fernández et al. 2008). It takes place in four stages: hydrolysis, acidogenesis, acetogenesis, and methanogenesis, and is the consequence of a series of metabolic interactions among bacterial and archaeal groups. The first group of microorganisms secretes enzymes which hydrolyze polymers to monomers, such as glucose, amino acids, lactate, and ethanol. These are subsequently converted by acidogenic and acetogenic bacteria to H2 and volatile fatty acids. Finally, methanogenic Archaea convert H2, formate, and acetate to CH4 and CO2.
Biogas produced during methanogenesis is a natural source of energy. It can be collected and used to generate heat energy and electricity, reducing the consumption of fossil fuels, and commercialized as carbon credits in accordance with the Kyoto protocol (Yadvika et al. 2004).
The anaerobic process is very fragile and during digestion; it is necessary to maintain the balance among different microbial populations. The use of molecular tools to biomonitor digesters helps to know the microorganisms present during the process which later can be useful improving the efficacy of organic matter degradation, methane production, and stability of the process (Talbot et al. 2007).
The use of both 16S rRNA genes and functional gene clone libraries is a culture-independent approach that has been applied to detect the diversity of prokaryotes from different environmental samples. Previous culture-independent analyses of anaerobic reactors have been based on phylogenetic markers like the 16S rRNA and mcrA genes (Sasaki et al. 2006, 2007; Krause et al. 2008; Rastogi et al. 2008). The mcrA gene encodes a subunit of methyl-coenzyme M reductase, a key enzyme in the methane production pathway (Banning et al. 2005). However, little information on the dynamics of prokaryote communities from organic household waste treated in anaerobic reactors is available. In this study, the bacterial and archaeal diversity of organic household waste (OHW), leachate (LC, start-up inoculum), and OHW at two stages of treatment in a mesophilic anaerobic reactor was investigated by analyzing sequences of 16S rRNA genes and mcrA functional gene clone libraries, thus extending our understanding of the involvement of these microorganisms in the operation of anaerobic reactors.
Materials and methods
Reactor conditions and chemical analysis
Two continuous, 20 L reactors, with an internal diameter of 30 cm and a height of 40 cm, were built at the DEFLOR LTDA Company (Contagem, state of Minas Gerais, Brazil) to treat OHW. The reactors were operated for 90 days at 35°C (mesophilic), and pH was maintained around 6.5 to 7.5 by neutralizing with sodium carbonate. The first reactor was fed with 40% (w/v) OHW (fruit and meal leftovers, cut into pieces) only at the beginning of the treatment, and 60% (v/v) of leachate (used as start-up inoculum), collected from an OHW anaerobic reactor after 90 days of treatment, which had been previously inoculated with anaerobic sludge collected from the anaerobic reactor UASB of the Arrudas wastewater treatment station (Contagem, Minas Gerais state, Brazil). These conditions were similar to those of the first reactor previously described. The volume of mass (OHW+LC) in the reactor was 16 L, and pH was adjusted early on to 7.5. Prokaryote community structure was analyzed in the LC, OHW, and OHW treated for 44 and 90 days (OT44 and OT90, respectively). OHW+LC and OT90 samples were decanted and analyzed for pH, total solids (TS), fixed solids (FS), and volatile solids (VS). The measured cumulative production of biogas was investigated after treatment. Methane and carbon dioxide content was measured by gas chromatography.
DNA extraction
DNA was extracted from approximately 10 g (wet weight) of LC, OHW, OT44, and OT90 residues using the MegaPrep Power Max ™ Soil KIT (Mo Bio Laboratories) according to the manufacturer's instructions. This procedure is effective for removing polymerase chain reaction (PCR) inhibitors. Total DNA from samples was quantified by absorbance at 260 nm using a NanoDrop Spectrophotometer (NanoDrop Technologies). DNA purity was assessed with the A260/A280 and A260/A230 ratios. Isolated DNA was stored at −20°C until further processing.
PCR amplification and clone library construction
Bacterial and archaeal 16S rDNA and mcrA genes fragments were amplified by touchdown PCR. Reactions were carried out with the bacterial-targeted primer set 8F (5′-AGAGTTTGATYMTGGCTCAG-3′) and 907R (5′-CCGTCAATTCMTTTRAGT-3′; Lane 1991); and archaea-targeted primer set Arc21F (5′-CCGGTTGATCCYGCCGGA-3′) and Arc958R (5′-YCCGGCGTTGAMTCCAATT-3′; Delong 1992). To amplify the mcrA gene fragments from methanogenic Archaea, we used primer set mcrAF (5′-GGTGGTGTMGGATTCAC-3′) and mcrAR (5′-TTCATTCGRTAGTTWGGRTAGTT-3′; Luton et al. 2002). Polymerase chain reaction mixtures (20 μL) consisted of 10 μL of Hot Star Taq Master Mix KIT® 24 (Quiagen, CA 2005), 0.5 μM of each primer, and 20 ng of DNA. PCR was conducted with a Mini-cyclerTM PTC-100 (MJ Research Inc., Waltham, MA) with an initial cycle of 5 min at 94°C, followed by 18 cycles of denaturation at 94°C for 1 min, annealing beginning at 57°C and ending at 49°C for 1 min, and extension at 72°C for 3 min. Annealing temperature was lowered by 1°C every two cycles until it reached 49°C; 12 additional cycles were carried out at the last annealing temperature. The cycles were finished with an elongation step of 10 min at 72°C. Gene amplification was achieved after 30 thermal cycles for bacterial and archaeal 16S rRNA genes, whereas amplification of the mcrA gene was achieved using a two-round approach. Thus, a 1-μL aliquot of the first round PCR product was subjected to a second round of PCR. Amplicons were visualized on 1% agarose gel stained with ethidium bromide and purified directly with the QIAquick Gel extraction kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions.
For cloning, the purified amplicons were cloned into the PCR 4-TOPO vector from a TOPO TA Cloning Kit (Invitrogen, EUA) as per the manufacturer's instructions and transformed into electro competent Escherichia coli DH5α. At least 800 positive clones from each clone library were transferred to 96-well plates with Luria–Bertani medium containing ampicillin (100 μg mL−1) and 20% glycerol, incubated overnight at 37°C and stored at −80°C. Plasmids containing 16S rDNA and mcrA inserts were extracted, using a standard alkaline lysis method (Ausubel et al. 1990), and the inserts were subsequently PCR-amplified with vector-specific primers M13F (5′-GTAAAACGACGGCCAG-3′) and M13R (5′-CAGGAAACAGCTATGAC-3′).
16S rRNA, mcrA gene sequencing, and phylogenetic analysis
Sequencing was performed in a MegaBace 1.000 capillary sequencer (Amersham Biosciences) using standard protocols with a DYEnamic ET dye terminator kit (Amersham Biosciences). The sequences (800 bp for bacterial and archaeal 16S rDNA) were compared against sequences held in the Ribosomal Database Project (RDP) using Library Compare and against sequences held in GenBank using BLASTN to search for similar homologous sequences of 16S rRNA gene partial sequences in clone libraries. The mcrA gene (490 bp) was compared only to GenBank sequences. To accomplish this, the 16S rRNA gene sequences were base-called, checked for quality, aligned, and analyzed with Phred v.0.020425, Phrap v.0.990319 and Consed 12.0. RDP's CHECK-CHIMERA program (Maidak et al. 2001) was used to detect and omit chimeric DNAs. Phylogenetic relationships were inferred by MEGA 3.1 (Kumar et al. 2004) using the neighbor-joining method (Saitou and Nei 1987) and Kimura's 2-P model of sequence evolution. DOTUR (Schloss and Handelsman 2005) was used to assign sequences to operational taxonomic units (OTUs). Sequences were grouped into an OTU at distance values of 0.03 (≥97% sequence similarity), which correspond to species level (Stackebrandt and Goebel 1994). Nucleotide sequences generated were deposited in the GenBank database under accession numbers FJ98280–FJ982838, FJ982839–FJ982863, FJ982864–FJ982879, and FJ945735–FJ945748 for 16S rRNA gene sequences from LC, OHW, OT44, and OT90 Bacteria, respectively. Accession numbers FJ982799 and FJ982800 correspond to 16S rRNA gene sequences from LC Archaea, and FJ982880–FJ982898, FJ982899–FJ982904, and FJ982905–FJ982912 correspond to mcrA gene sequences from LC, OHW, and OT44, respectively.
Estimation of bacterial diversity and clone library similarity
The DOTUR program was applied to calculate rarefaction, richness, and diversity, as well to estimate the representation of the OTUs and to characterize the bacterial and archaeal diversity in the samples. The diversity indices included nonparametric richness estimators abundance-based coverage estimator (ACE) and Chao1 (Chao 1984), Shannon diversity index (1/D), and Shannon–Weaver diversity index. Rarefaction curves with 95% confidence intervals were constructed by comparing the number of clones in each 16S rRNA and mcrA gene library. The coverage (C) of each clone library, a measure of captured diversity, was calculated as: C = 1 − (n/N), where n is the number of different OTU types from a clone library that were encountered only once (Henriksen 2004), and N is the total sequenced number of clones in the library (Good 1953).
Phylogenetic diversity in the bacterial libraries was compared using LIBSHUFF (Singleton et al. 2001; Hughes and Bohannan 2004) and S-LIBSHUFF (Schloss et al. 2004). These programs use Good's coverage formula (Good 1953) to generate homologous and heterologous coverage curves (C X and C XY, respectively) for two 16S rDNA clone libraries (X and Y) at a similarity level or evolutive distance (D). To determine whether coverage curves C X (D) and C XY (D) were significantly different, the Crámer-von Mises (Pettitt 1982) statistic test was used to determine the distance between the two curves. Curves were considered significantly different if P < 0.05.
Results
Characterization of the samples and biogas production
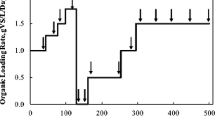
Total solids, volatile solids, fixed solids, and pH of the samples OHW+LC and OT90, before and after treatment in the mesophilic anaerobic reactor, are shown in Table 1. Biogas yield in the reactor reached 570 L over 90 days (Fig. 1). Methane and carbon dioxide content in the produced biogas at the end of the process were 37.5% and 42.5%, respectively.

Temporal variation of greenhouse gases production in an anaerobic reactor having organic household waste as substrate and leachate as inoculum
PCR amplification and clone library analysis
To determine the composition of the bacterial and archaeal communities, clone libraries were constructed from PCR-amplified 16S rRNA gene (bacterial and archaeal) and methanogenic Archaea mcrA gene fragments retrieved from the four samples. The archaeal 16S rRNA gene yielded a PCR product only for DNA from LC and Halobacterium salinarum ATCC 19700 (positive control); consequently, archaeal clone libraries could not be constructed for the samples from OHW, OT44, and OT90. Clone libraries were constructed where PCR amplification was successful. However, no OT90 clone library was obtained using the mcrA gene partial amplicon, despite four attempts to do so. Therefore, eight clone libraries from Bacteria (OHW, LC, OT44, and OT90), Archaea (LC), and methanogenic Archaea (OHW, LC, and OT44) were generated. The number of clones obtained in each library ranged from 800 to 2,000. A total of 601 clones from the eight aforementioned libraries were randomly picked and sequenced. Twenty-seven chimeric sequences were excluded from further analyses (Table 2).
Bacterial 16S rDNA clone library diversity
Phylogenetic analysis of the nucleotide sequences revealed a significant degree of bacterial diversity in the four clone libraries (Figs. 2, 3, 4, and 5). The LC library was most diverse, with few or no OTUs in common with the other libraries (Table 2, Figs. 2 and 6, and Electronic supplementary material, Table S 1). The bacterial libraries contained 93 distinct OTUs, which were affiliated to 13 different phyla. Most of the sequences had high identity to those of uncultured organisms obtained from various environments (Electronic supplementary material, Table S 1). OTUS from four libraries were affiliated with Firmicutes (47%), Proteobacteria (31%), and Bacteroidetes (11%), followed by Actinobacteria, Thermotogae, Lentisphaerae, Acidobacteria, Chloroflexi, Cyanobacteria, Synergistetes, Spirochaetes, Deferribacteres, and Deinococcus-Thermus detected at low frequency and exclusively in a single library (Fig. 6 and Electronic supplementary material, Table S 1).

Neighbor-joining tree deduced from bacterial 16S rRNA gene sequences of clone library of the LC sample. The numbers at the nodes indicate percentages of occurrence in 1,000 bootstrapped trees. Halobacterium salinarum (AJ496185) was used as an outgroup

Neighbor-joining tree deduced from bacterial 16S rRNA gene sequences of clone library of the OHW sample. The numbers at the nodes indicate percentages of occurrence in 1,000 bootstrapped trees. H. salinarum (AJ496185) was used as an outgroup

Neighbor-joining tree deduced from bacterial 16S rRNA gene sequences of clone library of the OT44 sample. The numbers at the nodes indicate percentages of occurrence in 1,000 bootstrapped trees. H. salinarum (AJ496185) was used as an outgroup

Neighbor-joining tree deduced from bacterial 16S rRNA gene sequences of clone library of the OT90 sample. The numbers at the nodes indicate percentages of occurrence in 1,000 bootstrapped trees. H. salinarum (AJ496185) was used as an outgroup

Phylogenetic distribution of bacterial 16S rDNA sequences generated from LC (a), OHW (b), OT44 (c), and OT90 (d) samples
The 44 OTUs affiliated with Firmicutes were represented by classes Clostridia and Bacilli in all libraries. Among the identified genera, namely Sporanaerobacter, Thermacetogenium, Clostridium, Leuconostoc, Bacillus, and Lactobacillus, the latter was most abundant (Figs. 2, 3, 4, and 5 and Electronic supplementary material, Table S 1).
All classes of Proteobacteria were present. The predominant γ-proteobacterial OTUs from a variety of environments were reported, as well as some that are closely related to cultured representatives of the genera Providentia, Klebsiella, Schineria, Escherichia, and Enterobacter. Other proteobacterial classes (α, β, δ, and ε) were represented by a small number of sequences (Figs. 2, 3, 4, and 5 and Electronic supplementary material, Table S 1).
The Bacteroidetes phylum was represented by ten OTUs, two of which belong to the genera Empedobacter and Flavobacterium. OTUs affiliated with the phyla Actinobacteria (two OTUs), Thermotogae, Spirochaetes, Lentisphaerae, Acidobacteria, Chloroflexi, Deferribacteres, Cyanobacteria, Synergistetes, and Deinococcus-Thermus (one OTU each) were all detected at low frequency (Figs. 2, 3, 4, and 5 and Electronic supplementary material, Table S 1).
Archaeal clone libraries diversity
Phylogenetic analysis of methanogenic archaeal (mcrA gene) and archaeal (16S rRNA gene) libraries (Figs. 7 and 8) revealed 33 and two distinct OTUs, respectively, that were assigned to the phylum Euryarchaeota (Fig. 9). Two hydrogenotrophic methanogenic orders, Methanomicrobiales and Methanobacteriales, are phylogenetically associated with this phylum. Most of the sequences had high identity to those of uncultured organisms obtained from biogas plants (Electronic supplementary material, Table S 2).

Neighbor-joining tree deduced from methanogenic Archaea mcrA gene sequences of clone libraries of the LC, OHW, and OT44 samples. The numbers at the nodes indicate percentages of occurrence in 1,000 bootstrapped trees. H. salinarum (AJ496185) was used as an outgroup

Neighbor-joining tree deduced from archaeal 16S rRNA gene sequences of clone library of the LC sample. The numbers at the nodes indicate percentages of occurrence in 1,000 bootstrapped trees. Methanopyrus kandleri (U57340) was used as an outgroup

Phylogenetic distribution of archaeal sequences generated from mcrA gene LC library (a), 16S rRNA gene archaeal LC library (b), mcrA gene OHW library (c), mcrA gene OT44 library (d)
Twenty-one out of 33 OTUS from the methanogenic archaeal libraries were affiliated to Methanomicrobiales, and all of the sequences were closely related to uncultured representatives of the genus Methanoculleus, whereas the OT44-6 OTU was affiliated with the species Methanoculleus bourgensis. Methanobacteriales was represented by 11 OTUs closely related to uncultured representatives with sequences similar to the genera Methanosphaera and Methanobacterium. Two OTUs (LC8 and LC12) were closely related to the species Methanosphaera stadtmanae (Fig. 7 and Electronic supplementary material, Table S 2).
The A-LC2 OTU from the LC archaeal library was related to an uncultured archaeal clone (EU591668, no accession), with 91% identity to Methanofollis formosanus, as inferred by BLAST analysis. However, it was not grouped with the clade of M. formosanus by phylogenetic tree analysis and was not related to any of the five orders of methanogenic Archaea (Fig. 8). Thus, this archaeon may potentially be classified as a new order. Another OTU (A-LC1) identified in this library was closely related to an uncultured Euryarchaeote clone with 99% identity to M. bourgensis (Fig. 8 and Electronic supplementary material, Table S 2).
Species richness and diversity
Rarefaction curves were obtained by plotting the number of OTUs observed against the number of clones sequenced for each of the eight libraries (supplementaryElectronic supplementary material, Fig. S 3A, B). A decrease in the rate of OTU detection was observed on every curve, indicating that the majority of diversity in every library might have been detected. This result was further supported by calculating the coverage of the libraries, which was >83% (Table 2).
The Shannon–Weaver and Simpson diversity indices, ACE, and Chao1 estimator values obtained for the eight libraries showed that the mcrA and bacterial 16S rRNA gene libraries from the LC sample had the highest diversity among the libraries (Table 2).
Comparison among the bacterial clone libraries
We applied the LIBSHUFF algorithm to assess whether observed differences in the LC and OHW bacterial community composition were statistically significant (supplementary Electronic supplementary material, Fig. S 4A, B). Pairwise comparisons by LIBSHUFF revealed that the bacterial community composition of the OHW and LC libraries differed significantly (OHW P = 0.005 and LC P = 0.002 for each combination). More information on the differences between clone libraries was obtained by examining the distribution of (C X − C XY)2 as a function of evolutionary distance (D). The coverage curves for representative pairs of clone libraries clearly showed major differences at low levels of genetic distance (D < 0.2). LIBSHUFF analysis also indicated that the LC library was characterized by a deeper divergence than the OHW library (Electronic supplementary material, Fig. S 4A, B).
Analysis using S-LIBSHUFF (Electronic supplementary material, Table S 5) to compare all bacterial libraries also showed a significant difference between the LC and OHW libraries (P < 0.003), in accordance with the LIBSHUFF analysis, whereas the remaining matchings were not significantly different from each other (P > 0.08).
Discussion
Work by several authors has revealed that the culture-independent approach provides insight into microbial community structure. In the present study, phylogenetic analysis of 16S rDNA and mcrA sequences was used to compare the microbial diversity of LC, OHW, and organic household waste treated at different stages in a mesophilic anaerobic reactor, detecting great bacterial diversity. To the authors' knowledge, this is the first phylogenetic evaluation of OHW and its degradation in an anaerobic reactor operating for 90 days.
Previous studies about microbial diversity in organic household waste anaerobic reactors have been reported (Tang et al. 2004; Levén et al. 2007; Weiss et al. 2008; Xian Qu et al. 2009). In our samples, the bacteria were associated to 13 phyla (Fig. 6). In other studies, using denaturing gradient gel electrophoresis (Weiss et al. 2008) and clone libraries construction followed by sequencing (Levén et al. 2007), the bacteria were associated with four and nine phyla, respectively. Lentisphaerae, Acidobacteria, Deferribacteres, and Deinococcus-Thermus were detected in our analysis but were not observed in the analyses of Levén et al. (2007). In addition to these phyla, Proteobacteria, Bacteriodetes, Chloroflexi, Cyanobacteria, and Spirochaetes found in our study were not detected by Weiss et al. (2008). In contrast, the candidate divisions of OD1 and OP9 were detected only by Levén et al. (2007). On the other hand, Methanosaeta and Methanosarcina, which are common in anaerobic reactors (Levén et al. 2007; Weiss et al. 2008; Xian Qu et al. 2009), were not identified in our archaeal and methanogenic archaeal libraries (Fig. 7 and 8). The direct analysis of 16S rRNA and mcrA genes through a PCR-based approach can potentially produce biased diversity estimations and underestimate the genetic diversity present in a given habitat (Moyer et al. 1994). Obviously, some factors such as the choice of primers and the proportion of 16S rRNA and mcrA gene sequences in a library may produce results that do not necessarily correspond to the actual proportion of these sequences in a natural community, but the qualitative complexity of a microbial community can be assessed (Suzuki et al. 1998).
In the bacterial 16S rDNA clone libraries, Firmicutes predominated in LC, OHW, and OT90 (Figs. 2, 3, and 5), whereas Proteobacteria occurred most frequently in the OT44 library (Fig. 4). This result could be compared to previous works (Lozada et al. 2004; Tang et al. 2004; Figuerola and Erijman 2007) which suggested that Firmicutes and Proteobacteria could be the most dominant groups in anaerobic reactors. Classes Clostridia and Bacilli were the most abundant in our samples (Figs. 2, 3, 4, and 5 and Electronic supplementary material, Table S 1). Bacilli are Gram-positive lactic acid bacteria, tolerant to low pH, which convert carbohydrates into organic acids, mainly lactic acid (Filya 2003). Clostridia ferment carbohydrates and cellulose, hydrolyze proteins, and grow in association with some methanogenic Archaea (Wiegel et al. 2005; Krause et al. 2008). Although most OTUs of Firmicutes and Proteobacteria from LC, OHW, and OT90 clone libraries are distinct, representatives of these phyla take part in the same stages of organic matter anaerobic digestion (hydrolysis and fermentation).
Proteobacteria includes several bacterial species with diverse phenotypes, lifestyles, and trophic capabilities (Kersters et al. 2003). γ-Proteobacteria predominated among the different proteobacterial classes (Figs. 2, 3, 4, and 5). It is worth noting that some members of the β-Proteobacteria that are known to be responsible for fixing nitrogen and oxidizing ammonia produced in large quantities in anaerobic reactors (Bond et al. 1995) were found in small numbers in our samples (Figs. 2, 3, and 4).
Bacteroidetes, which are typically strictly anaerobic degraders, are ubiquitous in a variety of environments and have been observed in wastewater reactors (Figuerola and Erijman 2007) and an agricultural biogas reactor (Krause et al. 2008). Deinococcus-Thermus, which was detected in the OT90 sample (Fig. 5), is known to be able to reduce compounds such as iron and manganese, and low concentrations of these metals are required for methanogenic Archaea to produce methane (Slobodkin 2005; Lovley 1991). The phylum Thermotogae, which was identified in the LC library (Fig. 2) and encompasses carbohydrate-degrading thermophilic species, was also detected at very low frequency in the mesophilic temperature reactor by Levén et al (2007). Other phyla were specific to a single library, such as Cyanobacteria (Fig. 3), Deferribacteres, Spirochaete, and Chloroflexi (Fig. 2), which were also identified in a household waste reactor by Levén et al. (2007). Lentisphaera and Acidobacteria (Fig. 2), which are found in acidic environments, have been identified in wastewater anaerobic reactors (Figuerola and Erijman 2007).
Phylogenetic analysis revealed greater diversity within the mcrA library than in the 16S rDNA library and showed that the archaeal clones were closely related to the hydrogenotrophic methanogens (Figs. 7 and 8). The differences in diversity within the mcrA and 16S rDNA libraries may be caused by an inadequately designed 16S rDNA primer set for detection of some species, although the archaeal 16S rDNA primer set used is potentially able to amplify representatives from all archaeal groups. In a balanced anaerobic reactor, the majority of methanogenic Archaea present are acetotrophic methanogenic Archaea, mainly Methanosaeta sp., and not hydrogenotrophic methanogenic Archaea, which are usually observed only in anaerobic reactors in an imbalanced condition. However, hydrogenotrophic methanogens appear to be important during the initial phase of waste treatment in anaerobic reactors (Montero et al. 2007). Identification only of this group was also reported in anaerobic reactors treating industrial waste under imbalanced conditions (Schnurer et al. 1999). This result can probably be explained by an inhibition of aceticlastic methanogens due to high accumulation of ammonium (Karakashev et al. 2005) and volatile fatty acids from hydrolysis of long-chain fatty acids and subsequent production of methane by a less sensitive hydrogenotrophic mechanism (Schnurer et al. 1999; Lalman and Bagley 2001). According to Speece (1983), aceticlastic and hydrogenotrophic methanogenic Archaea contribute with 70% and 30%, respectively, of methane production in industrial wastewater treatment. However, Angenent et al. (2002) reported that the major route of methane production is through a syntrophic relationship between acetate-oxidizing bacteria and hydrogenotrophic methanogens. Some members of the genera Thermacetogenium and Clostridium, which were identified in our libraries, are syntrophic bacteria and oxidize acetate in co-culture with hydrogenotrophic methanogens, as observed by Schnuërer et al. (1996) and Hattori et al. (2001).
In this study, the identification of Methanobacterium, Methanosphaera, and Methanoculleus, which were detected in great number in our libraries (Figs. 7 and 8), agrees with other community analyses in anaerobic reactors (Leclerc et al. 2004; Tang et al. 2004), suggesting that these microorganisms were responsible for hydrogenotrophic methanogenesis in such systems. The absence of acetotrophic methanogens could probably be caused by acid (acetate, propionate, and butyrate) and ammonium concentrations in the reactor that were above the threshold level for acetotrophic methanogen growth. Furthermore, the genera Methanoculleus, Methanosphaera, and Methanobacterium might compete for acetate with acetotrophic methanogens, given that hydrogenotrophic methanogens require acetate as their sole energy source for biomass production, despite not catabolizing acetate into methane (Boone et al. 2001).
The diversity indices (Table 2) indicated a high diversity level in the bacterial and methanogenic archaeal communities represented by the LC library and are in accordance with the rarefaction curves (Electronic supplementary material, Fig. S 3A, B). The results suggest that the number of analyzed clones encompassed the majority of the bacterial and archaeal diversity present in every sample. The S-LIBSHUFF and LIBSHUFF analyses showed a deeper divergence between the OHW and LC libraries. Unlike these results, according to the S-LIBSHUFF comparisons, these libraries did not show significant P values for the OT44 and OT90 samples nor between libraries OT44 and OT90, since the bacterial communities of the OT44 and OT90 samples were obtained from LC and OHW initial samples (Electronic supplementary material, Fig. S4A, B and Table S 5).
The microbial community changed during anaerobic digestion, and the results suggest that the production of methane was carried out mainly using CO2 as electron acceptor and hydrogen as electron donor via the hydrogenotrophic pathway. The fact that aceticlastic methanogens could not be identified, the low abundance of methanogens, the low percentage of methane in the biogas produced, and the low organic matter degradation in the anaerobic reactor treating OHW suggest that this reactor could have been under imbalanced conditions, which might have been caused by its being fed only at treatment start-up, as occurs in batch reactors.
References
Abegglen C, Ospelt M, Siegrist H (2008) Biological nutrient removal in a small-scale MBR treating household wastewater. Water Res 42:338–346
Angenent LT, Sung S, Raskin L (2002) Methanogenic population dynamics during startup of a full-scale anaerobic sequencing batch reactor treating swine waste. Water Res 36:4648–4654
Ausubel F, Brent R, Kingston R, Moore D, Seidman J, Smith H, Strujil K (eds) (1990) Current Protocols in Molecular Biology, vol 1. Greene Publishing Associates and Wiley–Interscience, New York
Banning N, Brock F, Fry JC, Parkes RJ, Hornibrook ER, Weightman AJ (2005) Investigation of the methanogen population structure and activity in a brackish lake sediment. Environ Microbiol 7:947–960
Bond PL, Hugenholtz P, Keller J, Blackall LL (1995) Bacterial community structures of phosphate-removing and non-phosphate-removing activated sludges from sequencing batch reactors. Appl Environ Microbiol 61:1910–1916
Boone DR, Whitman WB, Koga Y (2001) Methanomicrobiales. In: Boone DR, Castenholz RW (eds) Bergeys manual of systematic bacteriology, vol1. The archaea and the deeply and phototrophic bacteria, 2nd edn. Springer, New York, pp 246–267
Chao A (1984) Non-parametric estimation of the number of classes in a population. Scand J Stat 11:265–270
Delong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci 89:5685–5689
Fernández N, Díaz EE, Amils R, Sanz JL (2008) Analysis of microbial community during biofilm development in an anaerobic wastewater treatment reactor. Microb Ecol 56:121–132
Figuerola ELM, Erijman E (2007) Bacterial taxa abundance pattern in an industrial wastewater treatment system determinated by the full rRNA cycle approach. Environ Microbiol 7:1780–1789
Filya I (2003) The effect of Lactobacillus buchneri and Lactobacillus plantarum on the fermentation, aerobic stability, and ruminal degradability of low dry matter corn and sorghum silages. J Dairy Sci 86:3575–3581
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–262
Hattori S, Luo H, Shoun H, Kamagata Y (2001) Involvement of formate as an interspecies electron carrier in a syntrophic acetate oxidizing anaerobic microorganism in coculture with methanogens. J Biosci Bioeng 91:294–298
Hughes JB, Bohannan BJM (2004) Application of ecological diversity statistics in microbial ecology. In: Kowalchuk GA, de Bruijn FJ, Head IM, Akkermans AD, van Elsas JD (eds) Molecular microbial ecology manual, 2nd edn. Kluwer Academic, London, pp 1321–1344
Henriksen JR (2004) webLIBSHUFF (http://libshuff.mib.uga.edu)
Karakashev D, Batstone DJ, Angelidaki I (2005) Influence of environmental conditions on methanogenic compositions in anaerobic biogas reactors. Appl Environ Microbiol 71:331–338
Kersters K, De Vos P, Gillis M, Swings J, Vandamme P, Stackebrandt E (2003) Introduction to the Proteobacteria. In Dworkin M et al. (eds) The prokaryotes: an evolving electronic resource for the microbiological community, 3rd ed., release 3.12, 28 March 2003 Online Springer, New York, N.Y. http://link.springer-ny.com/service/books/10125
Krause L, Diaza NN, Edwardse RA, Gartemanna KH, Krömekea H, Neuwegera H, Pühlerd A, Runte KJ, Schlüterd A, Stoyea J, Szczepanowski R, Taucha A, Goesmanna A (2008) Taxonomic composition and gene content of a methane-producing microbial community isolated from a biogas reactor. J Biotechnol 136:91–101
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Lalman JA, Bagley DM (2001) Anaerobic degradation and methanogenic effects of oleic and stearic acids. Water Res 35:2975–2983
Lastella G, Testa C, Cornacchia G, Notornicola M, Voltasio F, Sharma VK (2002) Anaerobic digestion of semi-solid organic waste: biogas production and its purification. Energy Convers Manag 43:63–75
Lane DJ (1991) 16S/23S rDNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York, pp 115–148
Leclerc M, Delgenes JP, Godon JJ (2004) Diversity of the archaeal community in 44 anaerobic digesters as determined by single strand conformation polymorphism analysis and 16S rDNA sequencing. Environ Microbiol 6:809–819
Levén L, Anders R, Ericksson B, Schurer A (2007) Effect of process temperature on bacterial and archaeal communities in two methanogenic bioreactors treating organic household waste. FEMS Microbiol Ecol 59:683–693
Lozada M, Itria RF, Figuerola ELM, Babay PA, Gettar RT, de Túlio LA, Erijman L (2004) Bacterial community shifts in nonylphenoll polyethoxylates-enriched activated sludge. Water Res 38:2077–2086
Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction. Microbiol Mol Biol Rev 55:259–287
Luton PE, Wayne JM, Sharp RJ, Riley PW (2002) The mcrA gene as an alternative to 16S rRNA in the phylogenetic analyses of methanogen populations in landfill. Microbiology 148:3521–3530
Maidak BL, Cole JR, Lilburn TG, Parker CT Jr, Saxman PR, Farris RJ (2001) The RDP-II (Ribosomal database project). Nucleic Acids Res 29:173–174
Montero B, Garcia-Morales JL, Sales D, Solera R (2007) Evolution of microorganisms in thermophilic-dry anaerobic digestion. Bioresour Technol 99:3233–3243
Moyer GL, Dobbs FC, Karl DM (1994) Estimation of diversity and community structure through restriction fragment length polymorphism distribution analysis of bacterial 16S rRNA genes from a microbial mat at an active, hydrothermal vent system, Loihi Seamount, Hawaiit. Appl Environ Microbiol 60:871–879
Pettitt AN (1982) Cramer-von Mises statistic. In: Kotz S, Johnson NL (eds) Encyclopedia of statistical sciences. Wiley-Interscience, New York, pp 220–221
Rastogi G, Ranade DR, Yeole TY, Patole MS, Shouche YS (2008) Investigation of methanogen population structure in biogas reactor by molecular characterization of methyl-coenzyme M reductase A (mcrA) genes. Bioresour Technol 99:5317–5326
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sasaki K, Haruta S, Ueno Y, Ishii M, Igarashi Y (2006) Archaeal population on supporting material in a methanogenic packed-bed reactor. J Biosci Bioeng 102:244–246
Sasaki K, Haruta S, Ueno Y, Ishii M, Igarashi Y (2007) Microbial population in the biomass adhering to supporting material in a packed-bed reactor degrading organic solid waste. Appl Microbiol Biotechnol 75:941–952
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schloss PD, Larget BR, Handelsman J (2004) Integration of microbial ecology and statistics: a test to compare gene libraries. Appl Environ Microbiol 70:5485–5192
Schnuërer A, Schink B, Svensson BH (1996) Clostridium ultunense sp. nov., a mesophilic bacterium oxidizing acetate in syntrophic association with a hydrogenotrophic methanogenic bacterium. Int J Syst Bacteriol 46:1145–1152
Schnurer A, Zellner G, Svensson BH (1999) Mesophilic syntrophic acetate oxidation during methane formation in biogas reactors. FEMS Microbiol Ecol 29:249–261
Singleton DR, Furlong MA, Rathbun SL, Whitman WB (2001) Quantitative comparisons of 16S rDNA sequence libraries from environmental samples. Appl Environ Microbiol 67:4373–4376
Slobodkin AI (2005) Thermophilic microbial metal reduction. Microbiology 74:5501–5514
Speece RE (1983) Anaerobic biotechnology for industrial wastewater-treatment. Environ Sci Technol 17:416–427
Stackebrandt E, Goebel BM (1994) Taxonomic note: a place for DNA: DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J System Bacteriol 44:846–849
Suzuki M, Rappe MS, Giovannoni S (1998) Kinetic bias in estimates of coastal picoplankton community structure obtained by measurements of small-subunit rRNA gene PCR amplicon length heterogenity. Appl Environ Microbiol 64:4522-4529
Talbot G, Topp E, Palin MF, Massé DI (2007) Evaluation of molecular methods used for establishing the interactions and functions of microorganisms in anaerobic bioreactors. Water Res 3:513–537
Tang Y, Shigematsu T, Ikbal Morimura S, Kida K (2004) The effects of micro-aeration on the phylogenetic diversity of microorganisms in a thermophilic anaerobic municipal solid-waste digester. Water Res 38:2537–2550
Xian Qu Mazéas L, Vavilin VA, Epissard J, Lemunier M, Mouchel JM, Pin-jing He, Bouchez T (2009) Combined monitoring of changes in δ13CH4 and archaeal community structure during mesophilic methanization of municipal solid waste. FEMS Microbiol Ecol 68:236–245
Yadvika SH, Sreekrishnan TR, Kohli S, Rana V (2004) Enhancement of biogas production from solid substrates using different techniques—a review. Bioresource Technol 95:1–10
Weiss A, Jérôme V, Freitag R, Mayer HK (2008) Diversity of the resident microbiota in a thermophilic municipal biogas plant. Appl Microbiol Biotechnol 81:163–173
Wiegel J, Tanner R, Rainey FA (2005) An introduction to the family Clostridiaceae. In: Dworkin M (ed) The prokaryotes: an evolving electronic resource for the microbiological community, 3rd edn. Springer, New York
Acknowledgments
We appreciate the financial support provided by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) in the form of a scholarship to Juliana Cardinali Rezende. We are grateful to Defesa Florestal Limitada (DEFLOR/LTDA) for their technical support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table S1
(DOC 142 kb)
Supplementary Table S2
(DOC 71 kb)
Supplementary Table S5
(DOC 28 kb)
Supplementary Fig. 3A
Rarefaction curves of (A) archaeal and bacterial 16S rRNA genes in LC, OHW, OT44, and OT90 libraries and (B) archaeal mcrA gene in LC, OHW, and OT44 libraries. Clones were grouped into OTUs at a level of sequence similarity of ≥97%. A and B denote bacterial and archaeal clones, respectively. (GIFF 106 kb)
Supplementary Fig. 3B
Rarefaction curves of (A) archaeal and bacterial 16S rRNA genes in LC, OHW, OT44 and OT90 libraries and (B) archaeal mcrA gene in LC, OHW, and OT44 libraries. Clones were grouped into OTUs at a level of sequence similarity of ≥97%. A and B denote bacterial and archaeal clones, respectively. (GIFF 93 kb)
Supplementary Fig. 4A
Results of LIBSHUFF comparisons of bacterial clone libraries from LC and OHW samples. Homologous and heterologous coverage curves for bacterial 16S rRNA gene sequences are presented. (A) OHW as homologous coverage and LC as heterologous coverage; (B) LC as homologous coverage and OHW as heterologous coverage. Taxonomic distances (D) were calculated using the Jukes–Cantor correction. (GIFF 60 kb)
Supplementary Fig. 4B
Results of LIBSHUFF comparisons of bacterial clone libraries from LC and OHW samples. Homologous and heterologous coverage curves for bacterial 16S rRNA gene sequences are presented. (A) OHW as homologous coverage and LC as heterologous coverage; (B) LC as homologous coverage and OHW as heterologous coverage. Taxonomic distances (D) were calculated using the Jukes–Cantor correction. (GIFF 55 kb)
Rights and permissions
About this article
Cite this article
Cardinali-Rezende, J., Debarry, R.B., Colturato, L.F.D.B. et al. Molecular identification and dynamics of microbial communities in reactor treating organic household waste. Appl Microbiol Biotechnol 84, 777–789 (2009). https://doi.org/10.1007/s00253-009-2071-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2071-z