
Abstract
In tropical soils, diversity and biotechnological potential of symbiotic diazotrophic bacteria are high. However, the phylogenetic relationships of prominent strains are still poorly understood. In addition, in countries such as Brazil, despite the broad use of rhizobial inoculants, molecular methods are rarely used in the analysis of strains or determination of inoculant performance. In this study, both rep-PCR (BOX) fingerprintings and the DNA sequences of the 16S rRNA gene were obtained for 54 rhizobial strains officially authorized for the production of commercial inoculants in Brazil. BOX-PCR has proven to be a reliable fingerprinting tool, reinforcing the suggestion of its applicability to track rhizobial strains in culture collections and for quality control of commercial inoculants. On the other hand, the method is not adequate for grouping or defining species or even genera. Nine strains differed in more than 1.03% (15) nucleotides of the 16S rRNA gene in relation to the closest type strain, strongly indicative of new species. Those strains were distributed across the genera Burkholderia, Rhizobium, and Bradyrhizobium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Within the large family of plants Leguminosae (Fabaceae in the USA), there are many species capable of establishing symbiotic associations with bacteria which results in biological nitrogen (N2) fixation, a process responsible for the wide adoption of legumes as food crops, forages, green manures, and in forestry (Allen and Allen 1981; Polhill and Raven 1981). From the 1970s, the green revolution resulted in an increased use of N-fertilizers in agriculture, and applied research on N2 fixation went through some decades of relative ostracism. Exceptions were found in countries where N-fertilizers have always been very expensive and were usually related to the economical pressure for large-scale production of cash crops, as is the case of soybean in South America (Hungria et al. 2005, 2006a; Hungria and Campo 2007). However, an increased interest in biological N2 fixation is restarting and should increase in the coming years, due to concerns about greater water pollution by nitrate, ideals of a more sustainable agriculture, and especially, their increasing higher cost of N fertilizers. The use of N2-fixing legumes is also expected to increase, not only as cash crops or biofuel but also in the recovery and improvement of soil fertility; therefore, it is timely to obtain new information regarding legumes and diazotrophic symbiotic bacteria.
Searching for the most effective rhizobial strains for each legume is a labor- and time-consuming process involving the production of dozens of rhizobial cultures, greenhouse experiments, and field trials. In Brazil, considerable efforts have been expended for more than 40 years in selecting effective strains for several legumes. Efforts have also been made to create strong legislation to control the use of rhizobial strains in commercial inoculants, aiming at avoiding the spread of noneffective strains. Since 1975, inoculants commercialized in the country can only contain strains recommended by Brazilian public research institutions (Hungria and Campo 2007). To enforce the strain recommendation, a network of microbiology laboratories and inoculant industries was created in 1985, with the objective of identifying the most effective rhizobial strains for each legume species. Since then, the maintenance of the strains, and their distribution to the inoculant industry has been a responsibility of the “Rhizobium Culture Collection SEMIA” (Seção de Microbiologia Agrícola; IBP World Catalog of Rhizobium Collections #443 in the WFCC World Data Center on Microorganisms), at the Fundação Estadual de Pesquisa Agropecuária (FEPAGRO), Porto Alegre, Brazil.
Presently, there are in the SEMIA collection 142 elite rhizobial strains officially recommended for inoculant production in Brazil (MAPA 2006), but despite the economical importance, their genetic characterization is still very poor. The strains are classified only as Rhizobium and Bradyrhizobium, based on the host legume specificity and on the acid/alkaline reaction and fast/slow growth in medium-containing mannitol as carbon source (FEPAGRO 1999). In a first effort to characterize this collection, the 16S rRNA gene of 68 rhizobial strains was sequenced, and 49 of them were reclassified at the genus and/or species level (Menna et al. 2006).
In this study, 54 of the remaining elite strains contained in the SEMIA collection, officially authorized for the use in commercial inoculants for 47 legumes used as forages, grains, and trees were investigated, with two main objectives. The first was to confirm if the rep-PCR technique shows discriminatory power and reproducibility for fingerprinting rhizobia, helping in the program of quality control of culture collections and of inoculants. The second objective was to investigate the phylogeny and taxonomic position of those elite strains based on the sequencing of the 16S rRNA gene.
Materials and methods
Strains
Fifty-four strains from the Brazilian (SEMIA) culture collection of rhizobia were selected. Table 1 provides information about both the strains, and the 47 legume hosts for which they are recommended. Strains were provided by FEPAGRO, and their purity was verified on yeast extract–mannitol agar (YMA) medium (Vincent 1970) containing Congo red (0.00125%). Stocks were prepared on YMA and kept at −70℃ (under 30% glycerol) for long-term storage and at 4℃ as source cultures.
In the rep-PCR analysis, 14 reference strains were included, as follows: Bradyrhizobium japonicum strains USDA 6T (= LMG 6,138, = NZP 5,549, = ATCC 10,324, = DSM 30,131, = 3I1b6, = SEMIA 5,052, = RCR 3,425, = ACCC 15,032) and USDA 110; Bradyrhizobium liaoningense LMG 18230T (= 2,281, = USDA 3,622); Bradyrhizobium elkanii USDA 76T (= LMG 6,134, = NZP 5,531, = ATCC 49,852, = DSM 11,554); Rhizobium tropici type A CFN 299 (= USDA 9,039, = LMG 9,517, = UMR1026); R. tropici type B CIAT 899T (= USDA 9,030, = SEMIA 4,077, = UMR1899, = TAL 1,797, = HAMBI 1,163, = CM01, = ATCC 49,672, = BR322); R. leguminosarum strains USDA 2370T (= ATCC 10,004, = LMG 14,904), and USDA 2,671 (= RCR 3,644); Rhizobium etli CFN 42T (= USDA 9,032, = ATCC 51,251, = DSM 11,541); Ensifer (= Sinorhizobium) fredii USDA 205T (= ATCC 35,423, = DSM 5,851, = PRC 205); Ensifer meliloti USDA 1002T (= ATCC 9,930, = DSM 30,135); Mesorhizobium loti USDA 3471T (= ATCC 33,669, = ATCC 700,743, = DSM 2,626, = NZP 2,213); M. ciceri USDA 3383T (= ATCC 51,585, = DSM 11,540, = LMG 14,989, = UPM-Ca7), and Azorhizobium caulinodans ORS 571T (= USDA 4,892, = DSM 5,975, = LMG 6,465, = ORS 571). They were provided by USDA, Beltsville, USA and by the Centro de Ciencias Genómicas, Cuernavaca, Mexico. All strains from this study are deposited at the “Diazotrophic and Plant Growth Promoting Bacteria Culture Collection” of Embrapa Soja (http://bmrc.lncc.br) and also at the “Rhizobium Culture Collection SEMIA” (IBP World Catalog of Rhizobium Collections #443 in the WFCC World Data Center on Microorganisms) at the FEPAGRO.
DNA extraction and rep-PCR (BOX) genomic fingerprinting
Total genomic DNA of each strain was extracted as described by Kaschuk et al. (2006) and amplified by PCR with the primer BOX A1R (5′-CTACGGCAAGGCGACGCTGACG-3′; Versalovic et al. 1994). Amplification procedures were performed as described by Kaschuk et al. (2006), except for the reduction from 35 to 30 cycles of amplification. The 1-kb DNA marker (Invitrogen™) was included on the left, right, and in the center of each gel. The amplified fragments were separated by horizontal electrophoresis on 1.5% agarose gel, stained with ethidium bromide, visualized under UV radiation, and photographed.
Sequencing analysis of the DNA region coding for the 16S rRNA gene
The procedure for amplification and sequencing analysis of the 16S rDNA was performed as previously described (Menna et al. 2006). The sequencing analysis was performed on a MegaBACE 1,000 DNA Analysis System (Amersham Biosciences).
The high-quality sequences obtained for each strain were assembled into contigs as described before (Menna et al. 2006) and sequences confirmed in the 3′and 5′ directions were submitted to the GenBank database (http://www.ncbi.nlm.nih.gov/blast) to seek significant alignments. Accession numbers given to the 16S rRNA sequences of the fifty-four strains are listed in Table 2.
Cluster analyses
For the rep-PCR analysis, first, the sizes of the fragments in all analyses were normalized according to the sizes of the DNA markers. Cluster analyses of the BOX-PCR profiles were performed using the Bionumerics program (Applied Mathematics, Kortrijh, Belgium, version 4.6), with the UPGMA algorithm (unweighted pair-group method, with arithmetic mean; Sneath and Sokal 1973) and the Jaccard coefficient (Jaccard 1912), considering the optimum values indicated by the Bionumerics program for the tolerance and the optimization parameters.
For the 16S rRNA analysis, the sequences obtained were aligned pairwise and compared to those of the following type/reference strains (accession numbers of the GenBank Data Library in parentheses): B. elkanii USDA 76T (U35000); B. japonicum USDA 6T (U69638); B. liaoningense LGM 18230T (AF208513); B. canariense BC-C2T (AY577427); B. yuanmingense CCBAU 10,071 T (= CFNEB 101, = B071) (AF193818); B. betae PL7HG1T (= LMG 21,987, = CECT 5,829; AY372184); Burkholderia cepacia ATCC 53867T (AY741356); Mesorhizobium amorphae ACC 19665T (= RCAN13; DQ022832); M. ciceri USDA 3383T (U07934); M. loti USDA 3471T (X67229); M. tianshanense USDA 3592T (= A-1BS; AF041447); Methylobacterium nodulans ORS 2060T (= CNCM I 2,342, = LMG 21,967; AF220763); R. leguminosarum USDA 2370T (U29386); R. mongolense USDA 1844T (U89817); Rhizobium (= Agrobacterium) rhizogenes ATCC 11325T (= 163C, = DSM 30,148, = IFO 13,257, = IMET 11,180; AY945955.1); R. tropici CIAT 899T (U89832); R. lusitanum P1-7T (= CECT 7,016, = LMG 22,705; AY738130); E. fredii USDA 205T (X67231), E. meliloti USDA 1002T (X67222). Methanococcus maripaludis strain C6 (U38487) was used as an outgroup strain.
Multiple alignments were performed with ClustalX version 1.83 (Thompson et al. 1997). Phylogenetic trees were generated using MEGA version 3.1 (Kumar et al. 2004) with default parameters, K2P distance model (Kimura 1980), and neighbor-joining algorithm (Saitou and Nei 1987). Azospirilum brasiliense strain A154 was used as an outgroup for 16S rDNA phylogenies. Statistic support for tree nodes was evaluated by bootstrap (Felsenstein 1985) analyses with 1,000 samplings (Hedges 1992).
Results
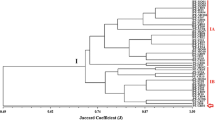
Complex fingerprinting patterns with multiple distinct bands of various intensities were obtained in the BOX-PCR analysis of the 54 elite rhizobial strains and allowed the identification of numerous well-defined groups with a high level of inter- and intraspecific diversity (Fig. 1). Furthermore, it is worth mentioning that the strains were analyzed three times, starting from the DNA extraction and resulted in band profiles showing good agreement, confirming the robustness of the method.

Cluster analysis (UPGMA with the coefficient of Jaccard) of the products obtained by BOX-PCR analysis. Type strains are labeled and SEMIA strains are classified according to the sequencing analysis of the 16S rRNA gene, as described on Table 2
Among the high diversity detected in the BOX-PCR analysis in this study, only two pairs of strains showed identical (100% similarity) fingerprints: (1) B. elkanii SEMIAs 6,389, recommended for Acacia podalyriifolia (subfamily Mimosoideae) and 6,403, recommended for four hosts, Enterolobium cyclocarpum, Pithecellobium guachapele, Samanea saman (Mimosoideae), and Poecilanthe parviflora (Papilionoideae) and (2) Rhizobium sp. SEMIAs 6,436 (host, Acacia farnesiana, Mimosoideae) and 6,438 (Adesmia latifolia, Papilionoideae). In addition, highly similar profiles (95% of similarity or higher) were obtained for SEMIA 6,437 (Adesmia latifolia) and SEMIAs 6,436 and 6,438, as well as for B. cepacia strains 6,417 and 6,422, both recommended for Mimosa flocculosa (Fig. 1).
In the cluster analysis of BOX-PCR profiles, all strains were grouped at a very low level of similarity, of about 20% (Fig. 1), highlighting the high genetic diversity of our rhizobial collection. The assigned genera and species in Fig. 1 were based on the 16S rRNA genes, as shown in Table 2. The comparison of BOX-PCR profiles (Fig. 1) with the classification based on the 16S rRNA (Table 2) shows a poor correlation between both sets of data. It is noteworthy that even strains belonging to the same genus showed high genetic variability in BOX-PCR profiles such that some clusters included strains occupying completely different taxonomic positions.
Similar to our previous definition that strains differing by more than 1.03% (15) nucleotides from the closest type strain could represent new species (Menna et al. 2006), nine strains from this study were denominated as “sp.” and some of them have differed by up to 35 nucleotides in relation to the closest type strain (Table 2).
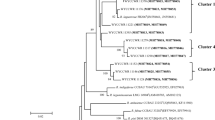
The phylogenetic tree obtained with the 16S rRNA-aligned sequences of the 54 rhizobial strains, as well as of the type and reference strains used in this study, is shown in Fig. 2. The strains were clustered into six main phylogenetic branches or well-defined main clusters (I–VI), with bootstrap support of 99%. In addition, there were some strongly bootstrap-supported subclusters and also a few isolated strains. Evidence of a new Rhizobium species was confirmed in the subcluster III.4, a cluster including strains SEMIAs 6,436, 6,437, and 6,438. In addition, Rhizobium sp. strains SEMIAs 6,411 and 6,435 were found to be closer to the R. tropici branch, showing differences of 32 and 23 bp in relation to the type strain, respectively. Cluster I grouped Methylobacterium strains, but clearly SEMIA 6,407 is highly different from M. nodulans type strain, a bacterium capable of fixing N2. However, strain SEMIA 6,407 has shown 99% similarity of bases with Methylobacterium mesophilicum (BLASTN), formerly classified as Pseudomonas mesophilica and not reported as being a diazotrophic bacterium; therefore, pathogenic and symbiotic genes of these strains should be investigated in more detail. Another interesting group of strains deserving further study is that formed by SEMIAs 6,396, 6,154, 6,395, and 6,439, positioned within subcluster II.2 (Fig. 1). These strains showed differences in relation to the closest type strains ranging from 12 to 16 bp, corresponding to 0.81% to 1.08% of the nucleotides (Table 2) and may well represent a new Bradyrhizobium species. Finally, Burkholderia sp. SEMIAs 6,410, 6,385, and 6,413 were grouped with strong bootstrap support (99%) as a subcluster of cluster VI also showing differences in relation to the closest type strain (Fig. 2).

Phylogenetic tree based on the 16S rRNA sequences of 54 strains, N2-fixing symbionts isolated from 47 legumes and officially recommended for the use in Brazilian commercial inoculants. GeneBank accession numbers of SEMIA strains (Table 2) and of type strains (“Materials and methods”) are given in the text
Discussion
In this study, we investigated a collection of 54 elite rhizobial strains, selected as the most effective in fixing N2 with 47 legume hosts. Some of the strains are characterized by high rates of N2 fixation with more than one legume; thus, our collection represents a total of 69 recommendations for the production of commercial inoculants in Brazil and in some other countries of South America.
The BOX-PCR technique has been broadly used (e.g., de Bruijn 1992; Alberton et al. 2006; Kaschuk et al. 2006; Barcellos et al. 2007; Batista et al. 2007; Menna et al. 2009) as a powerful fingerprinting tool, revealing strong genetic diversity among rhizobial strains. Indeed, also in this study, a very high level of diversity among the strains was detected in the BOX-PCR analysis, such that they were all joined at a final level of similarity of only 20%. In addition, high reproducibility of the BOX-PCR fingerprinting protocol was recently confirmed using a variety of rhizobial strains (Menna et al. 2009) and also confirmed in this study. Therefore, we validate a previous suggestion (Menna et al. 2009) that the method is reliable and represents an important tool for purposes other than detecting genetic diversity, including tracking of rhizobial stains in culture collections and programs of quality control of commercial inoculants. Indeed, based on the results from our group, including this study, the Brazilian government has decided to adopt BOX-PCR fingerprinting for such purposes.
Most rhizobial strain-selection programs worldwide have been performed based exclusively on symbiotic properties, partially because genetic methods for characterization of elite strains are rarely available. It is noteworthy that although some strains showing identical or very similar BOX-PCR profiles in this study were identified as the most very effective in fixing N2 for dissimilar legumes, some belong to different subfamilies of the Leguminosae family. In addition, the cluster analysis of BOX-PCR products showed no correlation with the host legumes, in accordance with previous reports from our group (Germano et al. 2006; Menna et al. 2006, 2009). Differences in the symbiotic plasmids or islands should be further investigated, but apparently, many tropical rhizobia and legumes are highly promiscuous.
Although the BOX-PCR analysis was highly effective in detecting genetic diversity, the correlation with the classification based on the 16S rRNA sequences was very poor. Therefore, the results from this study confirm previous reports (Laguerre et al. 1997; Mostasso et al. 2002; Grange and Hungria 2004; Hungria et al. 2006b; Menna et al. 2009) that BOX-PCR is a powerful means of fingerprinting and detecting high genetic diversity among rhizobial strains, but it is inadequate for grouping or defining species or even genera. However, we should mention that for other genera and species, that assumption might not be true, and one example was shown with Yersinia species, in which 16S rRNA sequencing and BOX-PCR were not efficient in differentiating Yersinia pseudotuerculosis from Yersinia pestis, while a clearer definition was achieved with REP- and ERIC-PCR (Kim et al. 2003). Finally, a recent suggestion for improving definition of the rhizobial genotypes and taxonomic groups that includes a polyphasic approach of BOX-PCR/16S rRNA (20/80%; Menna et al. 2009) deserves further consideration and could also apply to the definition of other genera and species.
Considering that strains differing by more than 1.03% (15) nucleotides from the closest type strain could represent new species (Menna et al. 2006), nine strains from this study might represent new species, including three Burkholderia sp., five Rhizobium sp., and one Bradyrhizobium sp. strain. The phylogenetic tree built with the 16S rRNA sequences highlights these putative new species.
It has long been suggested that rhizobia are more diverse in tropical than in temperate soils (e.g., Oyaizu et al. 1992; Urtz and Elkan 1996; Vinuesa et al. 1998, 2005; Doignon-Bourcier et al. 1999; Germano et al. 2006; Menna et al. 2006), and the fingerprinting and 16S rRNA results obtained in this study suggest that much diversity in symbiotic tropical diazotrophic bacteria remains to be revealed. However, in practical terms, the results from our study also point out the feasibility of recognizing and tracking rhizobial strains—including those commercially used in inoculants—by using BOX-PCR fingerprinting. In addition, the sequencing analysis of the 16S rRNA genes of the 54 strains contributed to understanding of the phylogenetic relationships among tropical rhizobia and revealed putative new species that will be subject to further study. The broad distribution of both rhizobia and legume hosts from this study across phylogenetic branches and the lack of relationship among micro and macrosymbiont clusters confirm previous observations (Germano et al. 2006; Menna et al. 2006) that the evolution of ribosomal and symbiotic genes might have occurred independently at least for the strains from this study.
References
Alberton O, Kaschuk G, Hungria M (2006) Sampling effects on the assessment of genetic diversity of rhizobia associated with soybean and common bean. Soil Biol Biochem 38:1298–1307
Allen ON, Allen E (1981) The Leguminosae: A source book of characteristics, uses and nodulation. The University of Wisconsin Press, Madison
Barcellos FG, Menna P, Batista JSS, Hungria M (2007) Evidence of horizontal transfer of symbiotic genes from a Bradyrhizobium japonicum inoculant strain to indigenous Sinorhizobium (Ensifer) fredii and Bradyrhizobium elkanii in a Brazilian savannah soil. Appl Environ Microb 73:2635–2643
Batista JSS, Hungria M, Barcellos FG, Ferreira MC, Mendes IC (2007) Variability in Bradyrhizobium japonicum and B. elkanii seven years after introduction of both the exotic microsymbiont and the soybean host in a cerrados soil. Microb Ecol 53:270–284
de Bruijn FJ (1992) Use of repetitive (repetitive extragenic palindromic and enterobacterial repetitive intergenic consensus) sequences and the polymerase chain reaction to fingerprint the genomes of Rhizobium meliloti isolates and other soil bacterial. Appl Environ Microb 58:2180–2187
Doignon-Bourcier F, Sy A, Willems A, Torck U, Dreyfus B, Gillis M, de Lajudie P (1999) Diversity of bradyrhizobia from 27 tropical Leguminosae species native of Senegal. Syst Appl Microbiol 22:647–661
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
FEPAGRO (Fundação Estadual de Pesquisa Agropecuária) (1999) Culture Collection Catalogue, 8th edn. FEPAGRO, Porto Alegre
Germano MG, Menna P, Mostasso FL, Hungria M (2006) RFLP analysis of the rRNA operon of a Brasilian collection of bradyhizobial strains from 33 legumes species. Int J Syst Evol Micr 56:217–229
Grange L, Hungria M (2004) Genetic diversity of indigenous common bean (Phaseolus vulgaris) rhizobia in two Brazilian ecosystems. Soil Biol Biochem 36:1389–1398
Hedges SB (1992) The number of replications needed for accurate estimation of the bootstrap p-value in phylogenetic studies. Mol Biol Evol 9:366–369
Hungria M, Campo RJ (2007) Inoculantes microbianos: situação no Brasil. In: Izaguirre-Mayoral ML, Labandera C, Sanjuan J (eds) Biofertilizantes en Iberoamérica: Visión Técnica, Científica y Empresarial. Cyted/Biofag, Montevideo, pp 22–31
Hungria M, Franchini JC, Campo RJ, Graham PH (2005) The importance of nitrogen fixation to soybean cropping in South America. In: Werner W, Newton WE (eds) Nitrogen fixation in agriculture, forestry, ecology and the environment. Springer, Dordrecht Amsterdam, pp 25–42
Hungria M, Chueire LMO, Megías M, Lamrabet Y, Probanza A, Guttierrez-Mañero FJ, Campo RJ (2006a) Genetic diversity of indigenous tropical fast-growing rhizobia isolated from soybean nodules. Plant Soil 288:343–356
Hungria M, Campo RJ, Mendes IC, Graham PH (2006b) Contribution of biological nitrogen fixation to the N nutrition of grain crops in the tropics: the success of soybean (Glycine max L. Merr.) in South America. In: Singh RP, Shankar N, Jaiwal PK (eds) Nitrogen nutrition and sustainable plant productivity. Studium Press LLC, Houston, pp 43–93
ILDIS (International Legume Database & Information Service) (2005) Retrieved April 28th. http://www.ildis.org
Jaccard P (1912) The distribution of flora in the alpine zone. New Phytol 11:37–50
Kaschuk G, Hungria M, Andrade DS, Campo RJ (2006) Genetic diversity of rhizobia associated with common bean (Phaseolus vulgaris L.) grown under no-tillage and conventional systems in Southern Brazil. Appl Soil Ecol 32:210–220
Kim W, Song M, Song W, Kim K, Chung S, Choi C, Park Y (2003) Comparison of 16 S rDNA analysis and rep-PCR genomic fingerprinting for molecular identification of Yersinia pseudotuberculosis. Ant van Leeuwenhoek 83:125–133
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar S, Tamura K, Nei M (2004) MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Laguerre G, van Berkum P, Amarger N, Prevost D (1997) Genetic diversity of rhizobial symbionts isolated from legume species within the genera Astragalus, Oxytropis, and Onobrychis. Appl Environ Microb 63:4748–4758
MAPA (Ministério da Agricultura, Pecuária e Abastecimento) (2006) Instrução normativa N 10, Retrieved March 21th. http://extranet.agricultura.gov.br/sislegis-consulta/consultarLegislacao.do?operacao=visualizar&id=16735
Menna P, Hungria M, Barcellos FG, Bangel EV, Hess PN, Martinez-Romero E (2006) Molecular phylogeny based on the 16S rRNA gene of elite rhizobial strains used in Brazilian commercial inoculants. Syst Appl Microbiol 29:315–332
Menna P, Pereira AA, Bangel EV, Hungria M (2009) rep-PCR of tropical rhizobia for strain fingerprinting, biodiversity appraisal and as a taxonomic and phylogenetic tool. Symbiosis 48 (1-3) in press
Mostasso L, Mostasso FL, Dias BG, Vargas MAT, Hungria M (2002) Selection of bean (Phaseolus vulgaris L.) rhizobial strains for the Brazilian Cerrados. Field Crop Res 73:121–132
Oyaizu H, Naruhashi N, Gamou T (1992) Molecular methods of analysing bacterial diversity: The case of rhizobia. Biodivers Conserv 1:237–249
Pinto FGS, Hungria M, Mercante FM (2007) Polyphasic characterization of Brazilian Rhizobium tropici strains effective in fixing N2 with common bean (Phaseolus vulgaris L.). Soil Biol Biochem 39:1851–1864
Polhill RM, Raven PH (1981) Advances in legume systematics. Royal Botanic Gardens, Kew
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sneath PBA, Sokal RR (1973) Numerical taxonomy. WH Freeman & Co, San Francisco
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–7882
Urtz BE, Elkan GH (1996) Genetic diversity among Bradyrhizobium isolates that effectively nodulate peanut (Arachis hypogaea). Can J Microbiol 42:1121–1130
Versalovic J, Schneider M, de Bruijn FJ, Lupski JR (1994) Genomic fingerprinting of bacteria using repetitive sequence based PCR (rep-PCR). Meth Mol Cell Biol 5:25–40
Vincent JM (1970) Manual for the practical study of root nodule bacteria. Blackwell, Oxford
Vinuesa P, Rademaker JLW, de Bruijn FJ, Werner D (1998) Genotypic characterization of Bradyrhizobium strains nodulating endemic woody legumes of the Canary Islands by PCR-Restriction Fragment Length Polymorphism analysis of genes encoding 16S rRNA (16S rDNA) and 16 S–23S rRNA intergenic spacers, repetitive extragenic palindomic PCR genomic fingerprinting, and partial 16S rRNA sequencing. Appl Environ Microbiol 64:2096–2104
Vinuesa P, Leon-Barrios M, Silva C, Willems A, Jarabo-Lorenzo A, Perez-Galdona R, Werner D, Martinez-Romero E (2005) Bradyrhizobium canariense sp. nov., an acid-tolerant endosymbiont that nodulates endemic genistoid legumes (Papilionoideae: Genisteae) from the Canary Islands, along with Bradyrhizobium japonicum bv. genistearum, Bradyrhizobium genospecies alpha and Bradyrhizobium genospecies beta. Int J Syst Evol Microbiol 55:569–575
Acknowledgments
The work was partially supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil), projects 552393/2005-3 and 577933/2008-6, PQ (300698/2007), and PNPD (558455/2008-5). The authors thank also Ligia Maria Oliveira Chueire (Embrapa Soja) for help in several steps of this work and Dr. Allan R.J. Eaglesham for helpful discussion. P. Menna acknowledges fellowships from CNPq (projects 505946/2004-1 and 558455/2008-5), and F.G. Barcellos acknowledges a pro-doc fellowship from CAPES.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Binde, D.R., Menna, P., Bangel, E.V. et al. rep-PCR fingerprinting and taxonomy based on the sequencing of the 16S rRNA gene of 54 elite commercial rhizobial strains. Appl Microbiol Biotechnol 83, 897–908 (2009). https://doi.org/10.1007/s00253-009-1927-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-1927-6