
Abstract
Based on the comparative study of the DNA extracts from two soil samples obtained by three commercial DNA extraction kits, we evaluated the influence of the DNA quantity and purity indices (the absorbance ratios A260/280 and A260/230, as well as the absorbance value A320 indicating the amount of humic substances) on polymerase chain reaction (PCR)-based denaturing gradient gel electrophoresis (DGGE) and a functional gene microarray used in the study of microbial communities. Numbers and intensities of the DGGE bands are more affected by the A260/280 and A320 values than by the ratio A260/230 and conditionally affected by the DNA yield. Moreover, we demonstrated that the DGGE band pattern was also affected by the preferential extraction due to chemical agents applied in the extraction. Unlike DGGE, microarray is more affected by the A260/230 and A320 values. Until now, the successful PCR performance is the mostly used criterion for soil DNA purity. However, since PCR was more influenced by the A260/280 ratio than by A260/230, it is not accurate enough any more for microbial community assessed by non-PCR-based methods such as microarray. This study provides some useful hints on how to choose effective DNA extraction method for the subsequent assessment of microbial community.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the past decade, a significant number of studies dealing with microbial diversity utilized molecular tools, among which DNA-based characterization techniques were very popular (Morris et al. 2002; Dorigo et al. 2005). These approaches are required to obtain DNA with sufficient quantity and purity, characterized by the absorbance ratios A260/280 and A260/230. However, the environmental samples, such as soil and sediment, often contain a high level of organic matters. Their DNA extraction presents a great challenge itself (Ogram 2000; LaMontagne et al. 2002). The most critical step for extracting DNA from such samples is the separation of DNA from coextracted humic substances since both of these are acid macromolecules (Niemi et al. 2001). As the widely reported impurities in nucleic acid extracts from soil and sediment (Wilson 1997), humic substances can inhibit the activity of the Taq DNA polymerase in polymerase chain reaction (PCR) (Tebbe and Vahjen 1993; Al-Soud and Radström 1998; Fortin et al. 2004) and reduce the specificity of DNA hybridization (Zhou et al. 1996; Cho and Tiedje 2001; Wu et al. 2001; Zhou and Thompson 2002). Therefore, the spectrometric value A320, which reflects the amount of coextracted humic substances (Miller 2001), is also an important purity index for DNA extracts while studying the soil and sediment samples.
To improve extraction and purification of total microbial community DNA from soils and sediments, numerous new methods have been investigated (Zhou et al. 1996; Kuske et al. 1998; Krsek and Wellington 1999; Miller et al. 1999; Bürgmann et al. 2001; Hurt et al. 2001; Roose-Amsaleg et al. 2001; Dong et al. 2006; Arbeli and Fuentes 2007). In recent years, commercial kits have also been increasingly utilized for DNA extraction and purification from soils and sediments (Martin-Laurent et al. 2001; Orphan et al. 2001; Tiquia et al. 2002; Kormas et al. 2003; Mumy and Findlay 2004; Klerks et al. 2006; Whitehouse and Hottel 2007; Mitchell and Takacs-Vesbach 2008). Due to the fact that the currently widely used approaches in the microbial community study, such as denaturing gradient gel electrophoresis (DGGE) analyses, often rely on PCR amplification (Dorigo et al. 2005), the evaluation of the suitable DNA extracts for subsequent assessment methods is mostly based on its suitability for PCR amplification. However, few studies have reported how the purity indices and quantity of DNA extracts, such as A260/280, A260/230, A320, and DNA yield, influence the PCR and, therefore, also the microbial community study.
Besides the PCR-based methods, microarray technology has shown, in recent years, a great potential to analyze microbial communities and metabolic functions (Stine et al. 2003; Taroncher-Oldenburg et al. 2003; Zhou 2003; Stahl 2004; Wu et al. 2004; Franke-Whittle et al. 2005; Schadt et al. 2005; He et al. 2007). Since total genomic DNA is generally used as target for microarray studies, effective and repeatable DNA extraction from the environment is, therefore, a key step (Schadt et al. 2005). Similar as in the PCR-based methods assessing the microbial community, few studies were carried out to evaluate the influence of the DNA purity indices and quantity on microarray analyses.
In this study, using three commercial DNA extraction kits, we isolated from two soil samples several DNA extracts representing different purities (characterized by A260/280, A260/230, and A320) and quantities and compared their representativeness of microbial populations by PCR-based DGGE and a functional gene microarray. Three independent replicates were included in each analysis, and the results were examined separately. Based on this comparative study, we evaluated the different influences of the purity indices and quantity of the DNA extracts on the PCR-based method (DGGE) and the microarray analyses in microbial community studies.
Materials and methods
Sampling
Soil samples were taken from a pilot-scale constructed wetland at the experimental site in Bitterfeld (Germany) for treating groundwater contaminated with complex organic matters. The hydrogeochemical characteristics of the study site, the process scheme, and the operation conditions of the constructed wetland were described previously (Weiss et al. 2001; Vogt et al. 2002; Braeckevelt et al. 2007). Briefly, this subsurface constructed wetland was composed of two tank segments filled with autochthonous quaternary aquifer material predominantly containing Bitterfeld mica sand (25%) and gravel (67%), which was embedded in lignite (10%) with an effective porosity of 28%. One of the tank segments was cultivated with Phragmites australis, while the other one was vegetation-free. The soil samples were collected from both segments 1 m from the inflow. The samples were stored at −80°C until analyses.
DNA extraction
For the two soil samples, three commercial DNA extraction kits were applied. All DNA extractions were performed following the manufacturer’s instructions with variations as indicated below.
The Bio101 extracts were obtained by using FastDNA Spin Kit for Soil (MP Biomedicals, LLC, Solon, OH, USA). To enhance the ability of the purification, 350 μL of Protein Precipitation Solution (PPS) was used, instead of 250 μL as indicated in the manufacturer’s protocol.
The UltraClean and PowerSoil extracts were obtained by using UltraClean Soil DNA Isolation Kit and PowerSoil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA). The PowerSoil method distinguishes itself from the UltraClean method by a humic substance/brown color removal procedure.
In addition, the unplanted soil sample was also extracted by using a modified protocol of PowerSoil in which the cells were disrupted by the FastPrep instrument used in the Bio101 kit. The rest of the DNA extraction steps were performed according to the original manufacturer’s protocol.
Prior to the subsequent molecular analyses, the DNA extracts from Bio101 and UltraClean were purified with the Wizard DNA Clean-Up System (Promega, Madison, WI, USA).
DNA quantification
The DNA purity indices, such as A260/280, A260/230, and A320, were quantified by the NanoDrop Spectrophotometer (ND-1000, NanoDrop Technology, Wilmington, DE, USA).
DNA yield was quantified by the PicoGreen assay (Molecular Probes, Eugene, OR, USA). The PicoGreen assay uses an ultrasensitive fluorescent nucleic acid stain for quantifying double-stranded DNA in the DNA extracts (Ahn et al. 1996; Rengarajan et al. 2002). A standard curve was constructed by a dilution series of 0, 0.004, 0.01, 0.04, 0.1, and 0.4 ng/μL of standard lambda phage DNA (100 ng/μL). For each sample, triplicate 50-μL aliquots of the DNA extracts were transferred to a Costar Plate (NUNC, Roskilde, Denmark). The plates were sealed and shaken (IKA–Schüttler MTS 4, AL-Labortechnik, Amstetten, Germany) for 5 min. After adding 50 μL of 1x PicoGreen solution to the DNA extracts, the plate was sealed and shaken again for 5 min. After incubation, fluorescence of DNA extracts was measured at 520 nm after excitation at 480 nm using a CARY Eclipse fluorescence photometer (Varian, Mulgrave, Australia). DNA concentration was calculated from the standard curve. During the whole procedure, all samples were protected from light to avoid photobleaching.
PCR amplification of general 16S rRNA gene fragments for DGGE analysis
The 16S rRNA gene fragments were amplified using the primer pair F968GC (5′-CGCCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACGGGGGGAACGCGAAGAACCTTAC-3′) and L1401 (5′-CGGTGTGTACAAGACCC-3′) (Nübel et al. 1996). Because of the lower purity, PCR of the DNA extracts from the unplanted soil was performed with a dilution series (0, 1:5, 1:10, and 1:100). According to Liebich et al. (2003), the reaction mixture (50 μL) was composed of 1 μL template DNA, 5 μL 10x Thermo-Start Standard buffer (ABgene), 2 μL 20 mM dNTP Mix (ABgene), 2.5 μL dimethyl sulfoxide (Sigma), 2 μL each primer (10 pmol/μL, MWG Biotech), 0.4 μL Thermo-Start DNA Polymerase (5 U/μL, ABgene), and 35.1 μL sterile water (18.2 MΩcm, Milli-Q 185 Plus, Millipore). After 10 min of denaturation at 95°C, 35 cycles of 1 min at 95°C, 1 min at 54°C, and 1 min at 72°C were carried out. A final extension step of 7 min at 72°C was used to complete the reaction. Products were controlled by electrophoresis in 1.2% agarose gels after ethidium bromide staining. For the following DGGE investigation, the PCR products were purified with the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany).
Denaturing gradient gel electrophoresis
The DGGE system (Dcode universal mutation Detection System; Bio-Rad, Hercules, CA, USA) was used according to the manufacturer’s instructions. Denaturing gradients of 40% to 60% denaturant (100% denaturant correspond to 7 M urea plus 40% [v/v] of deionized formamide) were prepared. The 6% acrylamide gels were polymerized for 3–4 h. Purified PCR products (100 ng) were loaded onto the gel and DGGE was performed in 1x TAE buffer at 60°C and a constant voltage of 70 V for 16 h. Gels were silver-stained according to the following protocol: After three times repeated fixation in 250 mL buffer containing absolute ethanol (10%, v/v) and acetic acid (0.5%, v/v) for 3 min, the gels were rinsed thoroughly with Milli-Q water. The gels were submersed in 250 mL of a 0.1% (w/v) silver nitrate solution for 10 min and rinsed twice with Milli-Q water to remove excess silver. The silver-stained gels were developed in 250 mL of a solution containing 1.5% NaOH (w/v), 0.01% NaBH4 (w/v), and 0.15% formaldehyde (v/v), until the bands were visible (∼20 min). To stop further staining, the gels were fixed with an aqueous solution of 0.75% Na2CO3 (w/v) for 10 min. Finally, the gels were scanned using ScanMaker i900 (Microtek, Hsinchu, Taiwan) and analyzed by Quantity One (Bio-Rad, Hercules, CA, USA).
To compare the similarity of the DGGE band patterns of the extracts from the same soil sample, the number of the bands was counted and their similarity indices were calculated with Dice’s similarity coefficients (Suchodolski et al. 2004).
Microarray techniques
Microarray chips
The functional gene microarray used in this study consisted of about 2,000 oligonucleotide probes of 50 base pair length. The microarray was designed following the procedure described by Schadt et al. (2005). Briefly, probes targeting key microbial functional genes involved in carbon, nitrogen, sulfur, and phosphorus metabolism as well as organic contaminant degradation and heavy metal resistance were designed from sequence information in the National Center for Biotechnology Information database or from clone libraries at the Environmental Science Division of Oak Ridge National Laboratory (Oak Ridge, TN, USA) using a modified version of the software PRIMEGENS (Xu et al. 2002). Based on global optimal alignments, segments of 50 bases length which had <90% nucleotide identity to the corresponding aligned regions of any of the Basic Local Alignment Search Tool hit sequences were selected as potential probes, considering also predicted probe–target melting temperature, probe self-complementarities. All probes met the probe design criteria as proposed by Liebich et al. (2006), which are the simultaneous consideration of percent similarity (≤90%), the length of identical sequence stretches (≤20 bases), and the binding free energy (≥−35 kcal mol−1). The oligonucleotide probes were synthesized by MWG Biotech (High Point, NC, USA) and were printed in duplicate spots onto aminopropyl silane-coated glass slides (UltraGAPS, Corning, Corning, NY, USA).
Hybridization
DNA extracted from the soil samples was labeled with Cy5-dUTP (GE Healthcare, Piscataway, NJ, USA) by using BioPrime Array CGH Genomic Labeling System (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols, which followed the principle of random priming labeling method. The hybridization was performed in hybridization chambers (Corning Incorporated, Acton, MA, USA). The prehybridization solution consisted of 50% formamide (v/v), 25% 20x SSC (v/v), 10 μL 10% sodium dodecyl sulfate (SDS), and 1% 10 mg/μL bovine serum albumin (v/v). Prehybridization was carried out at 45°C for 45 min followed by a washing procedure as described below. For hybridization of the wetland DNA, 10 μL formamide, 2.82 μL 20x SSC, 0.56 μL 10% SDS, 1.41 μL sperm DNA, 0.17 μL dithiothreitol, and 0.17 μL spermidine were combined with 4.86 μL labeled Cy5-DNA and denatured at 95°C for 5 min. The hot mixture was applied to the microarray slide. In the hybridization chamber, the mixture was incubated at 45°C overnight. In the posthybridization washing procedure, the arrays were washed at first in solution containing 2x SSC and 0.1% SDS at 45°C for 5 min with gentle shaking. After washing two times in a solution containing 0.1x SSC and 0.1% SDS at room temperature for 5 min with gentle shaking, the arrays were transferred to a solution containing 0.1x SSC and incubated at room temperature for 5 min with gentle shaking. Then the arrays were rinsed in a 0.01x SSC solution for 10 s and dried by blowing clean compressed nitrogen. The slides were scanned in a laser scanner (GenePix 4000, Epicentre) at 635 nm.
Data analysis
Data analysis was performed using ImaGene 6.0 (Biodiscovery, El Segundo, CA, USA) and Excel 2003 (Microsoft, Redmond, WA, USA). Using ImaGene 6.0, spots with signal-to-noise ratios ≥2 were considered positive. Correction for background signal intensity was performed using the mean value of the local background intensity of a spot. After adjustment of positive spots and background correction by using ImaGene 6.0, the data were transferred to Excel (version 2003) for data analysis. The spots in duplicate were chosen for the following analysis: mean intensity of all of the chosen positive spots, ratio of intensity of every chosen spots to mean intensity of all the spots, and mean value of the duplicate. To compare the different intensities of spots in both soil samples, the intensity of each spot of the planted soil was divided by its intensity of the unplanted soil. The spots were considered as significantly changed when the absolute value of the logarithmic ratio was greater than log 2.
Results
Purity and quantity of DNA extracts and their PCR performance
DNA was extracted from the soil samples of planted and unplanted segments in the constructed wetland in Bitterfeld (Germany) by three commercial DNA extraction kits (Bio101, UltraClean, and PowerSoil). To improve the purity of the original Bio101 and UltraClean extracts from both soils, they were additionally purified by the Wizard DNA purification kit. To increase the DNA yield of the PowerSoil extracts from the unplanted soil, DNA was also extracted by a modified method. Altogether, 11 different DNA extracts were obtained. Their DNA purity indices and quantity (A260/280, A260/230, A320, and DNA yield in Table 1) varied significantly not only with respect to the applied extraction kits but also to the analyzed soil samples.
Generally, the A320 value of the PowerSoil extracts was the lowest among all original and purified DNA extracts from the same soil sample. The Bio101 extracts were characterized by the high DNA yields among the extracts with the same treatment (original or purified). It is important to notice that, among the extracts applied later for the DGGE and microarray analyses, e.g., the original PowerSoil extracts and the purified Bio101 and UltraClean extracts from both soil samples, the Bio101 extracts had the highest A260/280 ratio and DNA yield, while the PowerSoil extracts the highest A260/230 and the lowest A320 value. Furthermore, the DNA yield of the PowerSoil extracts from the unplanted soil was increased threefold after using the modified method and reached the similar value (0.40 μg DNA/g soil) as the purified Bio101 extracts from the unplanted soil (0.41 μg DNA/g soil).
The PCR for all the extracts was performed with varying success (Table 1). According to the A260/280 ratio and the success of the PCR, the DNA extracts could be divided into three groups (Table 1): group I included the original PowerSoil extracts of the planted soil (A260/280 = 1.65) and the additionally purified Bio101 and UltraClean extracts of the planted soil (A260/280 = 1.74 and 1.56, respectively), as well as the modified PowerSoil extracts of the unplanted soil (A260/280 = 1.55). Their PCR amplification was performed successfully without any dilution. These extracts all had higher A260/280 ratio (≥1.55 in this study) and lower A320 value (<0.1 in this study). In group II, an additional dilution is required prior to PCR. The extracts in this group had an A260/280 ratio of about 1.5, such as the original Bio101 extracts of the planted soil with A260/280 ratio of 1.53, the original PowerSoil extracts of the unplanted soil with 1.47, and the additionally purified Bio101 and UltraClean extracts of the unplanted soil with 1.51 and 1.47, respectively. In this group, 1:5 dilution was sufficient for a successful PCR performance if the A320 value is relative low (<0.1 in this study). When the A320 value is relative high (≥0.15 in this study), higher dilution (such as 1:10) could also lead to a successful PCR in the extracts with high DNA yield, e.g., in the original Bio101 extracts from the planted soil (9.12 μg DNA/g soil) with 1:10 dilution. However, no successful PCR was achieved in the extracts with lower DNA yield under any dilutions, such as in the purified UltraClean extracts from the unplanted soil (0.14 μg DNA/g soil). For the extracts of group III, whose A260/280 ratios were less than 1.35, no PCR products could be observed.
Microbial composition assessed by DGGE
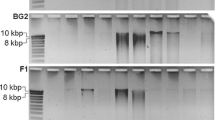
The PCR products of the original PowerSoil extracts and purified Bio101 and UltraClean extracts were further evaluated by DGGE. The DGGE patterns of each extracts are displayed in Fig. 1a, and their similarity is compared according to the numbers of detected bands (Table 2). Generally, DGGE patterns of the three independent replicate extracts were almost identical (D sc > 95%), indicating the reproducibility of the results for both DNA extraction and PCR amplification (Fig. 1a). Although the extracts were obtained by using different kits, all of them from the same soil sample were shown in a resembled pattern of the community profiles (D sc > 84% for the planted soil, D sc > 74% for the unplanted soil), especially between band patterns of UltraClean and PowerSoil extracts of the planted soil (D sc = 94%). However, differences on the band pattern among the used extraction kits were also observed. The Bio101 extracts always showed band patterns with different intensities in comparison to the UltraClean or PowerSoil extracts. Moreover, some additional bands were also observed in the band pattern of the Bio101 extracts.

DGGE of a DNA extracts of the planted and the unplanted soil obtained by Bio101 (Bio), UltraClean (UC), and PowerSoil (PS) (lanes 1–9 planted soil, lanes 1–3 Bio101, lanes 4–6 UltraClean, lanes7–9 PowerSoil, lanes 10–13 unplanted soil, lane 10 Bio101, lane 11 UltraClean, lanes 12–13 PowerSoil) and b extracts from the unplanted soil obtained by using different protocols (PowerSoil and PowerSoil_F); lane 1′ Bio101, lane 2′ UltraClean, lane 3′ PowerSoil, lane 4′ PowerSoil_F, lane 5′ PowerSoil_F with 1:5 dilution
To eliminate the differences possibly caused by the DNA yield, the DGGE pattern profiles were also compared among the modified PowerSoil extracts without and with 1:5 dilution, as well as other extracts from the unplanted soil (Fig. 1b). Between the modified PowerSoil extracts and its 1:5 diluted extracts, no significant differences were observed. Comparing to the DGGE band patterns of the original PowerSoil extracts, the modified PowerSoil extracts showed enhanced intensity of the bands, as well as some new bands, which were also observed in the band pattern of Bio101 (Fig. 1b). Therefore, the similarity of the DGGE band pattern between the Bio101 and the PowerSoil extracts was improved from 74% to 90% (Table 2) by using the modified method. However, some predominant bands in the Bio101 extracts were still missing in the extracts from the modified PowerSoil method. Thus, the Bio101 extracts comprised the broadest band pattern in this study.
Microbial diversity assessed by functional gene microarray
In addition to DGGE analyses, the microbial diversity of these extracts (without PCR) was also evaluated by a 50-mer functional gene microarray simultaneously. This microarray contained probes targeting more than 2,000 functional genes of various microorganisms, involved in carbon, nitrogen, sulfur, and phosphorus metabolism and in the degradation of organic contaminants. Positive spots were observed in each extract. The PowerSoil extracts contained more positive spots, 115 for planted and 80 for unplanted soil, while the two Bio101 extracts and two UltraClean extracts had 78, 58 and 49, 58 positive spots, respectively (Fig. 2). Analyses of these spots showed that the PowerSoil extracts nearly covered all of the positive spots detected in Bio101 and UltraClean extracts, over 97% (76 out of 78 and 49 out of 49) in planted soil and over 91% (53 out of 58 and 54 out of 58) in unplanted soil. Additionally, the PowerSoil extracts had about 30% (37 out of 115 and 24 out of 80) spots that could not be detected in the extracts by the other two kits. Thus, the PowerSoil extracts appears to be more suitable for the microarray analyses.

Venn diagram comparing the numbers of the total detected positive spots after microarray hybridization of the DNA extracts obtained by Bio101 (Bio), UltraClean (UC), and PowerSoil (PS); a planted soil, b unplanted soil
However, a high level of nonspecific hybridization could have occurred by using the random priming labeling method (Franke-Whittle et al. 2006). To neutralize the nonspecific hybridization, the extracts from the planted and unplanted soils obtained by the same kit were paired together for further analyses. A positive spot was selected when it was detected exclusively in one extract of the pair or when it could be detected in both extracts but with significantly different intensities. Altogether, 54 changed spots were detected in PowerSoil extracts, while only 24 and eight spots were found in Bio101 and UltraClean extracts, respectively (Table 3). Due to the low number (eight) of the changed spots in the UltraClean extracts, only the results of the PowerSoil and Bio101 extracts were used in further comparative analyses. Since multiple positive spots could correspond to the same gene and multiple genes belong to the same microbial population, the comparative analyses were performed according to the corresponding genes of the changed positive spots and to the possible microbial populations. The results are summarized in Figs. 3 and 4, respectively.

Comparison on the corresponding genes of the changed positive spots in the Bio101 (Bio) and the PowerSoil (PS) extracts according to four main gene categories according to their involved metabolic processes: carbon degradation, organic contaminant degradation, alternative respiration processes based on nitrate and sulfate as terminal electron acceptors (denitrification and sulfate reduction), and others including the genes involved in heavy metal resistance, carbon fixation, and phosphorous metabolism. Exclusive: spots detected only in the respective kit

Comparison on the possible changed microbial populations in the Bio101 (Bio) and the PowerSoil (PS) extracts, divided into two major groups (Actinobacteria and Proteobacteria) and others (Firmicutes, Cyanobaceria, and Deinocuccus–Thermus). Exclusive: spots detected only in the respective kit
In the PowerSoil and Bio101 extracts, 47 and 22 changed genes were detected, respectively. These genes were grouped into four gene categories according to their involved metabolic processes: carbon degradation, organic contaminant degradation, alternative respiration processes based on nitrate and sulfate as terminal electron acceptors (denitrification and sulfate reduction), and others including the genes involved in heavy metal resistance, carbon fixation, and phosphorous metabolism. Among all of the changed genes, 90% (47 out of 52) could be detected in the PowerSoil extracts but only 40% (22 out of 52) in the Bio101 extracts. Moreover, the superiority of the PowerSoil extracts in representing the changed genes between the two soils can be observed in all of the four gene categories (Fig. 3).
The analysis of microbial community revealed that 36 and 19 possible bacterial populations were found in PowerSoil and Bio101 extracts, respectively (Fig. 4). The superiority of PowerSoil extracts was also observed, in that it contained over 75% (15 out of 19) of microbial populations in the Bio101 extracts. Moreover, in the two detected major groups (Actinobacteria and Proteobacteria), at least 89% (eight out of nine) of the possible changed microbial populations were detected in the PowerSoil extracts while no more than 56% (five out of nine) in the Bio101 extracts. Therefore, the PowerSoil extracts can obviously demonstrate more variations with respect not only to the changed genes but also to the possible changed microbial populations. The PowerSoil extracts are more suitable for the comparative microarray analysis than the Bio101 extracts.
Discussion
In this study, the DNA was isolated from the planted and the unplanted soil samples in a constructed wetland by three commercial DNA extraction kits. DNA extracts with diverse purity indices and quantity were obtained and compared for the subsequent microbial community study assessed by DGGE and microarray. These different DNA extracts resulted in diverse outcomes in DGGE and microarray analyses.
The fact that DNA extraction could affect microbial community profiles of DGGE from soils and sediment was reported in the previous studies (De Lipthay et al. 2004; Carrigg et al. 2007) and confirmed in this study as well. Furthermore, the influence of DNA extraction on the microarray results was also observed in this study. It is interesting to notice that the influence of the DNA extraction on these two assessment methods, reflected by the suitability of the extracts, is quite different. While the purified Bio101 extracts resulted in the broadest band pattern of DGGE, the original PowerSoil extracts are more suitable for microarray. Therefore, the outcomes of the PCR-based DGGE and microarray analyses should be significantly affected due to the different purity indices and quantity of the DNA extracts.
Since DGGE analyses are usually performed after PCR amplification, the success of PCR is an important criterion that affects the microbial community studies while using this method. According to Krsek and Wellington (1999), the success of PCR amplification is more influenced by A260/280 than by A260/230. This was confirmed by group I and II extracts in this study (see the “Results” section). However, a higher A260/280 ratio does not always lead to successful PCR results; for example, no PCR products were obtained in the UltraClean extracts of the unplanted soil in any tested dilution (1:5, 1:10, and 1:100). This extract had the same A260/280 ratio as the PowerSoil extract for the same soil sample but a twofold higher absorption at 320 nm, suggesting that a low content of humic substances is also crucial for the success of PCR prior to DGGE.
Moreover, DNA yield is also frequently discussed in view of its representativeness to evaluate the microbial diversity of an environmental sample by DGGE. Some studies highlighted that the assessment of microbial diversity is strictly dependent on the total DNA yield recovered (Osborn et al. 2000; LaMontagne et al. 2002), while others confirmed that there was no direct implication in estimates of microbial diversity within a sample (Stach et al. 2001; Gabor et al. 2003; Luna et al. 2006). Based on our data, we conclude that the DNA yield may affect the analysis of the microbial community conditionally. For the group I extracts in this study (A260/280 > 1.55; A320 < 0.1), a successful PCR amplification can be achieved without any dilution. The representativeness on the microbial communities was not obviously affected by the DNA yield, even if the extract was diluted; for example, no significant differences were observed in the DGGE pattern between the modified PowerSoil extract and its 1:5 diluted extract (Fig. 1b). However, the influence of DNA yield on the representativeness of the microbial communities became noticeable, when the A260/280 ratio of the extracts reached a moderate level (about 1.5 in this study), just as the extracts in group II. In this case, the dilution of extracts is necessary for the success of PCR, but this could simultaneously result in either insufficient amount of the template DNA for the PCR, such as in the case of the purified UltraClean extracts from the unplanted soil, or DGGE profiles with fewer bands and weaker intensities, e.g., the original PowerSoil extracts. Thus, the DNA yield could also become critical for the microbial community investigations.
However, the discussion above could still not explain why some bands with low or moderate intensity in the Bio101 extracts were missing in the band patterns of the modified PowerSoil extracts. According to the study of Martin-Laurent et al. (2001), a preferential DNA recovery and/or preferential DNA amplification could cause the variations of band intensity in the fingerprint analysis due to using different DNA extraction kits. In this study, identical soil samples that were used for the extraction and cells from all microorganisms are lysed in equal proportions by using direct lysis (Reysenbach et al. 1992; Sipos et al. 2007). Since uniform method was utilized in PCR, differences caused by preferential amplification should not be observed in the DGGE band pattern. Thus, the main reason for the missing bands is due to preferential DNA recovery, which may be caused by the specificity (at strain or species level) of the different chemical agents applied in the DNA extraction (Wilson 1997; Al-Soud and Radström 1998; Theron and Cloete 2000; Lloyd-Jones and Hunter 2001; Bakken and Frostegard 2006). In this respect, the DGGE band patterns of the extracts from the planted soil obtained by the kits UltraClean and PowerSoil, whose methods differed mainly by a humic substances/brown color removal procedure, showed higher similarity (94%). Due to the preferential extraction of different extraction kits, the effort to remove coextracted humic substances as much as possible from the DNA extracts may not always be the best way to gain suitable DNA extracts for PCR-based fingerprint methods, like the PowerSoil extracts for DGGE analyses in this study.
According to previous publications (Zhou and Thompson 2002; Lemarchand et al. 2005; Eads et al. 2006), poor quality of extracted DNA may interfere with microarray hybridizations yielding high background and false positives. To the authors’ knowledge, few studies are available for a thorough discussion about the influence of DNA extraction on microarray analyses. In this study, we could conclude that the PowerSoil extracts with the highest A260/230 and the lowest A320 value were more suitable for the microarray. Due to the amount of the template DNA (2 μg) that was significant enough for the microarray study (Schadt et al. 2005), the different outcomes in microarray must be caused by the DNA purity indices. Therefore, the sample quality mentioned above, unlike in the PCR, was obviously less dependent on the A260/280, but more on the A260/230 and A320 values. Thus, using successful PCR amplification as a criterion of suitable soil DNA purity, as indicated by literature (Cullen and Hirsch 1998; Moreira 1998; Bürgmann et al. 2001; Lakay et al. 2007), is not accurate enough any more for the microarray analyses.
In conclusion, the outcomes of the PCR-based methods (DGGE in this study) and microarray hybridizations were affected by different purity indices and quantity of DNA extracts. The selection of a suitable DNA extraction kit (or method) does not only depend on the type of sample or the microbial population targeted, but also significantly on the applied subsequent assessment method. The success of PCR amplification, as the most used criterion for the purity of the DNA extracts in the previous publications, is not always suitable to decide the good DNA extracts for the microarray analysis. As a key step in the microarray studies, DNA extraction should continuously be improved in line with the development of the microarray technique.
References
Ahn SJ, Costa J, Emanuel JR (1996) PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res 24:2623–2625
Al-Soud WA, Rådström P (1998) Capacity of nine thermostable DNA polymerases to mediate DNA amplification in the presence of PCR-inhibiting samples. Appl Environ Microbiol 64:3748–3753
Arbeli Z, Fuentes CL (2007) Improved purification and PCR amplification of DNA from environmental samples. FEMS Microbiol Lett 272:269–275
Bakken LR, Frostegard A (2006) Nucleic acid extraction from soil. In: Nannipiere P, Smalla K (eds) Nucleic acids and proteins in soil. 1st edn. Springer, Berlin, pp 4–76
Bürgmann H, Pesaro M, Widmer F, Zeyer J (2001) A strategy for optimizing quality and quantity of DNA extracted from soil. J Microbiol Methods 45:7–20
Braeckevelt M, Rokadia H, Imfeld G, Stelzer N, Paschke H, Kuschk P, Kästner M, Richnow HH, Weber S (2007) Assessment of in situ biodegradation of monochlorobenzene in contaminated groundwater treated in a constructed wetland. Environ Pollut 148:428–437
Carrigg C, Rice O, Kavanagh S, Collins G, O’Flaherty V (2007) DNA extraction method affects microbial community profiles from soils and sediment. Appl Microbiol Biotechnol 77:955–964
Cho JC, Tiedje JM (2001) Bacterial species determination from DNA–DNA hybridization by using genome fragments and DNA microarray. Appl Environ Microbiol 67:3677–3682
Cullen DW, Hirsch PR (1998) Simple and rapid method for direct extraction of microbial DNA from soil for PCR. Soil Biol Biochem 30:983–993
De Lipthay JR, Enzinger C, Johnsen K, Aamand J, Sorensen SJ (2004) Impact of DNA extraction method on bacterial community composition measured by denaturing gradient gel electrophoresis. Soil Biol Biochem 36:1607–1614
Dorigo U, Volatier L, Humbert JF (2005) Molecular approaches to the assessment of biodiversity in aquatic microbial communities. Water Res 39:2207–2218
Dong D, Yan A, Liu H, Zhang X, Xu Y (2006) Removal of humic substances from soil DNA using aluminium sulfate. J Microbiol Methods 66:217–222
Eads B, Cash A, Bogart K, Costello J, Andrews J (2006) Troubleshooting microarray hybridizations. Methods Enzymol 411:34–49
Fortin N, Beaumier D, Lee K, Greer CW (2004) Soil washing improves the recovery of total community DNA from polluted and high organic content sediments. J Microbiol Methods 56:181–191
Franke-Whittle IH, Klammer SH, Insam H (2005) Design and application of an oligonucleotide microarry for the investigation of compost microbial communities. J Microbiol Methods 62:37–56
Franke-Whittle IH, Klammer SH, Mayrhofer S, Insam H (2006) Comparison of different labelling methods for the production of labelled target DNA for microarray hybridization. J Microbiol Methods 65:117–126
Gabor EM, de Vries EJ, Janssen DB (2003) Efficient recovery of environmental DNA for expression cloning by indirect extraction methods. FEMS Microbiol Ecol 44:153–163
He Z, Gentry TJ, Schadt CW, Wu L, Liebich J, Chong SC, Huang Z, Wu W, Gu B, Jardine P, Criddle C, Zhou J (2007) GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J 1:67–77
Hurt RA, Qiu X, Wu L, Roh Y, Palumbo AV, Tiedje JM, Zhou J (2001) Simultaneous recovery of RNA and DNA from soils and sediments. Appl Environ Microbiol 67:4495–4503
Kormas KA, Smith DC, Edgcomb V, Teske A (2003) Molecular analysis of deep subsurface microbial communities in Nankai Trough sediments (ODP leg 190, Site 1176). FEMS Microbiol Ecol 45:115–125
Klerks MM, van Bruggen AHC, Zijlstra C, Donnikov M (2006) Comparison of methods of extracting Salmonella enterica serovar enteritidis DNA from environmental substrates and quantification of organisms by using a general internal procedural control. Appl Environ Microbiol 72:3879–3886
Krsek M, Wellington EMH (1999) Comparison of different methods for the isolation and purification of total community DNA from soil. J Microbiol Methods 39:1–16
Kuske CR, Banton KL, Adorada DL, Stark PC, Hill KK, Jackson PJ (1998) Small-scale DNA sample preparation method for field PCR detection of microbial cells and spores in soil. Appl Environ Microbiol 64:2463–2472
LaMontagne MG, Michel FC, Holden PA, Reddy CA (2002) Evaluation of extraction and purification methods for obtaining PCR-amplifiable DNA from compost for microbial community analysis. J Microbiol Methods 49:255–264
Lakay FM, Botha A, Prior BA (2007) Comparative analysis of environmental DNA extraction and purification methods from different humic acid-rich soils. J Appl Microbiol 102:265–273
Lemarchand K, Berthiaume F, Maynard C, Harel J, Payment P, Bayardelle P, Masson L, Brousseau R (2005) Optimization of microbial DNA extraction and purification from raw wastewater samples for downstream pathogen detection by microarrays. J Microbiol Methods 63:115–126
Liebich J, Schäffer A, Burauel P (2003) Structural and functional approach to studying pesticide side-effects on specific soil functions. Environ Toxicol Chem 22:784–790
Liebich J, Chong SC, Schadt CW, He Z, Zhou J (2006) Improvement of oligonucleotide probe design criteria for the development of functional gene microarrays for environmental applications. Appl Environ Microbiol 72:1688–1691
Lloyd-Jones G, Hunter DWF (2001) Comparison of rapid DNA extraction methods applied to contrasting New Zealand soils. Soil Biol Biochem 33:2053–2059
Luna GM, Dell’Anno A, Danovaro R (2006) DNA extraction procedure: a critical issue for bacterial diversity assessment in marine sediments. Environ Microbiol 8:308–320
Martin-Laurent F, Philippot L, Hallet S, Chaussod R, Germon JC, Soulas G, Catroux G (2001) DNA extraction from soils: old bias for new microbial diversity analysis methods. Appl Environ Microbiol 67:2353–2359
Miller DN (2001) Evaluation of gel filtration resins for the removal of PCR-inhibitory substances from soils and sediments. J Microbiol Methods 44:49–58
Miller DN, Bryant JE, Madsen EL, Chiorse WC (1999) Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl Environ Microbiol 65:4715–4724
Mitchell KR, Takacs-Vesbach CD (2008) A comparison of methods for total community DNA preservation and extraction from various thermal environments. J Ind Microbiol Biotech 35:1139–1147
Moreira D (1998) Efficient removal of PCR inhibitors using agarose-embedded DNA preparations. Nucleic Acids Res 26:3309–3310
Morris CE, Bardin M, Berge O, Frey-Klett P, Fromin N, Girardin H, Guinebretiére MH, Lebaron P, Thiéry JM, Troussellier M (2002) Microbial biodiversity: approaches to experimental design and hypothesis testing in primary scientific literature from 1975 to 1999. Microbiol Mol Biol Rev 66:592–616
Mumy K, Findlay RH (2004) Covenient determination of DNA extraction efficiency using an external DNA recovery standard and quantitative-competitive PCR. J Microbiol Methods 57:259–268
Niemi RM, Heiskanen I, Wallenius K, Lindström K (2001) Extraction and purification of DNA in rhizosphere soil samples for PCR-DGGE analysis of bacterial consortia. J Microbiol Methods 45:155–165
Nübel U, Engelen B, Felske A, Snaidr J, Wieshuber A, Amann RI, Ludwig W, Backhaus H (1996) Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J Bacteriol 178:5636–5643
Ogram A (2000) Soil molecular microbial ecology at age 20: methodological challenges for the future. Soil Biol Biochem 32:1499–1504
Orphan VJ, Hinrichs KU, Usler W 3rd, Paull CK, Taylor LT, Sylva SP (2001) Comparative analysis of methane-oxidizing archaea and sulfate-reducing bacteria in anoxic marine sediments. Appl Environ Microbiol 67:1922–1934
Osborn AM, Moore ERB, Timmis KN (2000) An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ Microbiol 2:39–50
Rengarajan K, Cristol SM, Mehta M, Nickerson JM (2002) Quantifying DNA concentrations using fluorometry: a comparison of fluorophores. Mol Vis 8:416–421
Reysenbach AL, Giver LJ, Wickham GS, Pace NR (1992) Differential amplification of ribosomal-RNA genes by polymerase chain-reaction. Appl Environ Microbiol 58:3417–3418
Roose-Amsaleg CL, Garnier-Sillam E, Harry M (2001) Extraction and purification of microbial DNA from soil and sediment samples. Agric Ecosyst Environ Appl Soil Ecol 18:47–60
Schadt CW, Liebich J, Chong SC, Gentry TJ, He Z, Pan H, Zhou J (2005) Design and use of functional gene microarrays (FGAs) for the characterization of microbial communities. Methods Microbiol 34:329–365
Sipos R, Szekely AJ, Palatinszky M, Revesz S, Mairaligeti K, Nikolausz M (2007) Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targeting bacterial community analysis. FEMS Microbiol Ecol 60:341–350
Stach JEM, Bathe S, Clapp JP, Burns RG (2001) PCR-SSCP comparison of 16S rDNA sequence diversity in soil DNA obtained using different isolation and purification method. FEMS Microbiol Ecol 36:139–151
Stahl DA (2004) High-throughput techniques for analyzing complex bacterial communities. Adv Exp Med Biol 547:5–17
Stine OC, Carnahan A, Singh R, Powell J, Furuno JP, Dorsey A, Silbergeld E, Willianms HN, Morris JG (2003) Characterization of microbial communities from coastal waters using microarrays. Environ Monit Asses 81:327–336
Suchodolski JS, Ruaux CG, Steiner JM, Fetz K, Williams DA (2004) Application of molecular fingerprinting for qualitative assessment of small-intestinal bacterial diversity in dogs. J Clin Microbiol 42:4702–4708
Taroncher-Oldenburg G, Griner EM, Francis CA, Ward BB (2003) Oligonucleotide microarray for the study of functional gene diversity in the nitrogen cycle in the environment. Appl Environ Microbiol 69:1159–1171
Tebbe CC, Vahjen W (1993) Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and yeast. Appl Environ Microbiol 59:2657–2665
Theron J, Cloete TE (2000) Molecular techniques for determining microbial diversity and community structure in natural environments. Crit Rev Microbiol 26:37–57
Tiquia SM, Lloyd J, Daniel AH, Hoitink HAJ, Michel FC (2002) Effects of mulching and fertilization on soil nutrients, microbial activity and rhizosphere bacterial community structure determined by analysis of T-RFLPs of PCR-amplified 16S rRNA genes. Appl Soil Ecol 599:1–18
Vogt C, Alfreider A, Lorbeer H, Ahlheim J, Feist B, Boehme O, Weiss H, Babel W, Wuensche L (2002) Two pilot plant reactors designed for the in situ bioremediation of chlorobenzene-contaminated ground water: hydrogeological and chemical characteristics and bacterial consotia. Water Air Soil Pollut 2:161–170
Weiss H, Schirmer M, Teutsch G, Merkel P (2001) Sanierungsforschung in regional kontaminierten Aquiferen (SAFIRA)-2. Projektüberblick und Pilotanlage. Grundwasser 3:135–139
Whitehouse CA, Hottel HE (2007) Comparison of five commercial DNA extraction kits for the recovery of Francisella tularensis DNA from spiked soil samples. Mol Cell Probes 21:92–96
Wilson IG (1997) Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol 63:3741–3751
Wu LY, Thompson DK, Li GS, Hurt RA, Tiedje JM, Zhou J (2001) Development and evaluation of functional gene arrays for detection of selected genes in the environment. Appl Environ Microbiol 67:5780–5790
Wu L, Thompson DK, Liu XD, Fields MW, Bagwell CE, Tiedje JM, Zhou JZ (2004) Development and evaluation of microarray-based whole-genome hybridization for detection of microorganisms within the context of environmental applications. Environ Sci Technol 38:775–782
Xu D, Li G, Wu L, Zhou JZ, Xu Y (2002) PRIMEGENS: a computer program for robust and efficient design of gene-specific targets on microarrays. Bioinformatics 18:1432–1437
Zhou JZ (2003) Microarrays for bacterial detection and microbial community analysis. Curr Opin Microbiol 6:288–294
Zhou JZ, Thompson DK (2002) Challenges in applying microarrays to environmental studies. Curr Opin Biotechnol 13:204–207
Zhou JZ, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62:461–468
Acknowledgements
The manuscript is a joint approach within the working group “Biodiversity” of the Helmholtz Association, Germany. The authors would like to thank Eva Mareike Seeger, Mareike Braeckevelt, and Heidrun Paschke in the group of the Department of Bioremediation (UFZ, Germany) for the help during the sampling at the SAFIRA plant in Bitterfeld and the group in the Institute for Environmental Genomics (Oklahoma, USA) for providing the microarray chips, as well as Dr. Naixin Li for the valuable discussion. The construction of microarray chips and its functional gene array predecessors is supported by the Environmental Stress Pathway Project (ESPP) of the Virtual Institute for Microbial Stress and Survival (http://vimss.lbl.gov) supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, Genomics: GTL Program, through contract DE-AC02-05CH11231 with Lawrence Berkeley National Laboratory.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ning, J., Liebich, J., Kästner, M. et al. Different influences of DNA purity indices and quantity on PCR-based DGGE and functional gene microarray in soil microbial community study. Appl Microbiol Biotechnol 82, 983–993 (2009). https://doi.org/10.1007/s00253-009-1912-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-1912-0