
Abstract
Streptomyces albulus NBRC14147 produces ɛ-poly-l-lysine (ɛ-PL), which is an amino acid homopolymer antibiotic. Despite the commercial importance of ɛ-PL, limited information is available regarding its biosynthesis; the l-lysine molecule is directly utilized for ɛ-PL biosynthesis. In most bacteria, l-lysine is biosynthesized by an aspartate pathway. Aspartokinase (Ask), which is the first enzyme in this pathway, is subject to complex regulation such as through feedback inhibition by the end-product amino acids such as l-lysine and/or l-threonine. S. albulus NBRC14147 can produce a large amount of ɛ-PL (1–3 g/l). We therefore suspected that Ask(s) of S. albulus could be resistant to feedback inhibition to provide sufficient l-lysine for ɛ-PL biosynthesis. To address this hypothesis, in this study, we cloned the ask gene from S. albulus and investigated the feedback inhibition of its gene product. As predicted, we revealed the feedback resistance of the Ask; more than 20% relative activity of Ask was detected in the assay mixture even with extremely high concentrations of l-lysine and l-threonine (100 mM each). We further constructed a mutated ask gene for which the gene product Ask (M68V) is almost fully resistant to feedback inhibition. The homologous expression of Ask (M68V) further demonstrated the increase in ɛ-PL productivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The amino acid homopolymer antibiotic, ɛ-poly-l-lysine (ɛ-PL), consisting of 25–30 l-lysine residues with a linkage between the α-carboxyl group and the ɛ-amino group (Fig. 1) is produced by Streptomyces albulus NBRC14147 (Shima and Sakai 1977, 1981a, b). Because ɛ-PL exhibits antimicrobial activity against a wide spectrum of microorganisms, including Gram-positive and Gram-negative bacteria (Shima et al. 1984), as well as antiphage activity (Shima et al. 1982), and because it is both safe and biodegradable, ɛ-PL has been introduced as a food preservative in Japan, South Korea, the USA, and other countries. Despite the commercial importance of ɛ-PL, limited information is available regarding its biosynthesis. Kawai et al. (2003) have recently suggested the possibility that ɛ-PL is produced by nonribosomal peptide synthases. Shima et al. (1983) have reported that the l-lysine molecule is directly utilized in ɛ-PL biosynthesis.

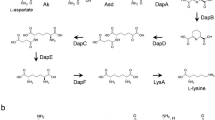
The amino acid biosynthetic pathway from l-aspartic acid (aspartate pathway). ɛ-PL ɛ-poly-l-lysine; AVG aminoethoxyvinylglycine; CPM-C cephamycin C
In most bacteria, l-lysine is biosynthesized by the amino acid biosynthetic pathway from l-aspartic acid (aspartate pathway; Fig. 1). This pathway is also involved in the formation of other amino acids (i.e., l-methionine, l-isoleucine, and l-threonine). The first two enzymes in this pathway are aspartokinase (Ask; EC.2.7.2.4), which catalyze the phosphorylation of l-aspartic acid to produce l-4-phospho aspartic acid, and aspartate semialdehyde dehydrogenase (Asd; EC.1.2.1.11), which reduces l-4-phospho aspartic acid into l-aspartate 4-semialdehyde. These two key enzymes are subject to complex regulation by the end-product amino acids. Because of the complexity of this pathway, different bacterial species have evolved diverse patterns of Ask regulation. For example, Escherichia coli and Bacillus subtilis have three separate Ask isozymes, each controlled by one of the end products of the aspartate pathways, diaminopimelic acid, lysine, threonine, and methionine (Hitchcock et al. 1980; Theze et al. 1974; Zhang et al. 1990; Zhang and Paulus 1990). In contrast, only one Ask has been described in antibiotic-producing actinomycetes. There are some reports of cloned ask genes from the rifamycin SV producer Amycolatopsis mediterranei (Zhang et al. 1999), from the cephamycin C (CPM-C) producers Amycolatopsis lactamdurans (Fig. 1; Hernando-Rico et al. 2001) and Streptomyces clavuligerus (Fig. 1; Tunca et al. 2004), and from the aminoethoxyvinylglycine (AVG) producer Streptomyces sp. NRRL5331 (Fig. 1; Cuadrado et al. 2004). These studies have shown that regulatory mechanisms can differ even among the actinomycetes. Zhang et al. (2000) have reported inhibition of “A. mediterranei” Ask by l-lysine alone, whereas Asks of A. lactamdurans and S. clavuligerus are feedback-regulated by the concerted action of l-lysine and l-threonine (Tunca et al. 2004; Hernando-Rico et al. 2001). Interestingly, these experimental observations indicate that the Asks of A. lactamdurans and S. clavuligerus (CPM-C producers) are slightly more resistant to concerted feedback inhibition than Ask IIIs of E. coli (Ogawa-Miyata et al. 2001) and B. subtilis (Kobashi et al. 2001; Table 1), although there is no discussion of this result in the respective reports. As such, the l-lysine productivity of these strains should be higher than those of E. coli and B. subtilis. This result could be due to the need for that, l-lysine, which is one of biosynthetic precursors of CPM-C, must be fully served to not only nascent protein biosynthesis but also the CPM-C biosynthesis (Fig. 1).
The ɛ-PL producer, S. albulus NBRC14147, can produce a large amount of ɛ-PL (usually 1–3 g/l). We therefore suspected that Ask(s) of S. albulus could also be potentially resistant to feedback inhibition by l-lysine and/or l-threonine to provide sufficient amounts of l-lysine for ɛ-PL biosynthesis, similar to those of A. lactamdurans and S. clavuligerus. In addition, because ɛ-PL is composed only of l-lysine molecules, ɛ-PL producer is a simple model microorganism to examine the metabolic flow of l-lysine between primary and secondary metabolism.
In this paper, we describe the cloning of the gene encoding Ask from ɛ-PL producer, S. albulus NBRC14147, and the regulation mechanism of the Ask. Furthermore, we also discuss the ɛ-PL productivity in a S. albulus transformant expressing mutated Ask enzyme, for which there is no regulation.
Materials and methods
Chemicals
S-(2-aminoethyl)-l-cysteine (AEC) and dl-3-hydroxynorvaline (HNV) acid was obtained from Sigma (Tokyo, Japan). All other chemicals were of analytical grade.
Bacterial strains, plasmids, media, and general techniques for DNA manipulation
The cryptic plasmid pNO33-curing S. albulus strain CR1 (Hamano et al. 2005), which is derived from S. albulus NBRC14147, was used as the DNA source for cloning of the ask gene and as a host strain for an experiment regarding ask gene homologous expression. The SLB medium and growth conditions used for S. albulus CR1 have been described previously (Hamano et al. 2005). E. coli XL1-Blue MRF′ (Toyobo, Osaka, Japan) and the plasmids, pUC18, pUC19, pGEM11Z (Promega, Tokyo, Japan), and pGEM-T(easy) (Promega), were used for the subcloning experiments and sequencing analysis. The cosmid SuperCos1 (Stratagene, La Jolla, CA, USA) was used for the preparation of a genomic library. The pQE30 plasmid and E. coli M15 (pREP4) (Qiagen, Tokyo, Japan) were used to overexpress the recombinant protein. The pLAE003 (Hamano et al. 2005) and pLAE006 (unpublished) plasmids were used for homologous expression of Ask and mutated Asks in S. albulus CR1. Isolation of genomic DNA from S. albulus CR1 that was cultivated in SLB medium was performed by the method of Kieser et al. (2000). Recombinant DNA procedures for the E. coli was performed using standard techniques (Sambrook and Russell 2001).
Cloning of the ask gene from S. albulus CR1
Based on the highly conserved amino acid sequence of the Asks from A. mediterranei (Zhang et al. 2000), A. lactamdurans (Hernando-Rico et al. 2001), S. clavuligerus (Tunca et al. 2004), and Streptomyces sp. NRRL5331 (Cuadrado et al. 2004), we designed primers 5′-CT(C/G)GT(C/G)GT(C/G)CAGAAGTACGG(C/G)GG-3′ and 5′-GATCTT(C/G)(C/G)C(C/G)A(C/T)CTGGTCGTCGTA-3′. Polymerase chain reaction (PCR) amplification using TaKaRa LA Taq with GC buffer (Takara Bio, Shiga, Japan) was performed under the following conditions: 30 cycles of denaturation at 94°C for 1 min, annealing at 50°C for 1 min, and elongation at 72°C for 1 min. An amplified DNA fragment was ligated into the plasmid pGEM-T(easy), and the DNA sequence of the insert was analyzed using an ABI Prism 3100 sequencer (Applied Biosystems, Foster City, CA, USA). This cloned PCR fragment was then used as a probe to isolate a cosmid clone containing the ask gene from a genomic library of S. albulus CR1. The genomic library of S. albulus CR1 was constructed as follows: The genomic DNA of S. albulus CR1 was partially digested with Sau3AI. Sau3AI fragments larger than 25-kbp were ligated into the BamHI site of the cosmid SuperCos1, and the ligation mixture was subsequently used for in vitro packaging with Gigapack III Gold Packaging Extracts (Stratagene). A cosmid clone pASKCOS2 was screened by colony hybridization using the PCR fragment as a probe. Several restriction enzyme-digested fragments, which hybridized to the probe, were subcloned into pUC18 or pUC19. After the construction of a series of plasmids, sequencing was carried out. Finally, the DNA sequences of the BamHI-Pst I 6.5-kb fragment were analyzed (Fig. 2a). The DNA sequence has been deposited in the DDBJ/EMBL/GenBank databank under accession number AB270718.

Schematic organization of the cloned ask gene. a The BamHI–PstI 6.5-kb fragment was cloned and sequenced in this study. b The ApaI 1.9-kb fragment carrying the ask genes (wild type and M68V mutant) was respectively cloned into the pLAE003 plasmid. The resulting plasmids were used for homologous expression in S. albulus CR1. c The PCR amplified fragments carrying the ask genes (wild type and M68V mutant) with their own ribosome binding site (RBS) were respectively cloned into the pLAE006 plasmid. The resulting plasmids were used for the homologous expression in S. albulus CR1
Construction of the His-tagged Ask (wild type) in E. coli
The following set of primers was designed and used to amplify the ask gene [open reading frame 5(ORF-5)]: 5′-GGGGGATCCGGCCTTGTCGTGCAGAAGTAC-3′ (forward primer; pr-askF) and 5′-ACCAAGCTTTCATCGCCCGGTGCCGCCGTA-3′ (reverse primer; pr-askR). Restriction enzyme sites (italic letters) and a stop codon (bold letters) were introduced for the in-frame expression of recombinant proteins in the pQE30 expression vector. The PCR was carried out under standard conditions. An amplified DNA fragment was digested with BamHI and HindIII, ligated into pUC119. After sequence confirmation of the resulting plasmid (pUC119-ask), a 1.3-kbp BamHI–HindIII fragment was inserted into the same site of pQE30. The resulting plasmid, pQE30-ask (wild), in which recombinant protein was expressed as N-terminal 6×His-tagged fusion proteins under control of the T5 promoter, was selected. E. coli M15(pREP4) harboring pQE30-ask (wild) was grown at 37°C in Luria–Bertani (LB) medium (Sambrook and Russell 2001) with ampicillin (100 μg/ml) and kanamycin (25 μg/ml). Expression of the recombinant protein was induced by adding 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when the optical density at 600 nm reached approximately 0.8. The cultivation was continued for an additional 18 h at 18°C. The cells from 50 ml culture broth were harvested by centrifugation at 6,000×g for 15 min. Samples were suspended in 5 ml of buffer A [50 mM KH2PO4–K2HPO4, 5 M NaCl, 5 mM 2-mercaptoethanol (pH 7.5)] containing 10 mM imidazole and sonicated on ice. Insoluble material was removed with centrifugation at 12,000×g for 15 min. A 1 ml column of nickel–nitriloacetic acid (Ni–NTA) Sepharose was pre-equilibrated with 5 ml of buffer A containing 10 mM imidazole and loaded with the supernatant of crude E. coli extracts. The column was washed with 5 ml of buffer A containing 20 mM imidazole. The recombinant N-terminal 6×His-tagged Ask (rAsk) was eluted with 1 ml of buffer A containing 250 mM imidazole, and was then used for the following enzyme assays.
rAsk assays
rAsk activity was assayed by measuring the amount of aspartyl-β-hydroxamate formed as described by Shiio and Miyajima (1969). The assay mixture contained in a 0.5 ml volume: 100 mM Tris–H2SO4 (pH 7.0), 600 mM (NH4)2SO4, 600 mM hydroxylamine–KOH (pH 7.0), 10 mM ATP, 10 mM MgSO4, 10 mM aspartic acid–KOH (pH 7.0), and 40 μg/ml rAsk. The reaction mixtures were incubated for 10 min at 30°C, and 0.75 ml ferric chloride solution (10% FeCl3·6H2O and 3.4% trichloroacetic acid in 0.7 N HCl) was added. After centrifugation, the A 540 was measured in the supernatant. Background activity was measured in the absence of aspartic acid. Kinetic assays were performed under conditions identical to those described above, except that the enzyme concentration (20 μg/ml) was reduced to enable measurement of steady-state kinetic parameters. All assays were carried out under linear conditions. The amount of aspartyl-β-hydroxamate was calculated from the molar extinction coefficient of 600. One unit of enzyme activity is defined as the amount of enzyme catalyzing the formation of 1 μmol aspartyl-β-hydroxamate per minute at 30°C. The protein concentrations were determined using a Bio-Rad Protein Assay Kit (Bio-Rad, California, USA). Bovine serum albumin was used as the standard protein. In the Ask assay of the S. albulus CR1 strain, the cell-free extract was prepared as follows: Cells were suspended in buffer A and sonicated. After centrifugation at 12,000×g for 15 min, soluble fraction was used as a cell-free extract in Ask assays.
Error-prone PCR random mutagenesis of the ask gene
The ask gene was amplified under optimized mutagenic PCR conditions with PCR primers (pr-askF and pr-askR described above). The PCR reaction mixtures with different concentrations of deoxyribonucleotide triphosphate (dNTP) were prepared as follows: 1× Mg2+ free rTaq DNA polymerase buffer (Toyobo), 10 ng/ml template plasmid (pASKCOS2), 1 μM of each primer, 0.5 U rTaq DNA polymerase (Toyobo), 1.5 mM MgCl2, 10%(v/v) dimethylsulfoxide, and dNTP (0.04 mM dATP plus 0.2 mM dGTP/dCTP/dTTP, 0.08 mM dGTP plus 0.2 mM dATP/dCTP/dTTP, 0.08 mM dCTP plus 0.2 mM dATP/dGTP/dTTP, or 0.04 mM dTTP plus 0.2 mM dATP/dGTP/dCTP). PCR was carried out with a program of 94°C for 2 min followed by 30 cycles of 94°C for 1 min, 65°C for 1 min, and 72°C for 1 min, and finally 72°C for 10 min. The 1.3-kb PCR products amplified under the four different dNTP concentrations were mixed, purified using a QIAquick gel extraction kit (Qiagen), digested with BamHI and HindIII, ligated into the corresponding sites of pQE30, and introduced into E. coli XL1-Blue MRF′. Transformants were plated on LB plates supplemented with 100 μg/ml ampicillin. After 18 h of incubation at 37°C, colonies were collected, and plasmids (mutated ask gene library) were purified. This resulting library was introduced into E. coli XL1-Blue MRF′ and plated on M9 minimum medium agar plates (Sambrook and Russell 2001) containing 8 mM AEC, 8 mM HNV, 100 μg/ml ampicillin, and 0.1 mM IPTG. Four transformants resistant to AEC and HNV were finally isolated, and the points of mutation of the mutated ask gene (ask-er2, ask-er13, ask-16, and ask-17) on the plasmids were investigated by sequencing analysis.
Site-directed mutagenesis of the ask gene
Mutations were introduced into the plasmid pUC119-ask by using the QuickChange XL Site-Directed Mutagenesis Kit (Stratagene). To construct rAsks, each of the resulting plasmids pUC119 possessing the mutated ask gene was digested with BamHI and HindIII, and each mutated ask gene was inserted into the corresponding site of pQE30 to yield pQE30-ask (I19V), pQE30-ask (M68V), pQE30-ask (T309A), pQE30-ask (I19V, M68V), pQE30-ask (I19V, T309A), and pQE30-ask (M68V, T309A). Expression and purification of these recombinant mutated Asks as well as Ask-er2 (I19V, M68V, T309A) were carried out by the same procedure as that described for rAsk (wild).
Homologous expression of Ask (wild) and Ask (M68V) in S. albulus CR1
A 1.9-kb ApaI fragment carrying the ask gene was inserted into the ApaI site of pGEM11Z. The BamHI–HindIII fragment carrying the 1.9-kb ApaI fragment of the resulting plasmid (pGEM11Z-ask) was subcloned into the same site of pLAE003 carrying the neomycin resistance gene to yield pLAE003-ask (wild; Fig. 2b). To construct pLAE003-ask (M68V; Fig. 2b), which carries the ask (M68V) gene, the following procedures were carried out: From pQE30-ask (M68V), the ClaI 0.2-kbp fragment carrying the mutation region of the ask (M68V) gene was introduced into the corresponding site within the ask gene on pGEM11Z-ask. The BamHI–HindIII fragment carrying the ask (M68V) gene was finally inserted into the same site of pLAE003. To construct pLAE006-ask (wild) and pLAE006-ask (M68V), in which the ask genes were expressed under the control of the ermE* constitutive promoter, the following set of primers was designed to amplify the ask gene region from a 20-bp upstream region of start codon (containing the ribosome binding site) to 50-bp downstream of the ask gene stop codon: 5′-GGGGGATCCTTGCGAGGAGCGCACGTGGGC-3′ (forward) and 5′-ACCAAGCTTGGGCCCCCGCCCCCCGCCGAAT-3′ (reverse). Restriction enzyme sites (shown in italics), BamHI and HindIII, were introduced into these primers. The PCR was carried out under standard conditions with pLAE003-ask (wild) or pLAE003-ask (M68V) as a template. After sequence confirmation of amplified fragments, the ask (wild) and ask (M68M) genes were ligated into pLAE006 carrying the ermE* promoter and neomycin resistance gene (Fig. 2c). Four plasmids thus constructed, pLAE003 (no insert), and pLAE006 (no insert) were introduced into the S. albulus CR1 strain by intergeneric conjugation from E. coli, as described previously (Hamano et al. 2005). The S. albulus CR1 harboring pLAE003 (no insert), pLAE006 (no insert), pLAE003-ask (wild), pLAE003-ask (M68V), pLAE006-ask (wild), and pLAE006-ask (M68V) were cultivated in ɛ-PL production medium (PLP medium; Hamano et al. 2006) containing 100 μg/ml neomycin by using a 5 l capacity bench scale jar-fermentor, as described previously (Kahar et al. 2001). The amount of ɛ-PL produced in the supernatant was determined according to the method reported by Itzhaki (1972).
Results
Cloning and functional analysis of the ask gene from S. albulus CR1
Based on the highly conserved amino acid sequences of the Asks from A. mediterranei (accession no. AF134837), A. lactamdurans (AJ298904), S. clavuligerus (AY112728), and Streptomyces sp. NRRL5331 (AJ437313), we designed PCR primers and carried out PCR using S. albulus CR1 chromosomal DNA as a template. A band with an expected size of approximately 1.0 kbp was readily amplified. The PCR product was cloned, and six randomly selected clones were sequenced. All six clones yielded an identical sequence (except differences resulting from primer utilization), which shows high homology to the actinomycete Asks. The PCR product was then used as a probe to screen a S. albulus CR1 cosmid library. From 12 positive clones, one cosmid clone (pASKCOS2) containing a 4.5-kb BamHI-hybridizing fragment was selected (Fig. 2a). Sequencing analysis of this fragment and its downstream region (BamHI–PstI 6.5 kbp) was carried out, and frame analysis with the codon usage for Streptomyces strains (Bibb et al. 1984) revealed five complete ORFs (ORF-2 to ORF-6) and two partial ORFs (ORF-1 and ORF-7; Fig. 2a). A database search with basic local alignment search tool (BLAST; Altschul et al. 1990) showed that the deduced amino acid sequence of ORF-5 has a significant similarity to the sequences of Ask α-subunits from Streptomyces sp. NRRL5331 (90% identity), A. mediterranei (67%), S. clavuligerus (66%), and A. lactamdurans (65%). ORF-6 has a significant similarity with Asd sequences from Streptomyces sp. NRRL5331 (70% identity), A. mediterranei (45%), S. clavuligerus (58%), and A. lactamdurans (55%).
The deduced amino acid sequences of the other four complete ORFs, ORF-2, ORF-3, and ORF-4, exhibited 78, 93, and 83% identity to a conserved hypothetical protein with unknown function (Gene ID no. SCO3619) from Streptomyces coelicolor A3(2) (accession no. AL939117), a putative recombination protein (RecD) (SCO3618) from S. coelicolor A3(2) (AL939117) and a hypothetical protein with unknown function (SAV4558) from Streptomyces avermitilis MA-4680 (BA000030), respectively. ORF-1 and ORF-7, whose deduced amino acids sequences were partially identified in the present study, have significant similarities with those of a DNA polymerase III subunit gamma (SCO4067) from S. coelicolor A3(2) (AL939117) and a RNA polymerase ECF-subfamily sigma factor (SAV4561) from S. avermitilis MA-4680 (BA000030), respectively.
To confirm that the cloned ask gene encodes a functional Ask, the enzyme activity of the rAsk of S. albulus CR1, which was overexpressed in E. coli and purified to near homogeneity, was assayed in vitro. An activity of Ask (3.1 ± 0.13 U/mg protein) was specifically detected with aspartic acid as a substrate.
Investigation of the feedback-inhibition profile in rAsk
The rAsk assays were carried out with reaction mixtures added by l-lysine and/or l-threonine (0–100 mM each). As shown in Fig. 3a, the 41% relative activity of rAsk was detected in assay mixtures containing both 10 mM l-lysine and 10 mM l-threonine, in which only 1% relative activity was reported in the “B. subtilis” Ask III (Kobashi et al. 2001; Table 1). In “E. coli” Ask III (Ogawa-Miyata et al. 2001), no activity was reported in the assay mixture containing both 5 mM l-lysine and 5 mM l-threonine (Table 1). As predicted, the Ask of S. albulus CR1 was thus revealed to be much more resistant to concerted feedback inhibition than Ask IIIs of E. coli and B. subtilis, similar to Asks of S. clavuligerus and A. lactamdurans, both of which are producers of CPM-C (l-lysine derived secondary metabolite). Compared with the Ask activity reported in S. clavuligerus (36% activity, Table 1) in the assay mixture containing both 5 mM l-lysine and 5 mM l-threonine (Tunca et al. 2004), the rAsk of S. albulus CR1 showed a 1.4-fold higher relative activity (approximately 50% activity, Fig. 3a). On the other hand, the rAsk relative activity of S. albulus CR1 was found to be 1.6-fold lower than that of A. lactamdurans (78% activity, Table 1) in this condition (5 mM l-lysine plus 5 mM l-threonine). It is, however, noteworthy that more than 20% activity in the “S. albulus CR1” rAsk was detected even with extremely high concentrations of l-lysine and l-threonine (100 mM each). Thus, Ask of S. albulus CR1 was found to be the feedback-resistant enzyme, but Ask of S. albulus CR1 is also subject to feedback inhibition by a mixture of l-lysine plus l-threonine of more than 1 mM each. l-Lysine or l-threonine alone has no effect on activity (Fig. 3a).

In vitro analysis of the feedback inhibition on rAsk. Purified rAsk was employed in the enzyme assays. The relative activities of rAsk in the presence of various concentrations of l-lysine and/or l-threonine (a) or AEC and/or HNV (b) were measured
Removal of the feedback inhibition from Ask of S. albulus CR1
In this strain, a point of interest is whether l-lysine accumulation in the cell leads to ɛ-PL overproduction. We therefore attempted to construct mutated genes encoding rAsks, for which there is no feedback inhibition, because it is known that overproduction of l-lysine can be achieved by mutagenesis leading to strains with a deregulated Ask, which is no longer sensitive to feedback inhibition by a mixture of l-lysine and l-threonine (Shiio 1982). It is well-known that such a mutant strain can easily be selected by their resistance to a mixture of the l-lysine-analog AEC and l-threonine-analog HNV (Shiio 1982). We first performed enzyme assay for the rAsk wild-type enzyme with these amino acids analogs. As shown in Fig. 3b, it was shown that, as expected, the rAsk of S. albulus CR1 tended to be synergistically inhibited by the addition of AEC plus HNV, AEC plus l-threonine, and l-lysine plus HNV. Furthermore, in vivo, it was shown that an additive mixture of 8 mM AEC plus 8 mM HNV was needed to inhibit growth of the E. coli strain harboring pQE30-ask, in which the ask gene of S. albulus CR1 was overexpressed, while a lower concentration of AEC plus HNV (4 mM each) inhibited the growth of the E. coli strain harboring pQE30 (no insert; Table 2). The supplements of l-lysine and/or l-threonine to M9 minimum medium containing a mixture of 8 mM AEC plus 8 mM HNV was able to complement the lack of growth of the E. coli strain harboring pQE30-ask (Fig. 4a), namely, the toxicities of these analogs were diluted by the natural amino acids. These results demonstrated that AEC and HNV indeed work as l-lysine and l-threonine analogs, respectively, in Ask of S. albulus CR1.

Removal of the feedback inhibition from Ask. a The susceptibilities to the amino acid analogs, AEC and HNV, were investigated in the E. coli transformant harboring pQE30-ask (wild) or pQE30-ask-er2. The 2 μl of the diluted culture broths (OD600 = 1 ∼ 1 × 10−4) of the E. coli transformant harboring pQE30-ask (wild) or pQE30-ask-er2 were spotted onto the M9 minimum medium agar plate with or without 8 mM AEC plus 8 mM HNV (photo: p1, p2, p6, and p7), and the plates were incubated for 2 days at 37°C. Three additional M9 minimum medium agar plates, to which the natural amino acid(s) (l-lysine and/or l-threonine) were added, were also used (photo: p3–p5). All M9 minimum medium agar plates used in these experiments were supplemented with 100 μg/ml ampicillin and 0.1 mM IPTG. b The relative activities of the mutated rAsks in the presence of various concentrations of l-lysine and l-threonine. Each value is represented as the mean of three experiments with standard deviations less than 4%
After the introduction of random mutagenesis on the ask gene by error-prone PCR, the mutated genes library was introduced into E. coli, plated on M9 minimum medium containing a mixture of 8 mM AEC plus 8 mM HNV (see “Materials and methods”). We successfully isolated four E. coli transformants exhibiting resistances against these amino acid analogs. From these four transformants, the plasmids, pQE30-ask-er2 (Fig. 4a), pQE30-ask-er13, pQE30-ask-16, and pQE30-ask-17, were respectively isolated to identify the points of mutations on the ask gene. By sequencing analysis, the points of mutation (I19V, M68V, T309A) and nucleotide changes were found to be identical among these four mutated ask genes thus obtained. One transformant harboring pQE30-ask-er2 was therefore selected for further experiments. Ask-er2 was overexpressed in E. coli, purified to near homogeneity (data not shown), and used for in vitro assays. The specific activity of Ask-er2 in the absence of a mixture of l-lysine and l-threonine was determined to be 5.4 ± 0.02 U/mg protein. In an assay mixture with 100 mM l-lysine plus 100 mM l-threonine, a more than 80% activity of Ask-er2 was detected, showing that the mutations (I19V, M68V, T309A) removed the regulation from the Ask wild type enzyme and conferred a feedback-inhibition resistance (Fig. 4b). To determine which substitutions of the amino acid residues were responsible for the feedback-inhibition resistance, we constructed six mutated rAsks as follows: rAsk (I19V), rAsk (M68V), rAsk (T309A), rAsk (I19V, M68V), rAsk (I19V, T309A), and rAsk (M68V, T309A). The enzyme assays with these mutated rAsks revealed that M68V mutation was found to be solely responsible for the feedback-inhibition resistance (Fig. 4b). The kinetic parameters are summarized and shown in Table 3. The \( K^{{Asp}}_{{\text{m}}} \) values of the rAsk (wild type) and rAsk (M68V) were calculated to be 50.1 ± 4.8 and 1.8 ± 0.3 mM for l-aspartic acid, respectively; this shows that the rAsk (M68V) has a higher affinity for aspartic acid. The calculated \( {V^{{Asp}}_{{\max }} } \mathord{\left/ {\vphantom {{V^{{Asp}}_{{\max }} } {K^{{Asp}}_{{\text{m}}} }}} \right. \kern-\nulldelimiterspace} {K^{{Asp}}_{{\text{m}}} } \) values of the reaction with rAsk (M68V) was approximately tenfold higher than that of the reaction with rAsk (wild type). No significant differences of kinetic parameters for ATP were observed between these two enzymes. Thus, the M68V mutation brought about not only such deregulation but also an increase in the catalytic activity.
Homologous expression of Ask (wild) and Ask (M68V) in S. albulus CR1
It is known that the addition of l-lysine to ɛ-PL production medium does not significantly increase ɛ-PL productivity (Shima and Sakai 1981a, b). Therefore, the intracellular metabolic flow of l-lysine between primary and secondary metabolism is of great interest in the S. albulus strain. To address this point, pLAE003-ask (wild) and pLAE003-ask (M68V; Fig. 2b), plasmids in which the ask genes were expressed under the control of their own promoters, were constructed and introduced into S. albulus CR1. Unexpectedly, however, no significant difference in ɛ-PL productivities were observed among the S. albulus CR1 strains harboring pLAE003 (no insert), pLAE003-ask (wild), and pLAE003-ask (M68V; data not shown). We therefore constructed additional plasmids, pLAE006-ask (wild) and pLAE006-ask (M68V; Fig. 2, C), in which the ask genes were expressed under the control of the ermE* constitutive promoter. As shown in Fig. 5, the ɛ-PL productivities of the S. albulus CR1 strains harboring pLAE006-ask (wild) and pLAE006-ask (M68V) were much higher than those of the strain possessing pLAE006 (no insert) throughout the entire cultivation period despite no significant differences of growth curves between these strains. Furthermore, the introduction of the deregulated ask gene (M68V) conferred the highest productivity to S. albulus CR1.

The ɛ-PL productivities in the S. albulus CR1 strains expressing Ask (wild) or Ask (M68V). The S. albulus CR1 strains harboring pLAE006 (no insert), pLAE006-ask (wild), and pLAE006-ask (M68V) were cultivated in PLP medium containing 100 μg/ml neomycin by using a 5-l capacity bench scale jar-fermentor
Discussion
In the cloning of the ask gene, we identified only one copy of ask gene from the genomic DNA of S. albulus CR1 in PCR with primers designed on the basis of the ask genes reported in the Streptomyces strains. There have been some reports of cloned ask genes from the rifamycin SV producer A. mediterranei (Zhang et al. 1999), from CPM-C producers A. lactamdurans (Hernando-Rico et al. 2001) and S. clavuligerus (Tunca et al. 2004), and from the AVG producer Streptomyces sp. NRRL5331 (Cuadrado et al. 2004). In these strains, only one ask gene was also identified. Furthermore, the recent sequencing of the S. coelicolor A(3)2 and S. avermitilis MA-4680 genomes also clarified that these two Streptomyces strains have only one ask gene. Therefore, S. albulus is strongly suggested to have one copy of the ask gene in its genome, with no additional copy of the ask gene existing for the exclusive use of ɛ-PL biosynthesis. As such, the feedback inhibition of this gene product was of interest, and the Ask assays were carried out by using the cell-free extract prepared from S. albulus CR1. However, the activities were not reproducibly detected, possibly due to strong phosphatase activity in cell-free extracts (data not shown). Therefore, the feedback inhibition of the Ask was investigated by using its recombinant enzyme purified to homogeneity. In vitro analysis, as predicted, revealed the feedback resistance of the rAsk; more than 20% activity of rAsk was detected in the assay mixture, even with a mixture of 100 mM l-lysine and 100 mM l-threonine (Fig. 3a). This result also suggested that the intracellular or extracellular l-lysine levels of the E. coli strain harboring pQE30-ask (wild), in which the ask gene of S. albulus CR1 was overexpressed, are likely higher than those of the E. coli strain harboring pQE30 (no insert). However, we could not clarify such a difference in l-lysine productivities between these E. coli strains because the amounts of l-lysine produced were below the detection limit of an amino acid analyzer (data not shown). Instead, it was found that the E. coli strain harboring pQE30-ask (wild) exhibited higher minimum inhibitory concentration (MIC) values of AEC and HNV than those obtained from the E. coli strain harboring pQE30 (no insert; Table 2). These results strongly suggest an increase in intracellular l-lysine levels because of the rAsk. We thus demonstrated that Ask of the ɛ-PL producer was partially resistant to feedback inhibition. Our attention was therefore turned to other ɛ-PL producers such as Streptomyces noursei NBRC15452 (unpublished observation). S. noursei NBRC15452 had been expected to exhibit a resistance against AEC and HNV at similar levels of S. albulus CR1. In MIC studies using AEC and HNV, S. albulus CR1 and S. noursei NBRC15452 indeed showed exactly the same MIC values (Table 2), whereas the different S. albulus strain, S. albulus NBRC13417, which is known to not produce ɛ-PL, showed significantly lower MIC values.
In corynebacteria, the concerted inhibition by l-lysine and l-threonine is exerted at a regulatory site that involves Ser301 of Ask (Kalinowski et al. 1991). A mutant resistant to AEC showed a Ser301 to Tyr301 change. Similarly, in Corynebacterium flavum N13, Gly345 of Ask has been reported to be involved in the concerted inhibition of Ask by l-lysine and l-threonine (Follettie et al. 1993). Based on these results, Hernando-Rico et al. (2001) have reported a construction of the feedback-resistant Ask of A. lactamdurans by substitutions of the two corresponding amino acid residues, Ser301 and Gly345. However, a homologous expression of the mutated Ask has not been carried out due to the extreme difficulty in transforming A. lactamdurans. On the other hand, because we recently developed a genetic system for S. albulus CR1, we decided to construct and introduce a deregulated Ask (feedback-inhibition resistant Ask) into this strain. By random mutagenesis of the ask gene with error-prone PCR and the subsequent site-directed mutagenesis, we successfully constructed the mutated Ask, rAsk (M68V), whose feedback-inhibition regulation was completely removed (Fig. 4). Although Ask of S. albulus CR1 also has Ser301 and Gly345, no mutated Ask with substitutions of these two amino acid residues were obtained in our screening system. Rather, it appeared to be more appropriate for Ask (M68V) to be expressed in S. albulus CR1 to investigate whether l-lysine accumulation in the cell leads to ɛ-PL overproduction, as the calculated \( {V^{{Asp}}_{{\max }} } \mathord{\left/ {\vphantom {{V^{{Asp}}_{{\max }} } {K^{{Asp}}_{m} }}} \right. \kern-\nulldelimiterspace} {K^{{Asp}}_{m} } \) values of rAsk (M68V) was tenfold higher than that of the rAsk (wild type). Compared with the productivity in the use of Ask (wild), the homologous expression of Ask (M68V) predictably conferred a higher productivity of ɛ-PL (Fig. 5), i.e., the l-lysine productivity was increased by Ask (M68V). However, intracellular and extracellular accumulations of l-lysine molecules were not observed (data not shown), suggesting the probable quick consumption for ɛ-PL biosynthesis.
The homologous expression of the wild type of Ask also increased ɛ-PL productivity only when the gene was introduced with ermE* constitutive promoter. This revealed that the expression of the ask gene with its own promoter is tightly regulated by repression. In E. coli, the ask genes are repressed by end-products of the aspartate pathway such as l-methionine, l-threonine, l-isoleucine, or l-lysine. However, in S. albulus, l-lysine could not cause this repression because it is quickly consumed in ɛ-PL biosynthesis. In fact, it is known that the addition of l-methionine, l-threonine, or l-isoleucine into the culture medium of S. albulus produces a negative effect for ɛ-PL production, despite there being no negative effects of l-lysine (Shima et al. 1983).
Taken together, in S. albulus, it was suggested that the metabolic flow of the l-lysine molecule, which is the end product of primary metabolism, should be directly linked to the biosynthesis of the secondary metabolite ɛ-PL. In addition, the Ask activity was found to be regulated by repression rather than the feedback inhibition, while the repression mechanisms remain unclear. More in-depth investigation of the repression mechanism(s) of the ask gene in the S. albulus CR1 is now in progress.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bibb MJ, Findlay PR, Johnson MW (1984) The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene 30:157–166
Cuadrado Y, Fernandez M, Recio E, Aparicio JF, Martin JF (2004) Characterization of the ask–asd operon in aminoethoxyvinylglycine-producing Streptomyces sp. NRRL 5331. Appl Microbiol Biotechnol 64:228–236
Follettie MT, Peoples OP, Agoropoulou C, Sinskey AJ (1993) Gene structure and expression of the Corynebacterium flavum N13 ask–asd operon. J Bacteriol 175:4096–4103
Hamano Y, Nicchu I, Hoshino Y, Kawai T, Nakamori S, Takagi H (2005) Development of gene delivery systems for the ɛ-poly-l-lysine producer, Streptomyces albulus. J Biosci Bioeng 99:636–641
Hamano Y, Yoshida T, Kito M, Nakamori S, Nagasawa T, Takagi H (2006) Biological function of the pld gene product that degrades å-poly-l-lysine in Streptomyces albulus. Appl Microbiol Biotechnol 72:173–181
Hernando-Rico V, Martin JF, Santamarta I, Liras P (2001) Structure of the ask–asd operon and formation of aspartokinase subunits in the cephamycin producer ‘Amycolatopsis lactamdurans’. Microbiology 147:1547–1555
Hitchcock MJ, Hodgson B, Linforth JL (1980) Regulation of lysine- and lysine-plus-threonine-inhibitable aspartokinases in Bacillus brevis. J Bacteriol 142:424–432
Itzhaki RF (1972) Colorimetric method for estimating polylysine and polyarginine. Anal Biochem 50:569–574
Kahar P, Iwata T, Hiraki J, Park EY, Okabe M (2001) Enhancement of ɛ-polylysine production by Streptomyces albulus strain 410 using pH control. J Biosci Bioeng 91:190–194
Kalinowski J, Cremer J, Bachmann B, Eggeling L, Sahm H, Puhler A (1991) Genetic and biochemical analysis of the aspartokinase from Corynebacterium glutamicum. Mol Microbiol 5:1197–1204
Kawai T, Kubota T, Hiraki J, Izumi Y (2003) Biosynthesis of ɛ-poly-l-lysine in a cell-free system of Streptomyces albulus. Biochem Biophys Res Commun 311:635–640
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces Genetics. The John Innes Foundation, Norwich, UK
Kobashi N, Nishiyama M, Yamane H (2001) Characterization of aspartate kinase III of Bacillus subtilis. Biosci Biotechnol Biochem 65:1391–1394
Ogawa-Miyata Y, Kojima H, Sano K (2001) Mutation analysis of the feedback inhibition site of aspartokinase III of Escherichia coli K-12 and its use in l-threonine production. Biosci Biotechnol Biochem 65:1149–1154
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Shiio I (1982) Metabolic regulation and over-production of amino acids. In: Krumphanzl V, Sikyta B, Vanek Z (eds) Overproduction of microbial products. Academic, London, pp 463–472
Shiio I, Miyajima R (1969) Concerted inhibition and its reversal by end products of aspartate kinase in Brevibacterium flavum. J Biochem (Tokyo) 65:849–859
Shima S, Sakai H (1977) Polylysine produced by Streptomyces. Agric Biol Chem 41:1807–1809
Shima S, Sakai H (1981a) Poly-l-lysine produced by Streptomyces. III. Chemical studies. Agric Biol Chem 45:2503–2508
Shima S, Sakai H (1981b) Poly-l-lysine produced by Streptomyces. II. Taxonomy and fermentation studies. Agric Biol Chem 45:2497–2502
Shima S, Matsuoka H, Sakai H (1982) Inactivation of bacteriopharges by ɛ-poly-l-lysine produced by Streptomyces. Agric Biol Chem 46:1917–1919
Shima S, Oshima S, Sakai H (1983) Biosynthesis of ɛ-poly-l-lysine by washed mycelium of Streptomyces albulus No. 346. Nippon Nogei KagakuKaishi 57:221–226
Shima S, Matsuoka H, Iwamoto T, Sakai H (1984) Antimicrobial action of ɛ-poly-l-lysine. J Antibiot (Tokyo) 37:1449–1455
Theze J, Margarita D, Cohen GN, Borne F, Patte JC (1974) Mapping of the structural genes of the three aspartokinases and of the two homoserine dehydrogenases of Escherichia coli K-12. J Bacteriol 117:133–143
Tunca S, Yilmaz EI, Piret J, Liras P, Ozcengiz G (2004) Cloning, characterization and heterologous expression of the aspartokinase and aspartate semialdehyde dehydrogenase genes of cephamycin C-producer Streptomyces clavuligerus. Res Microbiol 155:525–534
Zhang JJ, Paulus H (1990) Desensitization of Bacillus subtilis aspartokinase I to allosteric inhibition by meso-diaminopimelate allows aspartokinase I to function in amino acid biosynthesis during exponential growth. J Bacteriol 172:4690–4693
Zhang JJ, Hu FM, Chen NY, Paulus H (1990) Comparison of the three aspartokinase isozymes in Bacillus subtilis Marburg and 168. J Bacteriol 172:701–708
Zhang W, Jiang W, Zhao G, Yang Y, Chiao J (1999) Sequence analysis and expression of the aspartokinase and aspartate semialdehyde dehydrogenase operon from rifamycin SV-producing Amycolatopsis mediterranei. Gene 237:413–419
Zhang WW, Jiang WH, Zhao GP, Yang YL, Chiao JS (2000) Expression in Escherichia coli, purification and kinetic analysis of the aspartokinase and aspartate semialdehyde dehydrogenase from the rifamycin SV-producing Amycolatopsis mediterranei U32. Appl Microbiol Biotechnol 54:52–58
Acknowledgments
This work was supported in part by a grant from Chisso Corporation.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Hamano, Y., Nicchu, I., Shimizu, T. et al. ɛ-Poly-l-lysine producer, Streptomyces albulus, has feedback-inhibition resistant aspartokinase. Appl Microbiol Biotechnol 76, 873–882 (2007). https://doi.org/10.1007/s00253-007-1052-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1052-3