
Abstract
Broad specificity amino acid racemase (E.C. 5.1.1.10) from Pseudomonas putida IFO 12996 (BAR) is a unique racemase because of its broad substrate specificity. BAR has been considered as a possible catalyst which directly converts inexpensive l-amino acids to dl-amino acid racemates. The gene encoding BAR was cloned to utilize BAR for the synthesis of d-amino acids, especially d-Trp which is an important intermediate of pharmaceuticals. The substrate specificity of cloned BAR covered all of the standard amino acids; however, the activity toward Trp was low. Then, we performed random mutagenesis on bar to obtain mutant BAR derivatives with high activity for Trp. Five positive mutants were isolated after the two-step screening of the randomly mutated BAR. After the determination of the amino acid substitutions in these mutants, it was suggested that the substitutions at Y396 and I384 increased the Trp specific racemization activity and the racemization activity for overall amino acids, respectively. Among the positive mutants, I384M mutant BAR showed the highest activity for Trp. l-Trp (20 mM) was successfully racemized, and the proportion of d-Trp was reached 43% using I384M mutant BAR, while wild-type BAR racemized only 6% of initial l-Trp.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
d-Amino acids have been considered as biologically inactive substances, although l-amino acids are components of proteins and play important roles in the metabolism. However, almost all the bacteria synthesize and utilize d-amino acids, such as d-Ala, d-Glu, d-Val, and d-Phe, as the components of their cell membrane (Caparros et al. 1992; Schleifer 1975) or antibiotics (Macdonald 1960; Saito et al. 1995; Stein et al. 1995). Moreover, d-Asp was discovered in mammalian cells, and it is suggested that the formation of d-Asp concerned to aging (Fisher et al. 1991; Fujii et al. 1999; Long et al. 1998; McFadden and Clarke 1982). Therefore, d-amino acids are no longer biologically inactive but are important substances with diverse bioactivity. Moreover, d-amino acids, especially d-Phe and d-Trp, are considered as useful chiral building blocks for synthesis of pharmaceuticals, food additives, and agrochemicals. Indeed, d-Trp is the intermediate in the synthesis of a therapeutic drug for erectile dysfunction (Daugan et al. 2003). In spite of this usefulness, commercially available d-Trp is chemically synthesized and expensive. Therefore, an effective method to synthesize d-Trp is desired.
d-Trp can be synthesized in two-step enzymatic reactions with hydantoinase and carbamoylase from 5′-substituted d,l-hydantoin (Kim et al. 2000; Lee et al. 2001; Nozaki et al. 2005; Rodriguez et al. 2002). Although this process has been studied in detail and the synthetic yield of d-Trp is high (70%), the reaction with two enzymes is complicated, and 5′-substituted d,l-hydantoin, the starting material of this process, is supplied from chemical synthesis, resulting in high cost. d-Trp synthesis from indole pyruvic acid was catalyzed by d-amino acid aminotransferase utilizing d-Ala or d-Glu as amino donor (Galkin et al. 1997). d-Amino acid aminotransferase employs several α-keto acids as amino acceptors and is capable to synthesize several d-amino acids; however, α-keto acids are expensive substances, and subsequently, d-amino acid production using d-amino acid aminotransferase is difficult in the availability of starting material.
Then, we focused amino acid racemase as a capable enzyme to synthesize d-Trp effectively. Amino acid racemase catalyzes racemization of l-amino acid to dl-amino acid; subsequently, d-amino acid is obtained through the chiral selective degradation of l-amino acid in the racemic compound. Moreover, d-amino acid derivatives would be synthesized utilizing d-amino acid selective modification enzymes from dl-amino acid. Therefore, simple and cost-effective production processes of d-amino acid and its derivative could be established. Almost all l-amino acids are produced from glucose in the fermentative process; therefore, inexpensive starting materials could be utilized in dl-amino acid production by amino acid racemase. Although there have been few reports concerning to amino acid racemase acting on d-Trp, amino acid racemase (E.C. 5.1.1.10) from Pseudomonas putida IFO 12996 (BAR) is known to show broad substrate specificity for several amino acids (Badet et al. 1984; Kimuratesaki et al. 1984; Lim et al. 1993; Roise et al. 1984; Shen et al. 1983; Soda and Osumi 1969, 1971). In the previous study of partially purified BAR, they were reported that basic amino acids and a part of neutral amino acids were racemized effectively, and the best substrate of BAR was Lys. On the other hand, acidic amino acids and aromatic amino acids were scarcely recognized as substrates by BAR. However, it was expected that Trp racemization activity might be readily appended to BAR by mutagenesis because of its broad substrate specificity. Then, we have started to clone the gene encoding BAR, and subsequently, random mutagenesis was performed on cloned BAR to obtain the modified BAR with high Trp racemization activity.
Materials and methods
Materials
d-Amino acids were purchased from Kanto Kagaku (Tokyo, Japan) or Kokusan Kagaku (Tokyo, Japan). 1-Fluoro-2,4-dinitrophenyl-5-l-alanine amide and 4-amino antipyrine were from Sigma-Aldrich (St. Louis, MO, USA). His-Trap HP 1-ml column, PD-10 column, and ÄKTA explorer 10S were from Amersham Bioscience (NJ, USA). Model 550 microplate reader was purchased from Bio–Rad Laboratories (CA, USA). L-7000 series high-performance liquid chromatography (HPLC)-equipped WH-C18A (3 μm) column was from Hitachi High-Technologies (Tokyo, Japan). All other chemicals were of reagent grade and obtained from commercial sources.
Bacterial strains, plasmids, and culture conditions
P. putida IFO 12996 and Escherichia coli BL21 (DE3)/pLysS were purchased from the Institute of Fermentation Osaka (Osaka, Japan) and Merck KGaA (Darmstadt, Germany), respectively. E. coli JM 109 was supplied from Nippon Gene (Tokyo, Japan), and Trp auxotroph E. coli JM 101: ΔtnaA, ΔtrpABCDE was the thankful gift from Bio Frontier Laboratories, Kyowa Hakko Kogyo (Tokyo, Japan). Plasmid vector pET 21a (+) and pSTV 28 were from Merck KGaA and Takara Bio (Mie, Japan).
P. putida IFO 12996 and E. coli strains were grown in LB medium at 37°C, with 120 rpm rotary shaking for 16 h. In the case of bar expression, recombinant E. coli BL21 (DE3)/pLysS harboring bar was grown in LB medium containing 50 μg/ml ampicillin, 25 μg/ml chloramphenicol, and 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 30°C, with 120 rpm rotary shaking for 16 h.
Gene cloning of bar
The gene encoding BAR was amplified by polymerase chain reaction using KOD-plus DNA polymerase (Toyobo, Osaka, Japan) with NdeI and EcoRI restriction sites at upstream and downstream of bar, respectively. After the digestion by NdeI and EcoRI, amplified bar was ligated to pET 21a (+) vector using DNA Ligation kit version 2.1 (Takara Bio) and introduced into E. coli JM 109 for plasmid isolation. The isolated recombinant plasmid harboring bar was introduced into E. coli BL 21(DE3)/pLysS.
Purification procedure of His×6-tagged BAR
The recombinant BAR with His×6 tag was produced in recombinant E. coli BL 21(DE3)/pLysS cells. The cells were harvested at 3,000×g for 10 min, and the cell pellet was treated with 5 ml of BugBuster Protein Extraction Reagent (Merck) for 20 min, then centrifuged at 16,000×g for 30 min, and the supernatant was collected as cell-free extract.
The cell-free extract was applied onto a His-Trap HP 1-ml column equilibrated with 100 mM Tris–HCl (pH 8), 150 mM NaCl, and 10 mM imidazole, and the column was washed with 10 column volumes of the same buffer. The His×6-tagged BAR was eluted by linear gradient with 100 mM Tris–HCl (pH 8), 150 mM NaCl, and 500 mM imidazole and desalted on a PD-10 column equilibrated with 100 mM Tris–HCl (pH 8.5).
Random mutagenesis on bar and construction of the mutant library
Ramdom mutagenesis on bar was carried out by error-prone PCR method. The PCR reaction mixture consisted of 10 mM Tris–HCl (pH 8.3), 50 mM KCl, 1 mM MgCl2, 0.5 mM MnCl2, 0.2 mM each of dNTPs, 0.5 pmol/μl each of primers, 800 pg/μl template DNA, and 25 mU/μl Taq DNA polymerase (Takara Bio). The amplified PCR products with EcoRI and BamHI restriction sites at 5′ or 3′-terminus were restricted and ligated to pSTV 28 vector. Then, E. coli JM 101: ΔtnaA, ΔtrpABCDE was transformed with resulting plasmids.
Screening of mutant BAR with Trp racemase activity
First, E. coli JM101: ΔtnaA, ΔtrpABCDE harboring mutated bar were grown in M9 medium containing 5 mg/l d-Trp and 50 mg/l casamino acids at 37°C for 16 h, and then spread onto M9 agar plate containing the same additives and incubated at room temperature until colonies were formed. The colony-forming mutants were applied to the second screening.
In the second screening, the mutants were inoculated to 800 μl of LB medium containing 25 μg/ml chloramphenicol and 0.1 mM IPTG and incubated at 37°C for 16 h. The grown cells were harvested at 2,800×g for 10 min, washed twice with 100 mM Tris–HCl (pH 8.5), and resuspended to the same buffer. Portions (50 μl) of the cell suspensions were mixed with 50 μl of 30 mM l-Trp solution and incubated at 37°C for 16 h, then 100 μl of colorimetric assay mixture consisted of 10 mM Tris-HCl (pH 8.5), 1 U/ml d-amino acid oxidase, 100 U/ml peroxidase, 5 mM phenol and 1 mM 4-amino antipirine was added, and the absorbance at 490 nm was measured. The selected mutant bars from the second screening were subcloned into E. coli BL 21(DE3)/pLysS as mentioned above.
Site-directed mutagenesis
The full length of the recombinant plasmid harboring wild-type bar was amplified by PCR using KOD-plus DNA polymerase with a pair of mutagenic primers to introduce the site-directed mutagenesis. In this PCR amplification, each primer contained 20 bp of overlapping region and 10 bp of each annealing region, and one of them had mutation between overlapping region and annealing region. The amplified PCR product was directly introduced into the E. coli JM 109, and the linear PCR product was cyclized in the host cell. After the plasmid isolation, occurrence of mutation was confirmed by DNA sequencing.
Tertiary structure prediction
The amino acid sequence of wild-type or mutant BAR was submitted to ESyPred3D (Lambert et al. 2002) (http://www.fundp.ac.be/urbm/bioinfo/esypred/) to predict tertiary structure. The predicted tertiary structures were visualized using VMD molecular visualization program (Humphrey et al. 1996).
Results
Cloning of the gene encoding BAR
The amino acid sequence near the active center of BAR has been determined as LTAVLKADAYGXGIGL in previous study (Roise et al. 1984). Then, the regions showing homologies to the amino acid sequence were searched in the complete genome sequence of P. putida KT2440 whose genome was disclosed in the National Center for Biotechnology Information, and three homologous regions were picked up. The ORF 1, ORF 2, and ORF 3 contained the homologous regions of LCAVLKADAYGHGIG (14 identical residues), AVIKADAYGHG (nine identical residues), YGLGIGL (six identical residues), respectively, and the homologies of the three ORFs were compared with the already-known proteins by FASTA. As a result, ORF 1 and ORF 2 showed homologies to Ala racemase, and an ORF 3 showed no homology to other proteins. According to the number of the identical residues and the homology to Ala racemase, the ORF 1, which was composed of 1,230 bp, was cloned from the genome of P. putida IFO 12996.
The ORF 1 from P. putida IFO 12996 was overexpressed in E. coli BL 21(DE3)/pLysS. After Ni2+ affinity chromatography, the protein was purified to near homogeneity in SDS-polyacrylamide gel electrophoresis. The deduced molecular mass of the protein was 45 kDa, and it was agreed with the molecular mass of BAR (42 kDa) including His×6 tag (3 kDa). Besides, as shown in Table 1, the protein from ORF 1 from P. putida IFO 12996 showed apparent racemase activity for Lys and Arg which were excellent substrates of BAR. Therefore, it was concluded that the ORF 1 from P. putida IFO 12996 encoded BAR. In addition, it was revealed that BAR showed slight racemase activity toward some amino acids including Trp. The nucleotide sequence of BAR has already been submitted to DNA Data Bank of Japan (accession number; BD373122) as a part of a patent by other researchers (Ikeda et al. 2003); however, the slight racemase activity for Trp and some more amino acids was newly revealed in this study.
Introduction of random mutagenesis on bar
The mutant library of BAR was constructed by the method of error-prone PCR. Although the mutation ratio is generally used to evaluate the mutant library by checking DNA sequence, it is not suitable for evaluation of a number of mutants. Then, a shift of the l-Leu racemizing activity was measured to confirm introduction of random mutagenesis on bar. Although l-Leu was a poor substrate of BAR, racemizing rate of l-Leu was more suitable rather than that of good substrates for quantitative analysis of a number of mutants. As shown in Fig. 1, almost all the arbitrarily selected mutants showed shifted racemase activity compared with wild-type BAR; therefore, it was confirmed that mutations were certainly introduced to bar.

Inactivation ratio of randomly mutated BARs. Twenty mutants were selected from mutant library of BAR generated by random mutagenesis and applied to colorimetric assay. The absorbance of wild-type BAR on colorimetric assay is shown in dotted line
Screening of mutant BAR with high Trp racemase activity
In the first screening of mutant BAR, the complementation of l-Trp auxotrophy was employed as indication for the increased Trp racemase activity. On the M9 agar plate containing d-Trp, only the l-Trp auxotroph E. coli JM 101: ΔtnaA, ΔtrpABCDE harboring mutant bar with increased Trp racemase activity was able to convert sufficient amount of d-Trp to l-Trp which was required for their growth. Through this screening, 80 positive mutants were isolated from 30,000 mutants.
The isolated 80 positive mutants were applied onto the second screening mentioned in the “Materials and methods”. As shown in Fig. 2, there was a positive mutant with the highest increase of 210% activity. However, only one mutant was not enough to investigate the diversity of positive mutations; then we isolated five positive mutants whose Trp racemase activities markedly increased, and the mutants were named mutants 1, 2, 3, 4, and 5, respectively.

Colorimetric assay of positive mutants from the first screening. The relative activities indicate the absorbance of 80 positive mutants when that of wild-type BAR was regarded as 100%
Amino acid substitutions and specific activities of mutant BARs
The specific activities of the mutants for Trp, Phe, Lys, and Ala were measured, and the amino acid substitutions were determined by DNA sequencing (Table 2). The mutants showed five- to 20-fold increased specific activities for Trp; besides, the specific activities of mutant 4 were also increased when the other amino acids were employed as substrates. On the other hand, the specific activity for Trp was selectively increased in mutants 1, 2, 3, and 5. Therefore, it was suggested that there were two types of mutants. From the analysis of the amino acid substitutions, mutant 4 which only showed the increased specific activity for overall amino acids had I384M extra substitution, although all the mutants had the amino acid substitution at Y396.
Then, site-directed mutagenesis was performed at Y396 and I384. The generated Y396C, Y396H, and I384M single mutants showed the same tendencies in increase of activity (Table 3). Therefore, it was definitely confirmed that Y396C and Y396H substitutions concerned to the selective increase of specific activity for Trp, and I384M substitution concerned to the increase of specific activities for overall amino acids.
Racemization of l-Trp using modified BAR
The modified BARs with the high Trp racemizing activity were obtained by Y396 and I384 substitution, and then racemization of l-Trp was tested. As shown in Fig. 3, Y396C and wild-type BAR partially racemized l-Trp, and the proportions of d-Trp in the reaction mixture were 12.5 and 6%, respectively. On the other hand, I384M mutant BAR racemized almost all l-Trp to dl-Trp in the 24-h reaction.

l-Trp racemization with modified BAR. The modified and wild-type BARs were incubated with 20 mM l-Trp. After the incubation, racemized d-Trp was measured by HPLC. Symbols indicate the amount of produced d-Trp with I384M mutant, circle; Y396C mutant, triangle; and wild-type BAR, square
Discussion
In this study, the gene encoding broad specificity amino acid racemase from P.putida IFO 12996 was cloned. BAR showed high homology to bacterial Ala racemases; therefore, BAR was considered as a kind of Ala racemase. Bacterial Ala racemase catalyzes the racemization of l-Ala to dl-Ala, and subsequently, d-Ala is supplied to the biosynthesis of peptidoglycan. Therefore, BAR is considered to have the same function in P. putida IFO 12996. However, there is no report about the existence of d-amino acid except d-Ala or d-Glu in the cell wall of P. putida. On the other hand, it has been reported that l-Lys was partially metabolized through d-Lys and suggested that there was Lys racemase in P. putida (Revelles et al. 2005). Therefore, BAR, which showed the highest activity toward Lys, would participate in the l-Lys metabolic pathway. At least, it is still unclear why BAR shows such broad substrate specificity.
From the analysis of the positive mutants obtained from random mutagenesis, it was suggested that the amino acid substitutions at Y396 and I384 increased the Trp racemizing activity of BAR, but the increased activities were achieved in the different mechanism. In the comparison with I384M single mutant and the double (I384M, Y396C) mutant obtained from random mutagenesis, I384M single mutant showed the higher specific activity for Trp, and the apparent effect of Y396C substitution was not observed on the double mutant. This suggests that the effect of the amino acid substitution at I384 on the activity of BAR is much higher than that of the amino acid substitution at Y396. However, Y396 is the only residue concerning to the substrate specificity of BAR; therefore, the further amino acid substitutions at Y396 are expected to contribute to elucidate the substrate recognition mechanism of BAR.
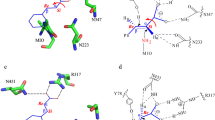
Among bacterial Ala racemases, the tertiary structures of Ala racemase from Bacillus stearothermophilus (Shaw et al. 1997) and Pseudomonas aeruginosa (LeMagueres et al. 2003) were reported so far (Fig. 4a,b). Both reported tertiary structures showed that the monomer of Ala racemase was consisted of two domains, and the N-terminal domain mainly constructed the active center. However, it was reported about Ala racemase from B. stearothermophilus that Y354, which is located in the region with tandem two α-helixes in C-terminal domain, was the residue to concern to construction of the active center, and the residue was considered to immobilize PLP cofactor. Because the Tyr residues were conserved in both Ala racemases from B. stearothermophilus and P. aeruginosa (Y341), it was suggested that other Ala racemases would have the same Tyr residue. In the C-terminal sequence of BAR, Y396 was the only Tyr residue, and it was supposed to be located in the same region as Tyr residues in other Ala racemases from the predicted tertiary structure (Fig. 4c). Then, it is considered that Y396 partially has interaction with PLP, and the substitution of Y396C or Y396H would change the conformation of active center to be more suitable for Trp. Besides, I384 was located at the same tandem two α-helixes; therefore, it is supposed that I384M substitution influences the formation of active center through the translocation of Y396 or the tandem two α-helixes themselves.

Tertiary structure of Ala racemase monomers. The Lys and Tyr residues which had interactions to PLP were pointed in the crystal structure of already-known Ala racemases from B. stearothermophilus (a) and P. aeruginosa (b), and their PDB IDs were 1SFT and 1RCQ, respectively. K75, Y396, and I384 were depicted in the predicted tertiary structure of BAR (c)
In the study of the l-Trp racemization, 20 mM l-Trp was partially racemized, and 2.5 mM d-Trp was formed by Y396C mutant BAR. On the other hand, almost all the initial l-Trp was racemized, and 8.6 mM d-Trp was formed by I384M mutant in 24 h. Although some slight racemization activities on Trp were reported, it was a novel report that I384M mutant BAR showed high racemizing activity of Trp as an enzyme. The d-Trp yield of 1.8 g/l was quite low compared to other d-Trp synthesizing processes utilizing hydantoinase–carbamoylase or d-amino acid aminotransferase. Besides, the product from the process utilizing BAR was a racemate. However, l-Trp is much more inexpensive than the starting materials of other processes, such as 5′-monosubstituted hydantoin or α-keto acid; therefore, chiral resolution utilizing l-Trp-specific degrading enzymes would be a cost-effective process to obtain optically pure d-Trp. Moreover, it is expected that I384M mutant BAR could be effectively utilized in the coupling with other enzymatic reactions to synthesize d-Trp derivatives.
References
Badet B, Floss HG, Walsh CT (1984) Stereochemical studies of processing of d- and l-isomers of suicide substrates by an amino acid racemase from Pseudomonas striata. J Chem Soc Chem Commun 1984:838–840
Caparros M, Pisabarro AG, De Pedro MA (1992) Effect of d-amino acids on structure and synthesis of peptidoglycan in Escherichia coli. J Bacteriol 174:5549–5559
Daugan A, Grondin P, Ruault C, Gouville ACLM, Coste H, Linget JM, Kirilovsky J, Hyafil F, Labaudiniere R (2003) The discovery of tadalafil: a novel and highly selective PDE5 inhibitor. 2:2,3,6,7,12,12a-hexahydropyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione analogues. J Med Chem 46:4533–4542
Fisher GH, D’Aniello A, Vetere A, Padula L, Cosano GP, Man EH (1991) Free d-aspartate and d-alanine in normal and Alzheimer brain. Brain Res Bull 26:983–985
Fujii N, Harada K, Momose Y, Ishii N, Akabosh M (1999) d-Amino acid formation induced by a chiral field within a human lens protein during aging. Biochem Biophys Res Commun 263:322–326
Galkin A, Kulakova L, Yoshimura T, Soda K, Esaki N (1997) Synthesis of optically active amino acids from alpha-keto acids with Escherichia coli cells expressing heterologous genes. Appl Environ Microbiol 63:4651–4656
Humphrey W, Dalke A, Schulten K (1996) VMD—Visual Molecular Dynamics. J Mol Graph 14:33–38
Ikeda H, Yonetani Y, Hashimoto S, Yagasaki M, Soda K (2003) Amino acid racemase having low substrate specificity and process for producing racemic amino acid. PCT Patent WO 2003/074690
Kim GJ, Lee DE, Kim HS (2000) Construction and evaluation of a novel bifunctional N-carbamylase-d-hydantoinase fusion enzyme. Appl Environ Microbiol 66:2133–2138
Kimuratesaki N, Tanaka H, Soda K (1984) Inactivation of amino acid racemase by S-(N-methylthiocarbamoyl)-d,l-cysteine. Agric Biol Chem 48:383–388
Lambert C, Leonard N, De Bolle X, Depiereux E (2002) ESyPred3D: prediction of proteins 3D structures. Bioinformatics 18:1250–1256
Lee CK, Lee ZS, Yang PF (2001) Effect of cell membrane of Agrobacterium radiobacter on enhancing d-amino acids production from racemic hydantoins. Enzyme Microb Technol 28:806–814
LeMagueres P, Im H, Dvorak A, Strych U, Benedik M, Krause KL (2003) Crystal structure at 1.45 Å resolution of alanine racemase from a pathogenic bacterium, Pseudomonas aeruginosa, contains both internal and external aldimine forms. Biochemistry 42:14752–14761
Lim YH, Yokoigawa K, Esaki N, Soda K (1993) A new amino acid racemase with threonine alpha-epimerase activity from Pseudomonas putida: purification and characterization. J Bacteriol 175:4213–4217
Long Z, Homma H, Lee JA, Fukushiia T, Santa T, Iwatsubo T, Yamada R, Imai K (1998) Biosynthesis of d-aspartate in mammalian cells. FEBS Lett 434:231–235
Macdonald JC (1960) Biosynthesis of valinomycin. Can J Microbiol 6:27–34
McFadden PN, Clarke S (1982) Methylation at d-aspartyl residues in erythrocytes: possible step in the repair of aged membrane proteins. Proc Natl Acad Sci USA 79:2460–2464
Nozaki H, Takenaka Y, Kira I, Watanabe K, Yokozeki K (2005) d-Amino acid production by E. coli co-expressed three genes encoding hydantoin racemase, d-hydantoinase and N-carbamoyl-d-amino acid amidohydrolase. J Mol Catal B Enzym 32:213–218
Revelles O, Espinosa-Urgel M, Fuhrer T, Sauer U, Ramos JL (2005) Multiple and interconnected pathways for l-lysine catabolism in Pseudomonas putida KT2440. J Bacteriol 187:7500–7510
Rodriguez SM, Vazquez FJLH, Jimenez JMC, Cazorla LM, Vico FR (2002) Complete conversion of d,l-5-monosubstituted hydantoins with a low velocity of chemical racemization into d-amino acids using whole cells of recombinant Escherichia coli. Biotechnol Prog 18:1201–1206
Roise D, Soda K, Yagi T, Walsh CT (1984) Inactivation of the Pseudomonas striata broad specificity amino acid racemase by d and l isomers of beta-substituted alanines: kinetics, stoichiometry, active site peptide, and mechanistic studies. Biochemistry 23:5195–5201
Saito M, Hori K, Kurotsu T, Kanda M, Saito Y (1995) Three conserved glycine residues in valine activation of gramicidin S synthetase 2 from Bacillus brevis. J Biochem (Tokyo) 117:276–282
Schleifer KH (1975) Chemical structure of the peptidoglycan, its modifiability and relation to the biological activity. Z Immunitatsforsch Exp Klin Immunol 149:104–117
Shaw JP, Petsko GA, Ringe D (1997) Determination of the structure of alanine racemase from Bacillus stearothermophilus at 1.9-Å resolution. Biochemistry 36:1329–1342
Shen SJ, Floss HG, Kumagai H, Yamada H, Esaki N, Soda K, Wasserman SA, Walsh CT (1983) Mechanism of pyridoxal phosphate-dependent enzymatic amino-acid racemization. J Chem Soc Chem Commun 1983:82–83
Soda K, Osumi T (1969) Crystalline amino acid racemase with low substrate specificity. Biochem Biophys Res Commun 35:363–368
Soda K, Osumi T (1971) Amino acid racemase (Pseudomonas striata). Methods Enzymol 17:629–636
Stein T, Kluge B, Vater J, Franke P, Otto A, Wittmann-Liebold B (1995) Gramicidin S synthetase 1 (phenylalanine racemase), a prototype of amino acid racemases containing the cofactor 4′-phosphopantetheine. Biochemistry 34:4633–4642
Acknowledgments
We are deeply grateful to Kyowa Hakko Kogyo for providing E. coli JM 101 ΔtnaA, ΔtrpABCDE, and Dr. Makoto Yagasaki in Bio Frontier Laboratories, Kyowa Hakko Kogyo for his helpful suggestions.
This study was supported in part by the 21C-COE Program “Practical Nano-Chemistry” from The Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and by a Waseda University Grant for Special Research Projects (2005B-142).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kino, K., Sato, M., Yoneyama, M. et al. Synthesis of dl-tryptophan by modified broad specificity amino acid racemase from Pseudomonas putida IFO 12996. Appl Microbiol Biotechnol 73, 1299–1305 (2007). https://doi.org/10.1007/s00253-006-0600-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0600-6