
Abstract
An extracellular endo-1,4-β-xylanase was purified from the culture supernatant of the ascomycete Aureobasidium pullulans ATCC 20524 grown on xylan. The purified enzyme was homogeneous as judged by sodium dodecyl sulphate–polyacrylamide gel electrophoresis and isoelectric focusing, which showed an apparent M r of 39 kDa and a pI of 8.9, respectively. Xylanase activity was optimal at pH 6.0 and 70°C. The genomic DNA and cDNAs encoding this protein were cloned and sequenced. The xylanase gene (xynII) encoded a 26 amino acid signal peptide and a 335 amino acid mature protein. DNA regions encoding the signal sequence and the mature protein were interrupted by introns of 56 and 73 bp, respectively. The xynII 5′-noncoding region had two consensus binding sites (5′-GCCARG-3′) for the transcription factor PacC mediating pH regulation. Quantitative real-time polymerase chain reaction analysis revealed that the transcription levels at pH 6.0 and 8.0 were 8-fold and 22-fold higher than that at pH 2.7, respectively. A cloned xynII cDNA was expressed and secreted in the yeast Pichia pastoris. Sequence alignment and phylogenetic analysis suggested that the XynII belongs to glycosyl hydrolase family 10 and that it is evolutionarily distant from two clusters formed by other family-10 xylanases.
Similar content being viewed by others

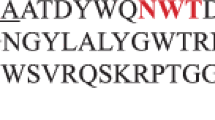

Avoid common mistakes on your manuscript.
Introduction
Xylan is a major constituent of hemicelluloses in the cell walls of monocots and hard woods (Puls and Schuseil 1993). It consists of a linear backbone of β-1,4-linked d-xylopyranose residues, which commonly contains side branches of arabinofuranose, glucuronic acid, and methylglucuronic acid. Efficient enzymatic hydrolysis of xylan is essential for the industrially viable process that utilizes cellulosic residues. The attention on the potential applications of xylan-degrading enzymes has led to a finding of numerous microbial enzymes that differ in biochemical properties. Two key reactions proceed during hydrolysis of xylan; endo-1,4-β-xylanases (xylanases; 1,4-β-d-xylan xylanohydrolase, EC 3.2.1.8) cleave internal linkages in the xylan backbone to yield oligosaccharides, which are further hydrolyzed by β-xylosidases (1,4-β-d-xylan xylohydrolase, EC 3.2.1.37) to xylose. Many fungi, bacteria, and actinomycetes secrete multiple isozymes of xylanase as distinct gene products. These xylanases have been classified into two glycosyl hydrolase families 10 (formerly F) and 11 (formerly G) on the basis of amino acid sequence similarities (Henrissat 1991; Henrissat and Bairoch 1993). Family 11 consists of xylanases with a relatively low M r ranging from 19 to 25 kDa, whereas the family 10 comprises xylanases with a higher M r of >30 kDa. A hierarchy among the family-11 and -10 xylanases was proposed for microbial degradation of plant cell wall xylan to xylose (Pell et al. 2004). Therefore, a molecular characterization of such multiple xylanases produced by a single microorganism is important for a better understanding of their physiological roles in xylan degradation process.
The ascomycete Aureobasidium pullulans (de Bary) Arnaud, a saprophyte that occasionally occurs on leaf surfaces of plants, is a promising candidate for the application of genetic engineering (Deshpande et al. 1992). A. pullulans has been reported to produce a family-11 xylanase exhibiting high specific activity (Leathers 1986; Leathers 1989; Li et al. 1993; Ohta et al. 2001). The A. pullulans xylanase gene was cloned and sequenced (Li and Ljungdahl 1994; Ohta et al. 2001), and its cDNA was expressed in the yeast Saccharomyces cerevisiae or the methylotrophic yeast Pichia pastoris (Li and Ljungdahl 1996; Tanaka et al. 2004). To date, however, there have been no reports of family-10 xylanases from A. pullulans.
In the present paper, we describe the purification and properties of an extracellular family-10 xylanase from the fungal strain and the cloning and sequencing of a genomic DNA and cDNAs encoding the enzyme. Moreover, we defined the phylogenetic position of the A. pullulans xylanase among homologous xylanases belonging to family 10.
Materials and methods
Fungal strain and culture conditions
A wild-type strain A. pullulans var. melanigenum ATCC 20524 (Ohta et al. 2001) was used in this study. Growth medium contained 0.67% (w/v) yeast nitrogen base (Difco Laboratories, Detroit, MI, USA) and 1.0% (w/v) oat-spelt xylan (Sigma Chemical Co., St. Louis, MO, USA) as the sole carbon source in 0.1 M phosphate buffer (pH 6.0), unless otherwise stated. Liquid cultures were grown on a rotary shaker (150 rpm) in 500-ml Erlenmeyer flasks containing 100 ml of medium at 30°C for the time specified in the text.
Enzyme and protein assays
The reaction mixture consisted of 0.2 ml of a 1.0% (w/v) suspension of oat-spelt xylan in deionized water and 0.2 ml of a suitably diluted enzyme solution in 0.1 M sodium-acetate-HCl buffer (pH 6.0). After incubation at 45°C for 30 min, reducing sugars were determined by the method of Somogyi (1952) and Nelson (1955). One unit (U) of xylanase activity was defined as the amount of enzyme that liberated 1 μmol of xylose equivalents from xylan per minute. To study the substrate specificity, the reaction mixture consisting of 0.5 ml of the enzyme solution and 0.5 ml of 10 mM p-nitrophenyl (pNP) glycosides (pNP-β-d-cellobioside, pNP-β-d-xylopyranoside, and pNP-α-l-arabinofuranoside; Sigma) was incubated at 45°C for 10 min. The reaction was stopped by the addition 1.0 ml of 1 M Na2CO3, and absorbance at 410 nm was measured. One unit (U) of enzyme activities toward pNP glycosides was defined as the amount of enzyme that liberated 1 μmol of p-nitrophenol per minute from each substrate. Protein concentrations were measured by the method of Lowry et al. (1951), using bovine serum albumin (Sigma) as the standard.
Enzyme purification
The submerged cultures grown for 5 days were centrifuged for 30 min at 6,000×g at 4°C. The culture supernatant (1,900 ml) was concentrated to one tenth of its original volume in dialysis tubing surrounded by a thick layer of dry polyethylene glycol 20,000. The sample was further concentrated in an ultrafiltration cell by passage through a 3×103 molecular weight cutoff membrane (Diaflo YM3 Amicon, Beverly, MA, USA). The concentrate was subjected to cation exchange chromatography on a CM-Cellulofine C-500 (Seikagaku Kogyo, Tokyo, Japan) column (2.6×45 cm) that had been equilibrated with 20 mM acetate buffer (pH 6.0). The adsorbed proteins were eluted at a flow rate of 1.0 ml/min with a linear gradient of 0 to 1.0 M NaCl in the same buffer. The fractions exhibiting the enzyme activity were pooled and further purified by gel permeation chromatography on a Superdex 75 pg (Amersham Biosciences, Piscataway, NJ, USA) column (1.6×60 cm) at a flow rate of 0.5 ml/min with 10 mM acetate buffer (pH 6.0) containing 0.15 M NaCl.
SDS-PAGE, N-terminal amino acid sequencing, and IEF
The purified enzyme was subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) according to the method of Laemmli (1970). Gels were stained for protein with Coomassie brilliant blue R-250. For the internal sequencing, the purified enzyme was cleaved by protease from Staphylococcus aureus V8 (Wako Pure Chemical Industries, Osaka, Japan). The resulting peptide fragments were separated by SDS-PAGE. The protein bands were electroblotted onto a polyvinylidene difluoride membrane (Bio-Rad Laboratories, Hercules, CA, USA) as described previously (Nakamura et al. 1997). The N-terminal amino acid sequences of the intact protein and the resulting peptide fragments were identified using a Procise 492 protein sequencing system (Applied Biosystems, Foster City, CA, USA).
Analytical isoelectric focusing (IEF) was performed with a Multiphor II electrophoresis system (Amersham Biosciences) using Ampholine PAGplate (pH 3.5 to 9.5) according to the manufacturer's instructions. An IEF calibration kit (Amersham Biosciences) was used to determine the pI value.
TLC analysis
The reaction mixture (50 μl) consisting of equal volumes of a 1% (w/v) solution of xylooligosaccharides (Megazyme, Wicklow, Ireland) or 3% (w/v) suspension of birch-wood xylan (Sigma) and the enzyme solution (20 mU/ml) in 50 mM phosphate buffer (pH 6.0) was incubated at 45°C. Hydrolysis was stopped by boiling for 10 min, and the hydrolysis products were analyzed for thin-layer chromatography (TLC) on silica gel plates (Merck AG, Darmstadt, Germany). The TLC plates were developed twice at room temperature with a solvent system of 1-butanol, pyridine, and water (3:2:1, v/v). Spots were stained by spraying the plates with orcinol-sulfuric acid reagent and then heating at 120°C for 5 min.
DNA manipulations and analyses
Restriction endonucleases and DNA-modifying enzymes were used as recommended by the supplier (Nippon Gene, Tokyo, Japan). Standard molecular cloning techniques were performed as described by Sambrook and Russel (2001). Polymerase chain reactions (PCRs) were done in a thermal cycler (Takara Bio, Otsu, Japan). The nucleotide sequences were determined with an ABI Prism 310 genetic analyzer using the BigDye Terminator version 3.1 cycle sequencing kit (Applied Biosystems). A neighbor-joining tree was constructed from the aligned amino acid sequences using the TREECON software package as described previously (Akimoto et al. 2000).
Construction of a xylanase-specific DNA probe
A pair of degenerate 20-mer oligonucleotides was designed and synthesized according to the N-terminal amino acid sequence GLAQAWT and the internal amino acid sequence MKWESTE of the purified enzyme (see Results): primer 1 (forward) (5′-GGY CTY GCY CAG GCY TGG AC-3′) and primer 2 (reverse) (5′-TCR GTR SWY TCC CAY TTC AT-3′). The primers amplified a 152-bp internal coding region of the potential xylanase gene, designated as xynII, from the A. pullulans genomic DNA as the template. The amplified fragment was cloned into pCR2.1-TOPO with the TOPO TA cloning kit (Invitrogen, Carlsbad, CA, USA) and sequenced to confirm its identity. The DNA fragment was then labeled with digoxigenin (DIG)-11-dUTP by the random-primed method using DIG DNA labeling and detection kit (Roche Diagnostics, Mannheim, Germany) for use as hybridization probe.
Isolation of poly(A)+ RNA and cDNA cloning
Mycelia were harvested from 72-h-old culture by centrifugation, and total RNA was isolated using an ISOGEN RNA isolation kit (Wako). Poly(A)+ RNA was obtained from total RNA by PolyATract mRNA Isolation System IV (Promega, Madison, WI, USA). The 5′ and 3′ ends of xynII transcripts were determined by 5′ and 3′ rapid amplification of cDNA ends (RACE) using a SMART RACE cDNA amplification kit (Clontech, Palo Alto, CA, USA) as described previously (Ohta et al. 2001). For 5′ RACE, primer 3 (NUP provided in the kit) and primer 4 [5′-TCG GTA GAC TCC CAC TTC AT-3′ complementary to nucleotides (nt) 285 to 304 relative to the A of the ATG start codon (see Fig. 3)] were used. For 3′ RACE, primer 5 (5′-AGA ATG CCA TGA AGT GGG A-3′ corresponding to nt 277 to 295) and primer 3 (see above) were used. The 5′ and 3′ RACE products were cloned into pCR2.1-TOPO and sequenced.
Quantitative real-time PCR
Mycelia were harvested from 72-h-old culture grown with pH control at pH 6.0 or 8.0 by 0.1 M phosphate buffer or without pH control (initial pH of 6.0). Total RNA was extracted from each mycelium as described above. First-strand cDNA was synthesized using total RNA as template and oligo d(T) primers by reverse transcriptase (RT) (ReverTra Ace, Toyobo, Osaka, Japan). The xynII transcripts were quantified by real-time PCR with an ABI Prism 7000 (Applied Biosystems) using the first-strand cDNA as the template and SYBR Premix Ex Taq (Takara) according to the manufacturer's instructions. Quantification was based on a 104-bp amplicon generated using the following gene-specific primers: primer 6 (forward) (5′-CCA CTG ACG CGA AGC TCA A-3′) and primer 7 (reverse) (5′-GGA AAC ACC CCA GAC AGT GAT AC-3′) (the coding region interrupted by intron). The expression of a housekeeping 18S rRNA gene of A. pullulans (Li et al. 1996) as an endogenous reference was measured in parallel PCR runs using primer 8 (forward) (5′-TTG TCT GCT TAA TTG CGA TAA CGA-3′) and primer 9 (reverse) (5′-GCT TGA GCC GAT AGT CCC TCT AA-3′). The thermal cycle program consisted of an initial step of 95°C for 10 s and 50 cycles of 95°C for 5 s and 60°C for 31 s. After determining amplifications, the target specificity was examined by the melting point analysis of RT-PCR products using a dissociation protocol, in which the temperature was raised linearly from 60°C. Each dissociation curve of the amplicons was observed as a single peak. In addition, amplicons of the expected size were confirmed by agarose gel electrophoresis. Real-time PCR data were processed using the relative quantitative method based on simulation of PCR kinetics (Liu and Saint 2002). Amplification efficiencies were calculated for each sample as
where R n is reporter fluorescence at cycle n, R n,A and R n,B are R n at arbitrary thresholds A and B in an individual curve, respectively, and C T,A and C T,B are the threshold cycles at these arbitrary thresholds. Normalization of xynII expression level to that of the rRNA gene was presented as
where R 0,xynII and R 0,rRNA are the initial copy numbers and C T,rRNA and C T,xynII are threshold cycles at the arbitrary thresholds. In this experiment, the xynII transcript level observed in A. pullulans grown without pH control was treated as the basal expression level.
Construction of yeast expression plasmid and P. pastoris transformation
The xynII cDNA encoding the precursor protein that includes a secretory signal sequence was amplified from the first-strand cDNA as the template with a pair of primers 5′-GGAATTCCATGCACTTCTCCACAATCAC-3′ (forward) and 5′-GGAATTCCT GAA TAC AAC TTG ATA CAT T-3′ (reverse) (letters in bold type indicate the xynII coding sequence) containing EcoRI sites (underlined). The PCR product was cloned into the integrative yeast expression vector pPIC3.5 (Invitrogen) at EcoRI site in the orientation of transcription from AOX1 promoter, yielding pXYN207. The pXYN207 was linearized with SacI, and the yeast P. pastoris GS115 (his4; Invitrogen) was transformed with the DNA fragment by electroporation as described previously (Tanaka et al. 2004).
Culture conditions for P. pastoris transformants and purification of recombinant enzyme
P. pastoris transformants were grown in 500-ml Erlenmeyer flask on an orbital shaker (150 rpm) at 30°C for 72 h. Each flask contained 100 ml of the buffered methanol complex medium as described previously (Tanaka et al. 2004). Additional methanol was supplied every 24 h to give a final concentration of 0.5% (v/v) during the culture period. Clear supernatant obtained from the contents of duplicate flasks was dialyzed against deionized water and lyophilized. The lyophilized sample was dissolved in 5 ml of 20 mM acetate buffer (pH 6.0) and subjected to purification by SP-Sepharose (Amersham Biosciences) and Superdex 75 pg column chromatographies.
Nucleotide sequence accession number
The nucleotide sequence of the 3,293-bp region containing the xynII gene will appear in the DDBJ/EMBL/GenBank nucleotide sequence databases with the accession number AB201542.
Results
Purification and properties of the xylanase
Table 1 summarizes the procedure for the purification of family-10 xylanase from A. pullulans. Cation exchange chromatography of the adsorbed proteins on a CM-Cellulofine column resulted in one major peak of xylanase with activity measured at pH 6.0. Meanwhile, most of the family-11 enzyme (XynI), the purification and characterization of which have been previously reported (Ohta et al. 2001), was not adsorbed onto the column. The family-10 xylanase was eluted from a Superdex 75 pg column as a single protein peak that coincided with the peak of enzyme activity. This protocol afforded a 48-fold purification of the xylanase from the culture supernatant with a yield of 27%. The enzyme was homogeneous as judged by SDS-PAGE (Fig. 1) and IEF (data not shown), which showed an apparent M r of 39 kDa and a pI of 8.9, respectively. The purified enzyme had a specific activity of 29.5 U/mg. The N-terminal amino acid sequencing identified the first 17 residues: S-Y-S-K-N-Q-G-L-A-Q-A-W-T-S-K-G-R. Internal sequences were identified for the two peptide fragments recovered after cleavage by V8 protease: N-A-M-K-W-E-S-T-E-P and L-D-V-R-F-T-T-P-A-T.

SDS-PAGE of purified native xylanase from A. pullulans and the recombinant xylanases expressed in P. pastoris. Protein was visualized by Coomassie brilliant blue R-250 staining. Lanes (1), standard proteins; (2), the native xylanase; (3), the recombinant xylanase P-I; and (4), the recombinant xylanase P-II
The xylanase activity was optimal at pH 6.0 and 70°C. The enzyme retained greater than 80% of the original activity between pH 4.0 and 10.0. The thermal stability was determined by incubating the enzyme solution in 0.1 M phosphate buffer (pH 6.0) at temperatures from 10 to 90°C for 30 min. The enzyme remained stable up to 70°C, but it lost the activity at 80°C. The xylanase activity was not influenced by ethylenediaminetetraacetic acid (112%), guanidine hydrochloride (110%), and p-chloromercuribenzoate (109%) at a final concentration of 1 mM, suggesting that any metal ions and sulfhydryl groups in the enzyme were not essential for the enzymatic reaction.
The purified xylanase was studied for its substrate specificity. The xylanase hydrolyzed birch-wood xylan (Sigma), with a similar activity toward oat-spelt xylan (Table 2). This enzyme also showed weak activities toward pNP-β-d-cellobioside and pNP-β-d-xylopyranoside, but no detectable activity toward carboxymethyl cellulose and pNP-α-l-arabinofuranoside. TLC analysis indicated that initial hydrolysis of birch-wood xylan by the xylanase yielded a series of oligosaccharides with a degree of polymerization of two and above (Fig. 2a). Xylotriose was produced as an intermediate that was eventually cleaved to xylobiose and xylose. The enzyme hydrolyzed xylotriose, xylotetraose, and xylopentaose, but not xylobiose (Fig. 2b).

Thin-layer chromatogram of hydrolysis products from birch-wood xylan (a) and xylooligosaccharides (b) by purified xylanase from A. pullulans. The enzyme reaction was done as described in the text. Xylose (X 1), xylobiose (X 2), xylotriose (X 3), xylotetraose (X 4), and xylopentaose (X 5) were used as standards (St)
Cloning of the xylanase gene
The nucleotide sequence of the 152-bp fragment amplified from the A. pullulans genomic DNA (see Materials and methods) showed that the deduced amino acid sequence included the N-terminal and internal sequences of the purified xylanase. Southern blots of the genomic DNA digested with various restriction enzymes were probed with the DIG-labeled 152-bp fragment. A hybridized 9.0-kbp EcoRI DNA fragment containing the xynII gene was cloned into the EcoRI site of pUC18 to generate a plasmid pXYN204. The 3.8-kbp XbaI-KpnI fragment from pXYN204 was subcloned into pUC18 to generate pXYN205.
Nucleotide sequences of the xynII gene and its cDNAs
The 3.8-kbp XbaI-KpnI insert in pXYN205 was sequenced for the first 3,293-bp from the KpnI site. The 3,293-bp sequence contained a complete xynII open reading frame and its flanking regions (Fig. 3). The first inflame ATG downstream of the transcription start points (see below) was the deduced start codon, which had a consensus A residue at the −3 position (Kozak 1989). A comparison of the xynII genomic and cDNA sequences showed that the regions encoding the signal sequence and the mature protein were interrupted by introns of 56 and 73 bp, respectively. Both introns fitted the GT/AG rule for 5′ and 3′ splicing sites (Gurr et al. 1987).

Nucleotide sequence of A. pullulans xynII gene and its flanking regions and deduced amino acid sequence. The noncoding sequences, including introns, are shown by lower case letters. Two transcription start points and three polyadenylation sites are indicated by open and solid arrowheads, respectively. Two putative PacC binding sites in the xynII promoter region and a putative polyadenylation signal are double and wavy underlined, respectively. Amino acid sequences underlined by thick line were identical to those found for the purified protein. The putative catalytic Glu residues are indicated by bold letters. Potential sites for N-linked glycosylation are underlined. The positions of gene-specific primers, P1 to P7 (excluding the universal primer P3), are indicated by horizontal arrows above the sequence
Two transcription start points were present at nt −60 (A) (three clones) and −54 (A) (three clones) as determined by sequence analysis of six independent cDNA clones of 5′ RACE products. The xynII 5′-noncoding region did not contain the TATAAA and CCAAT motifs. A consensus binding site for the CreA repressor (5′-SYGGRG-3′), which mediates carbon catabolite repression in Aspergillus nidulans (Cubero and Scazzocchio 1994), was present at nt −294 (GTGGGG). In addition, consensus and near-consensus binding sites for the zinc finger transcription factor PacC (5′-GCCARG-3′), which mediates gene expression by the ambient pH in A. nidulans (Pañalva and Arst 2002), were present at nt −832 (GCCAAG) and −1136 (GCCATG of the complementary strand). However, the binding motif 5′-GGCTAAA-3′ for the transcriptional activator XlnR of the xylanolytic system in Aspergillus niger (van Peij et al. 1998) was absent in the xynII promoter. Eight cDNA clones obtained by 3′ RACE were polyadenylated at three different positions, 114 bp (two clones), 119 bp (four clones), and 138 bp (two clones) downstream of the stop codon. The consensus sequence AATAAA preceding the polyadenylation sites was present 44 bp downstream of the stop codon.
Modulation of the xynII mRNA levels by ambient pH
To investigate whether the xynII expression was regulated by the ambient pH, A. pullulans was grown for 72 h with pH control by 0.1 M phosphate buffer of pH 6.0 or 8.0 and without pH control. The initial pH 6.0 decreased to 2.7 without pH control, and the culture showed abundant growth and became black due to dark hyphae. The initial pH 6.0 or 8.0 remained constant under pH-controlled conditions, and the cultures showed delayed growth from that observed for the pH-uncontrolled culture and remained yellow or cream. Quantitative real-time PCR showed that the transcription levels at pH 6.0 and 8.0 were 8-fold and 22-fold higher, respectively, than that at pH 2.7 reached in the culture without pH control.
Deduced amino acid sequence and enzymatic activity of the xynII gene product
The xynII gene encoded a precursor protein (XynII) of 361 amino acids with a calculated M r of 39,937 Da. The XynII contained sequences identical to the N-terminal and two internal amino acid sequences from the purified protein (see Fig. 3), indicating that the signal peptide of 26 amino acids was cleaved off during secretion. The XynII mature protein consisted of 335 amino acids with a calculated M r of 37,344 Da and a deduced pI of 8.44. The calculated M r was smaller than the 39 kDa estimated by SDS-PAGE, probably because the xylanase was a glycoprotein. In this regard, there were two potential N-linked glycosylation sites (Asn-X-Thr) in the deduced amino acid sequence (see Fig. 3). The deduced pI was consistent with the pI of 8.9 found by IEF. Four Cys residues were present at positions 110, 152, 304, and 310.
To verify the identity of the xynII as a xylanase gene and to produce the recombinant enzyme in high yields, the yeast P. pastoris was chosen as a suitable host system for heterologous expression of xynII cDNA because of the lack of xylanase activity and its high secretion efficiency (Cregg 1999). The pXYN207 transformant grown for 72 h showed xylanase activity of 0.12 U/ml in the culture supernatant, whereas the control strain transformed with the vector pPIC3.5 was devoid of the extracellular enzyme activity. Elution profile of the extracellular proteins from an SP-Sepharose column revealed two xylanase peaks, P-I and P-II, and pooled fractions from each peak were purified individually by a Superdex 75 pg column. SDS-PAGE analysis showed that the M r of the purified recombinant xylanase P-I was slightly larger than that of the P-II enzyme (Fig. 1). N-terminal amino acid sequence of P-II was identical to that of the native protein, whereas P-I contained additional six amino acid residues preceding the native cleavage site. Concentrations of the recombinant xylanases, P-I and -II, in the culture media were estimated to be 6.5 mg/l and 36 mg/l, respectively, on the basis of their specific activities. The results show that most of the XynII precursor protein was correctly recognized and processed by the P. pastoris secretory pathway.
Discussion
We observed that when A. pullulans was grown on xylan as the only carbon source under the control at pH 6.0, the family-10 xylanase (XynII) is secreted into the culture media. The purified enzyme produced xylooligosaccharides as intermediates of xylan hydrolysis. It released mainly xylobiose and xylose as end products from xylan but not hydrolyzed xylobiose. These results indicate that the enzyme is an endo-1,4-β-xylanase. Family-10 xylanases exhibit lower substrate specificity than family-11 enzymes (Biely et al. 1997). The A. pullulans enzyme showed the weak activity toward pNP-β-d-xylopyranoside. The family-10 xylanase was free of activity toward carboxymethyl cellulose. This advantageous for bleaching of pulps where purified cellulose is required.
The preferential transcription of the xynII gene at alkaline pH coincided with the presence of two PacC consensus sequences in the 5′-noncoding region. In contrast, the xynI gene encoding an acidophilic family-11 xylanase from the same A. pullulans strain (Ohta et al. 2001) was expressed at pH 2.7, but not or weakly expressed under the conditions of pH control at 6.0 or 8.0 (data not shown). Likewise, the A. nidulans xlnA encoding a neutral xylanase and possessing two consensus PacC binding sites was preferentially expressed at alkaline ambient pH, whereas the xlnB gene encoding an acidic xylanase from the same strain was preferentially expressed at acidic ambient pH despite the presence of a single consensus PacC binding site (MacCabe et al. 1998). These results strongly suggested that the PacC-like transcription factor participates in pH regulation of the xynII expression in A. pullulans. Thus, A. pullulans seems to have a regulatory system responsible for the high transcription level at the optimum pH for the enzyme activity of the gene product.
A BLAST search of A. pullulans XynII in the protein sequence database found significant degree of identity to the following family-10 xylanases of fungal and bacterial origin that can hydrolyze xylan and/or cellulose (Gilkes et al. 1991): 46% for Penicillium funiculosum (accession no. AJ635947) and Humicola grisea (Iikura et al. 1997); 43% for the basidiomycete fungus Agaricus bisporus (De Groot et al. 1998) and the bacterium Cellulomonas fimi (O'Neill et al. 1986); 42% for the actinomycete Thermobifida alba (Blanco et al. 1997); 41% for A. nidulans (MacCabe et al. 1996); 39% for Aspergillus kawachii (Ito et al. 1992) and Penicillium simplicissimum (Schmidt et al. 1998); 38% for Penicillium chrysogenum (Haas et al. 1993); and 37% for the fungal pathogen Magnaporthe grisea (Wu et al. 1995). The alignment of catalytic domain of the homologous sequences showed eight conserved regions (I to VIII) as previously described for family-10 xylanases (Baba et al. 1994; Fukumura et al. 1995), suggesting that the XynII would be the first xylanase from A. pullulans belonging to family 10. The Glu-160 in region III (WDVVNE) and Glu-276 in region VI (TELD) of A. pullulans XynII could be involved in catalytic reaction as an acid–base and as a nucleophile, respectively (Tull et al. 1991). Cys-304 and -310 among the four Cys residues were highly conserved in homologous xylanases. Cys-110 and -152 might form the disulfide bond as viewed by a comparative protein modeling using Swiss-Model (Guex and Peitsch 1997; Peitsch 1995; Schwede et al. 2003) (data not shown).
Interestingly, both the A. pullulans xynI and xynII genes encoding family-11 and -10 xylanases, respectively, had a single intron in the region encoding the secretory signal sequence, whereas this is not the case for other homologous fungal xylanases. In addition, the xynII gene had only one intron in mature protein coding region despite the fact that typically several introns exist in fungal family-10 xylanase genes. Introns in family-10 xylanase genes have been shown to play a key role in protein evolution as a mediator for module fusion and exon shuffling to facilitate slightly different substrate specificities to xylanases without changing the catalytic function (Sato et al. 1999).
A phylogenetic tree was constructed for the catalytic domain using the alignment, the part of which is shown in Fig. 4. As shown by Sato et al. (1999), the above-mentioned xylanases except the A. pullulans XynII were separated into two clusters, I and II (Fig. 5). Cluster I included bacterial and fungal xylanases in spite of their evolutionary distances and may have evolved through lateral or horizontal gene transfer of the catalytic domains, whereas cluster II consisted of those from only filamentous fungi. The XynII was phylogenetically located at an intermediate position between the two clusters.

Amino acid sequence alignment of the catalytic domain of A. pullulans XynII and other homologous xylanases. The alignment was done using the ClustalW program (version 1.81) and the aligned amino acid sequences depicted is the part encompassing the conserved regions V to VIII. Ap A. pullulans (this study), Ta T. alba (Z81013), Cf C. fimi (M15824), Pf P. funiculosum (AJ634957), Ab A. bisporus (Z83310), Hg H. grisea (AB001030), Ps P. simplicissimum (AF070417), Ak A. kawachii (D14847), Pc P. chrysogenum (M98458), An A. nidulans (Z49894), Mg M. grisea (L37530). Numbering of the amino acids shown in parentheses starts at the N-termini of the proteins. Gaps were introduced for optimal alignment and are indicated by dashes. Asterisks indicate identity, and single and double dots indicate semiconservative and conservative replacements. Conserved regions, V to VIII, are indicated by horizontal arrows. Inserted region A in A. pullulans xylanase and conserved regions B and C in xylanases of cluster I (see Fig. 5) are boxed. The putative catalytic Glu residues are indicated by bold letters

Unrooted tree showing phylogenetic relationships of catalytic domains between A. pullulans XynII and other homologous xylanases. Numbers on the branch represent the percentage of bootstrap confidence values based on 1,000 replications (values below 50 are not shown). The scale bar corresponds to 0.1 amino acid substitutions per site. For sources of sequence data, see Fig. 4
Sequence alignment (see Fig. 4) showed that cluster I xylanases and the XynII shared extra regions B and C as compared to those of cluster II and a further insertion of ten amino acid sequences was found in the XynII (region A between conserved regions V and VI in Fig. 4). Although limited availability of closely related fungal xylanase sequences makes interpretation of the phylogenetic analysis difficult, the XynII seemed to be deviated from the cluster I xylanases and to be phylogenetically distant from cluster II xylanases.
In conclusion, this is the first report of the purification of a family-10 xylanase from A. pullulans as well as cloning and characterization of the encoding gene xynII. The xynII gene was preferentially expressed at alkaline ambient pH in contrast to the acid-expressed xynI gene encoding a family-11 xylanase from the same strain. Further compilation of the relevant sequence data to be published might allow us to present a more complete picture of the evolutional relationship of the A. pullulans XynII with other family-10 xylanases.
References
Akimoto H, Kiyota N, Kushima T, Nakamura T, Ohta K (2000) Molecular cloning and sequence analysis of an endoinulinase gene from Penicillium sp. strain TN-88. Biosci Biotechnol Biochem 64:2328–2335
Baba T, Shinke R, Nanmori T (1994) Identification and characterization of clustered genes for thermostable xylan-degrading enzymes, β-xylosidase and xylanase, of Bacillus stearothermophilus 21. Appl Environ Microbiol 60:2252–2258
Biely P, Vršanská M, Tenkanen M, Kluepfel D (1997) Endo-β-1,4-xylanase families: differences in catalytic properties. J Biotechnol 57:151–166
Blanco J, Coque JJR, Velasco J, Martín JF (1997) Cloning, expression in Streptomyces lividans and biochemical characterization of a thermostable endo-β-1,4-xylanase of Thermomonospora alba ULJB1 with cellulose-binding ability. Appl Microbiol Biotechnol 48:208–217
Cregg JM (1999) Expression in the methylotrophic yeast Pichia pastoris. In: Fernandez JM, Hoeffler JP (eds) Gene expression systems. Academic Press, New York, pp 157–191
Cubero B, Scazzocchio C (1994) Two different, adjacent and divergent zinc finger binding sites are necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J 13:407–415
De Groot PWJ, Basten DEJW, Sonnenberg ASM, Van Griensven LJLD, Visser J, Schaap PJ (1998) An endo-1,4-β-xylanase-encoding gene from Agaricus bisporus is regulated by compost-specific factors. J Mol Biol 277:273–284
Deshpande MS, Rale VB, Lynch JM (1992) Aureobasidium pullulans in applied microbiology: a status report. Enzyme Microb Technol 14:514–527
Fukumura M, Sakka K, Shimada K, Ohmiya K (1995) Nucleotide sequence of the Clostridium stercorarium xynB gene encoding an extremely thermostable xylanase, and characterization of the translated product. Biosci Biotechnol Biochem 59:40–46
Gilkes NR, Henrissat B, Kilburn DG, Miller RC JR, Warren RAJ (1991) Domains in microbial β-1,4-glycanases: sequence conservation, function, and enzyme families. Microbiol Rev 55:303–315
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723
Gurr SJ, Unkles SE, Kinghorn JR (1987) The structure and organization of nuclear genes of filamentous fungi. In: Kinghorn JR (ed) Gene structure in eukaryotic microbes. IRL Press, Oxford, pp 93–139
Haas H, Friedlin E, Stöffler G, Redl B (1993) Cloning and structural organization of a xylanase-encoding gene from Penicillium chrysogenum. Gene 126:237–242
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Henrissat B, Bairoch A (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 293:781–788
Iikura H, Takashima S, Nakamura A, Masaki H, Uozumi T (1997) Cloning of a gene encoding a putative xylanase with a cellulose-binding domain from Humicola grisea. Biosci Biotechnol Biochem 61:1593–1595
Ito K, Ikemasu T, Ishikawa T (1992) Cloning and sequencing of the xynA gene encoding xylanase A of Aspergillus kawachii. Biosci Biotechnol Biochem 56:906–912
Kozak M (1989) The scanning model for translation: an update. J Cell Biol 108:229–241
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Leathers TD (1986) Color variants of Aureobasidium pullulans overproduce xylanase with extremely high specific activity. Appl Environ Microbiol 52:1026–1030
Leathers TD (1989) Purification and properties of xylanase from Aureobasidium. J Ind Microbiol 4:341–348
Li XL, Ljungdahl LG (1994) Cloning, sequencing, and regulation of a xylanase gene from the fungus Aureobasidium pullulans Y-2311-1. Appl Environ Microbiol 60:3160–3166
Li XL, Ljungdahl LG (1996) Expression of Aureobasidium pullulans xynA in, and secretion of the xylanase from, Saccharomyces cerevisiae. Appl Environ Microbiol 62:209–213
Li XL, Zhang ZQ, Dean JFD, Eriksson KEL, Ljungdahl LG (1993) Purification and characterization of a new xylanase (APX-II) from the fungus Aureobasidium pullulans Y-2311-1. Appl Environ Microbiol 59:3212–3218
Li S, Cullen D, Hjort M, Spear R, Andrews JH (1996) Development of an oligonucleotide probe for Aureobasidium pullulans based on the small-subunit rRNA gene. Appl Environ Microbiol 62:1514–1518
Liu W, Saint DA (2002) A new quantitative method of real time reverse transcription polymerase chain reaction assay based on simulation of polymerase chain reaction kinetics. Anal Biochem 302:52–59
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
MacCabe AP, Fernández-Espinar MT, de Graaff LH, Visser J, Ramón D (1996) Identification, isolation and sequence of the Aspergillus nidulans xlnC gene encoding the 34-kDa xylanase. Gene 175:29–33
MacCabe AP, Orejas M, Pérez-González JA, Ramón D (1998) Opposite patterns of expression of two Aspergillus nidulans xylanase genes with respect to ambient pH. J Bacteriol 180:1331–1333
Nakamura T, Shitara A, Matsuda S, Matsuo T, Suiko M, Ohta K (1997) Production, purification and properties of an endoinulinase of Penicillium sp. TN-88 that liberates inulotriose. J Ferment Bioeng 84:313–318
Nelson NJ (1955) Colorimetric analysis of sugar. Methods Enzymol 3:85–86
Ohta K, Moriyama S, Tanaka H, Shige T, Akimoto H (2001) Purification and characterization of an acidophilic xylanase from Aureobasidium pullulans var. melanigenum and sequence analysis of the encoding gene. J Biosci Bioeng 92:262–270
O'Neill G, Goh SH, Warren RAJ, Kilburn DG, Miller RC Jr (1986) Structure of the gene encoding the exoglucanase of Cellulomonas fimi. Gene 44:325–330
Pañalva MA, Arst HN Jr (2002) Regulation of gene expression by ambient pH in filamentous fungi and yeast. Microbiol Mol Biol Rev 66:426–446
Peitsch MC (1995) Protein modeling by e-mail. Biotechnology (N.Y.) 13:658–660
Pell G, Szabo L, Charnock SJ, Xie H, Gloster TM, Davies GJ, Gilbert HJ (2004) Structural and biochemical analysis of Cellvibrio japonicus xylanase 10C. J Biol Chem 279:11777–11788
Puls J, Schuseil J (1993) Chemistry of hemicelluloses: relationship between hemicellulose structure and enzymes required for hydrolysis. In: Coughlan MP, Hazlewood GP (eds) Hemicellulose and hemicellulases. Portland Press, London, pp 1–27
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Sato Y, Niimura Y, Yura K, Go M (1999) Module-intron correlation and intron sliding in family F/10 xylanase genes. Gene 238:93–101
Schmidt A, Schlacher A, Steiner W, Schwab H, Kratky C (1998) Structure of the xylanase from Penicillium simplicissimum. Protein Sci 7:2081–2088
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385
Somogyi M (1952) Notes on sugar determination. J Biol Chem 195:19–23
Tanaka H, Okuno T, Moriyama S, Muguruma M, Ohta K (2004) Acidophilic xylanase from Aureobasidium pullulans: efficient expression and secretion in Pichia pastoris and mutational analysis. J Biosci Bioeng 98:338–343
Tull D, Withers SG, Gilkes NR, Kilburn DG, Warren RAJ, Aebersold R (1991) Glutamic acid 274 is the nucleophile in the active site of a “retaining” exoglucanase from Cellulomonas fimi. J Biol Chem 266:15621–15625
van Peij NNME, Gielkens MMC, de Vries RP, Visser J, de Graaff LH (1998) The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl Environ Microbiol 64:3615–3619
Wu S-C, Kauffmann S, Darvill AG, Albersheim P (1995) Purification, cloning and characterization of two xylanases from Magnaporthe grisea, the rice blast fungus. Mol Plant-Microb Interact 8:506–514
Acknowledgements
We thank Prof. Tatsuo Nakayama of Miyazaki Medical College for the N-terminal amino acid sequencing. This study was supported in part by The Iwatani Naoji Foundation's Research Grant 2003.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tanaka, H., Muguruma, M. & Ohta, K. Purification and properties of a family-10 xylanase from Aureobasidium pullulans ATCC 20524 and characterization of the encoding gene. Appl Microbiol Biotechnol 70, 202–211 (2006). https://doi.org/10.1007/s00253-005-0045-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-0045-3