
Abstract
A recombinant xylanase gene (rxynUMB) from Ustilago maydis 521 was expressed in Pichia pastoris, and the enzyme was purified and characterized. Phylogenetic analysis demonstrated that rxynUMB belongs to glycosyl hydrolase family 11. The Trp84, Trp95, Glu93, and Glu189 residues are proposed to be present at the active site. The apparent molecular mass of the recombinant xylananse was approximately 24 kDa, and the optimum pH and temperature were 4.3 and 50 °C, respectively. Xylanase activity was enhanced by 166 and 115 % with Fe2+ and Mn2+, respectively. The biochemical properties of this recombinant xylanase suggest that it may be a useful candidate for a variety of commercial applications.
Similar content being viewed by others


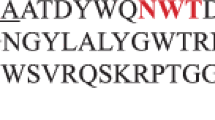
Avoid common mistakes on your manuscript.
Introduction
Xylan, the major component of plant hemicellulose, is the second most abundant renewable resource. It is present in plant cell walls and some algae and constitutes up to 39 % of the total dry weight of terrestrial plants (Shi et al. 2014). Hydrolysis of xylan is an important step in the degradation of lignocellulosic materials. However, industrial chemical hydrolysis of xylan is accompanied by the formation of toxic compounds that are hazardous to the environment. The use of site-specific microbial enzymes for xylan hydrolysis would ensure that the process is not hazardous to the environment (Mittal et al. 2012). Xylan can be hydrolyzed into xylooligosaccharides and xylose by several enzymes, among which, the most crucial one is β-1,4-xylanase (EC 3.2.1.8).
Xylanases have been widely used in various industrial processes, including biobleaching of wood pulp, treating animal feed to increase digestibility, processing food to increase clarification, and converting lignocellulosic biomass to feedstock and biofuel (Figueroa-Espinoza et al. 2004). Generally, the suitability of an enzyme for industrial application is dependent on its homogeneity, thermal stability, and other kinetic properties (Mittal et al. 2012). The market demand for xylanases has increased but the production of these enzymes requires the growth of many microorganisms. Therefore, heterologous expression has become the primary tool for the production of xylanases at an industrial level (Deesukon et al. 2013).
Ustilago maydis is a biotrophic pathogen that causes smut disease in maize. In the Ustilago maydis/maize interaction, the biotrophic interface of U. maydis acquires nutrients from maize cells to promote the formation of galls and tumors on the ears of maize (O’Connell and Panstruga 2006).
We produced a recombinant xylanase gene (rxynUMB) from U. maydis 521 according to codon usage preference and constructed a high secretion expression vector of Pichia pastoris. Here, we describe the biochemical properties of the recombinant enzyme expressed in P. pastoris, which suggest that it may be a useful candidate for a variety of commercial applications.
Materials and methods
Strains, vectors and chemicals
Pichia pastoris strain GS 115 (His-Mut+), used as a host for heterologous expression of xylanase, was from Invitrogen. Vector pYPX88 (GenBank accession no.: AY178045), used as expression vector, was prepared in our laboratory. The pGEM-T Easy vector was from Promega. Taq DNA polymerase, T4 DNA ligase and restriction endonucleases were purchased from Takara. Birchwood xylan and xylose and Ni2+-NTA agarose affinity column were from Sigma. All other chemicals were of analytical grade.
Xylanase gene synthesis and expression vector construction
According to the amino acid sequence (GenBank accession no.: XM757404.1), the xylanase gene from U. maydis 521 with the addition of a C-terminal 6× His tag sequence was optimized using codon usage bias and synthesized by successive PCR (Xiong et al. 2004). The amplified fragment was digested with BamHI and SacI and then inserted into pYPX88 vector, which contains a 357-bp fragment of the α-factor prepro-leader MF4I (GenBank accession no.: AY145833) with the preferred codon usage for P. pastoris, which substitutes the wild-type α-signal sequence to enhance the expression level (Xiong et al. 2003). Routine DNA manipulations were performed using a standard recombinant method.
Transformation and expression of the recombinant xylanase in Pichia pastoris
The recombinant plasmid was linearized by BglII digestion and transformed into P. pastoris GS115 competent cells using electroporation. Transformants were selected on histidine-deficient SD medium and incubated at 30 °C for 3 days. To screen for xylanase activity of the recombinant protein, the His+ transformants were streak-cultivated in BMGY at 28 °C, and then the cells were inoculated into 96-well plates containing 50 µl BMMY medium with 1 % (v/v) methanol and incubated for 12 h at 30 °C. Positive colonies were selected by enzymatic activity determination. For large-scale protein production and purification, a single colony with a high level expression was cultured in 50 ml BMGY until the OD600 reached 3. The cells were collected and resuspended in equal volume of BMMY and cultured at 30 °C for 3 days. Methanol was added to give 1 % (v/v) every 24 h. Aliquots of culture supernatant were taken daily and examined for protein production by SDS-PAGE, and the xylanase activity was assayed at the same time.
Purification and SDS-PAGE of rxynUMB xylanase
All purification steps were carried out at 4 °C. After centrifugation and removed of the cells, the supernatant was concentrated by ammonium sulfate precipitation. The recombinant protein was purified using Ni2+-NTA agarose affinity column. The purified production was checked using 12 % (w/v) SDS-PAGE. The spots of proteins were visualized by staining with Coomassie Brilliant Blue R-250.
Enzyme assay
Xylanase activity was measured by the dinitrosalicylic acid method (Bailey 1985). The reaction mixture in 200 µl contained 50 µl diluted enzyme solution and 100 µl 1 % (w/v) birchwood xylan in citrate buffer (pH 4.3). The reactions were carried out at 50 °C for 10 min. One unit of xylanase activity is defined as the amount of enzyme capable of generating 1 µmol reducing sugar equivalent to xylose per min under the above assay conditions. All the assays were performed with the purified xylanase. Each assay was done in triplicates.
Characterization of rxynUMB xylanase
The effect of pH, temperature and metal ions on rxynUMB activity was performed. Km and Vmax were assayed using birchwood xylan from 2 to 30 mg/ml. Kinetic parameters were determined using Lineweaver–Burk plots.
Xylanase phylogeny and sequence analysis
We chose several xylanase genes belonging to family GH11 to perform phylogenetic analysis. The sequences were aligned using ClustalW, and a phylogenetic tree was constructed using MEGA.
Alignment of protein sequences between rxynUMB and other published family 11 xylanase was performed with DNAMAN. Based on the alignment and known catalytic residues and substrate binding residues, we identified the active site of rxynUMB.
Results and discussion
Xylanase phylogeny
The result of sequence alignment showed that sequence identity between rxynUMB and other known characterization xylanases was not high (approximately 40 % identity). However, signature motifs and catalytic residues for xylanases from known glycosyl hydrolase family 11 were identical, indicating that rxynUMB belongs to glycosyl hydrolase family 11 (Fig. 1).

Schematic diagram of domain structure of rxynUMB xylanase. The signature motif is highlighted with a red box. The catalytic residues are highlighted in a thick box, and residues involved in substrate binding are in a thin box. The alignment was produced using DNAMAN
The amino acid sequence of rxynUMB was compared with that of published xylanases by phylogenetic analysis (Fig. 2). The phylogenetic tree showed that rxynUMB xylanase was most closely related to the xylanase from Aureobasidium pullulans (AF169630). Protein sequence alignment of available family 11 xylanases showed the presence of conserved residues at the active site. Also Trp84, Trp95 and Glu93, Glu189 residues were implicated in rxynUMB xylanase activity (Fig. 3). Based on the chemical modification studies of BadX xylanase from Bacillus agaradhaerens (Poon et al. 2003), Glu93 and Glu189 residues may be catalytic residues, and Trp84 and Trp95 residues may be involved in substrate binding. We used ClustalW to construct the three-dimensional structures of rxynUMB xylanase and BadX xylanase. There was high similarity between their structures, although there was only 34 % identity between the two protein sequences (Fig. 3). The conserved residues were all present at the active site. The sequence of rxynUMB xylanase was closest to that of BadX xylanase. The increase in BadX xylanase activity at high pH values was assigned to the nucleophile Glu94 and the loss of activity to Glu184. A similar conclusion was also obtained for BcX xylananse (McIntosh et al. 1996). Based on the findings of previously studies, it can be inferred that Glu93 in rxynUMB assumes the role of a nucleophile and Glu189 that of an acid/base residue.

Phylogenetic tree of rxynUMB xylanase from the U. maydis 521 and other published xylanases of family 11. The fungal xylanase from Aureobasidium pullulans (AF169630) and Helminthosporium turcium (AJ238895) are most closed to xynUMB xylanase. Two bacterial xylanases from Pseudomonas fluorescens (Z48927) and Phaedon cochleariae (Y17908) have a closer phylogenetic relationship than other bacterial xylanases (AF120156, AF198618, AF195421, U15985, D32065). The farthest Firmicutes group contains fungal xylanases from the rumen fungi (X82439, U57819, AB057588) and three xylanases from Bacillales and Clostridiales (U76545, AF220528, AF036925)

The stereo-view of the structure of rxynUMB xylanase and the xylanase from Bacillus Agaradhaerans. a rxynUMB xylanase; b xylanase from Bacillus Agaradhaerans; Glu93 and Glu189 residues were catalytic residues and Trp84 and Trp95 residues were involved in substrate binding
Codon optimization, gene cloning, and expression vector construction
According to the codon bias of P. pastoris, the rxynUMB gene without the signal peptide sequence and stop codon with an additional C-terminal 6× His tag sequence was optimally designed, in which 91 nucleotides of the native sequence were changed. The optimal sequence has 49.8 % GC content and shared 84 % similarity to the wild-type xylanase gene (Supplementary Fig. 1). The codon-optimized gene was subcloned into the pYPX88 vector, which contains the inducible promoter from the AOX1 gene, the chemically synthesized signal MF4I, and the native transcription termination and the polyadenylation signal from the AOX1 gene. Sequencing data showed that the expression plasmid was constructed correctly.
Expression and purification of rxynUMB xylanase
The recombinant pYPX88-xylanase plasmid was transformed and targeted into P. pastoris GS115 genome. Multiple plasmid integration events occurred spontaneously in P. pastoris at a frequency between 1 and 10 % of all His+ transformants. Small-culture analysis experiments and activity assays are an efficient method to screen for the best strain for expression of recombinant xylanase. After induction for 24 h in 96-well plates, 23 (23.9 %) of the tested transformants showed xylanase activity. The transformant rxynUMB#6 was selected because it synthesized rxynUMB xylanase with the highest activity. The maximal xylanase activity and protein concentration in the culture supernatant reached 110 U/ml and 0.042 mg/ml, respectively, after induction for 72 h.
Xylanase rxynUMB was purified from the culture supernatant using the Ni2+-NTA agarose affinity column. A distinctive band of purified recombinant xylanase with an apparent molecular mass of approximately 24 kDa was observed (Fig. 4).

SDS-PAGE pattern of the purified rxynUMB xylanase from P. pastoris. Lane M protein marker; Lane 1 purified rxynUMB xylanase by Ni2+-NTA agarose affinity column
Pichia pastoris is an efficient host for high-level heterologous expression. However, no xylanase gene from the basidiomucete U. maydis has been expressed in P. pastoris. We overexpressed the rxynUMB gene in P. pastoris, with a yield of 0.042 mg/ml, which was higher than that for the xylanase gene of Chartomium thermophilum (xyn669, 0.0075 mg/ml) expressed in P. pastoris (Ghaffar et al. 2011).
Characterization of rxynUMB xylanase
The optimum pH for xylanase activity was 4.3 (Fig. 5a), and the optimum temperature of the enzyme was 50 °C (Fig. 5b). rxynUMB had a half-live at 55 °C (40 min) but exhibited better thermal stability than Aspergillus usamii xylanase expressed in Escherichia coli, which had a half-life of 2.5 min at 55 °C (Zhou et al. 2008). Significant enzyme stability at higher temperatures is important for the industrial application of xylanase.

Effects of pH and temperature on the activity and stability of rxynUMB. a The optimal pH was assayed with pH varied from 2.2 to 10.6. pH stability was obtained after incubation at pH varied from 2.2 to 10.6 at 37 °C for 3 h. The buffers used were 0.2 M Na2HPO4–citric acid buffer (pH 2.2–7), and Tris–HCl buffer (pH 8–10.6). b The optimum temperature was assayed with temperature varied from 30 to 90 °C. Thermal stability was obtained after pre-incubation of the enzyme solution for 40 min at different temperatures. The activity under standard reaction conditions (50 °C, pH 4.3) without pre-incubation at different pH values was defined as 100 % (102.5 U mg−1). Values are means of three replications ± SD
As shown in Fig. 6, the activity of rxynUMB xynalase was most inhibited by Hg2+, while Cu2+ inhibited enzyme activity by approx. 30 %. In contrast, xylanase activity was enhanced by 166 and 115 % with Fe2+and Mn2+, respectively. Xylanase activity was strongly inhibited by Hg2+, which might be due to its interaction with sulfhydryl groups present on the enzyme, suggesting the need for a cysteine residue in or close to the active site of the enzyme. Two cysteine residues were detected in the rxynUMB sequence. However, further studies are needed to ascertain the mechanism.

Effects of different metal ions on rxynUMB activity. Activities was investigated after incubation of the enzyme with the metal ions for 24 h at room temperature. In the control, no metal ion was added to the reaction system. Activity under standard reaction conditions (50 °C, pH 4.3) without metal ions was defined as 100 % (102.5 U mg−1). Values are the means of three replications ± SD
The Km of the enzyme was 1.8 mg/ml, and the maximal velocity was 119 µmol min−1 mg−1. The Km was smaller than that of some xylanases (2.86–11 mg/ml) (Nair et al. 2008).
In summary, there has no report of the expression of the xylanase gene from the basidiomycete U. maydis in P. pastoris. We overexpressed the rxynUMB gene in P. pastoris using codon optimization, with a yield of 0.042 mg/ml. The characteristics of the purified enzyme, such as significant stability at higher temperatures and acidic pH, make it potentially effective for industrial applications.
References
Bailey P (1985) Microbial xylanolytic systems. Trends Biotechnol 3:286–290
Deesukon W, Nishimura Y, Sakamoto T, Sukhumsirichart W (2013) Purification, characterization of GH11 endo-β-1,4-xylanase from thermotolerant Streptomyces sp. SWU10 and overexpression in Pichia pastoris KM71H. Mol Biotechnol 54:37–46
Figueroa-Espinoza MC, Poulsen C, Soe JB, Zargahi MR, Rouau X (2004) Enzymatic solubilization of arabinoxylans from native, extruded, and high-shear-treated rye bran by different endo-xylanases and other hydrolyzing enzymes. J Agric Food Chem 52:4240–4249
Ghaffar A, Khan SA, Mukhtar Z, Rajoka MI, Latif F (2011) Heterologous expression of a gene for thermostable xylanase from Chaetomium thermophilum in Pichia pastoris GS115. Mol Biol Rep 38:3227–3233
McIntosh LP, Hand G, Johnson PE, Joshi MD, Korner M, Plesniak LA, Ziser L, Wakarchuk WW, Withers SG (1996) The pKa of the general acid/base carboxyl group of a glycosidase cycles during catalysis: A13C-NMR study of Bacillus circulans xylanase. Biochemistry 35:9958–9966
Mittal A, Nagar S, Kaur SJ, Kirti, Gupta VK (2012) Isolation, purification and characterization of alkali and thermostable xylanase from Bacillus sp. KS 09. Int J Res Dev Pharm Life Sci 1:63–68
Nair SG, Sindhu R, Shashidhar S (2008) Purification and biochemical characterization of two xylanases from Aspergillus sydowii SBS 45. Appl Biochem Biotechnol 149:229–243
O’Connell RJ, Panstruga R (2006) Inside a plant cell: establishing compatibility between plants and biotrophic fungi and oomycetes. New Phytol 171:699–718
Poon DK, Webster P, Withers SG (2003) Characterizing the pH-dependent stability and catalytic mechanism of the family 11 xylanase from the alkalophilic Bacillus agaradhaerens. Carbohydr Res 338:415–421
Shi H, Zhang Y, Zhong H, Huang YJ, Li X, Wang F (2014) Cloning, over-expression and characterization of a thermotolerant xylanase from Thermotoga thermarum. Biotechnol Lett 36:587–593
Xiong AS, Peng RH, Li X, Fan HQ, Yao QH, Guo MJ, Zhang SL (2003) Influence of signal peptide sequences on the expression of heterogeneous proteins in Pichia pastoris. Acta Biochim Biophys Sin 35:154–160
Xiong AS, Yao QH, Peng RH, Li X, Fan HQ, Li Y, Cheng ZM (2004) A simple, rapid, high fidelity and cost-effective PCR based two-step DNA synthesis (PTDS) method for long gene sequences. Nucleic Acids Res 32:e98
Zhou C, Bai J, Deng S, Wang J, Zhu J, Wu M, Wang W (2008) Cloning of a xylanase gene from Aspergillus usamii and its expression in Escherichia coli. Bioresour Technol 99:831–838
Acknowledgments
The research was supported by the International Scientific and Technological Cooperation (13440701700), Agriculture Science Technology Achievement Transformation Fund (133919N1300), Key Project Fund of Shanghai Minhang Science and Technology Committee (2012MH059), Young Foundation of Shanghai Academy of Agricultural Science (2010–2014). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supporting information
Supplementary Fig. 1 The optimal sequence derived for the wild-type xylanase gene, xynUMB, from Ustilago maydis 521.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Han, H., You, S., Zhu, B. et al. Characterization and high expression of recombinant Ustilago maydis xylanase in Pichia pastoris . Biotechnol Lett 37, 697–703 (2015). https://doi.org/10.1007/s10529-014-1716-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-014-1716-x