
Abstract
The polyene macrolide antibiotic nystatin, produced commercially by the bacterium Streptomyces noursei, is an important antifungal agent used in human therapy for treatment of certain types of mycoses. Early studies on nystatin biosynthesis in S. noursei provided important information regarding the precursors utilised in nystatin biosynthesis and factors affecting antibiotic yield. New insights into the enzymology of nystatin synthesis became available after the gene cluster governing nystatin biosynthesis in S. noursei was cloned and analysed. Six large polyketide synthase proteins were implicated in the formation of the nystatin macrolactone ring, while other enzymes, such as P450 monooxygenases and glycosyltransferase, were assumed responsible for ring “decoration”. The latter data, supported by analysis of the polyene mixture synthesised by the nystatin producer, helped elucidate the complete nystatin biosynthetic pathway. This information has proved useful for engineered biosynthesis of novel nystatin analogues, suggesting a plausible route for the generation of potentially safer and more efficient antifungal drugs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Polyene macrolide antibiotics are potent antifungal agents with a unique mode of action that are currently being used in human therapy. These antibiotics are generally composed of a large macrolactone ring containing 20–40 carbon atoms, and deoxysugar mycosamine (Omura and Tanaka 1984). Sets of four to eight conjugated double bonds within the macrolactone ring (hence the term “polyene”) are believed to be crucially important for the mode of action of polyenes since they are responsible for the interaction of antibiotics with sterols in fungal membranes (reviewed in Zotchev 2003). Somehow, upon such interaction, polyene macrolide molecules assemble themselves into pore-like structures within the membranes, leading to leakage of cell constituents, metabolic disruption and, ultimately, cell death. Unfortunately, polyene macrolides also have affinity, albeit lower, for the cholesterols in mammalian cell membranes, causing serious side effects in patients undergoing long-term treatments for fungal infections (Abu-Salah 1996). Despite this, polyene macrolides remain one of the most reliable fungicidal agents, as resistance of fungal pathogens to these antibiotics is relatively rare.
Nystatin, which was discovered by E.L. Hazen and R.F. Brown working at the New York State Department of Health, was the first useful polyene macrolide. Their early work showed that a potent antifungal agent is produced by Streptomyces No. 48240 isolated from soil at a farm owned by H. Nourse. Consequently, this steptomycete strain was classified by Ettlinger et al. (1958) as Streptomyces noursei. The antifungal antibiotic produced by this organism was itself later isolated, purified, characterised, and named nystatin, after New York State (Hazen et al. 1953). At least one other streptomycete, namely S. fungicidicus ATCC 27432 (Matsuoka 1960), has been shown to produce nystatin. S. albulus ATCC 12757 (Veiga and Fabregas 1983) is also referenced in the DSMZ (German Collection of Microorganisms and Cell Cultures) as nystatin producer strain DSM 40492. As of today, only S. noursei is used for commercial production of this antibiotic.
Nystatin is used mainly for topical treatment of oral, gastro-intestinal, and genital candidosis, and remains an important commercial product. This paper reviews the biosynthesis of nystatin and its regulation in the producing organism S. noursei, as well as possibilities for using nystatin biosynthetic genes for production of novel nystatin analogues.
Nystatin: chemical structure and occurrence in nature
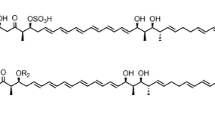
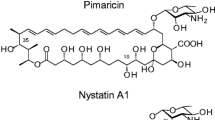
The complete stereochemical structure of nystatin A1 was solved in 1986 (Fig. 1; Lancelin and Beau 1989). The molecule consists of a 38-membered macrolactone ring with sets of two and four conjugated double bonds separated by a saturated bond (Fig. 1). Despite the presence of two sets of conjugated double bonds, nystatin is classified as tetraene, although it is sometimes referred to as “degenerated heptaene” (Ciftci et al. 1984). Like other polyene macrolide antifungals, the nystatin molecule also contains a mycosamine moiety linked to the macrolactone ring via a β-glycosidic bond, and exocyclic carboxy group, both of which appear to be important for biological activity and toxicity (Borowski 2000).

Chemical structures of some nystatin-related polyene macrolides produced by Streptomyces noursei ATCC 11455
The first studies on nystatin biosynthesis in S. noursei were published almost a decade after the discovery of this antibiotic. It immediately became apparent that S. noursei produces a complex of nystatin-related molecules, initially designated as fungicidin (Okami 1954). Beside fungicidins, several other metabolites with biological activities were isolated from this organism, e.g. antifungal cycloheximide (Roszkowski et al. 1972) and the antibacterials phalamycin (Brown et al. 1953) and nourseothricin (Roder et al. 1985). Later, fungicidins were shown to be represented by three related polyene macrolides, nystatin A1, A2, and A3, nystatin A1 being the major component (Mechlinski and Schaffner 1974). The chemical structure of nystatin A3 (Fig. 1), which contains an additional l-digitoxose moiety glycosidically bound to C35, has also been solved (Fig. 1; Zielinski et al. 1988), while the structure of nystatin A2 has not been published to date. Minor amounts of a heptaene polyene compound may also be found in products from S. noursei fermentations (Veiga and Fabregas 1983). S. noursei var polifungini produces polifungin, an antifungal complex containing the nystatins and polifungin B, which is closely related to nystatin A3 (Roszkowski et al. 1972).
Nystatin biosynthesis in S. noursei
Precursors for nystatin biosynthesis and growth factors affecting yield
The macrolactone ring of polyene antibiotics is synthesised principally by condensation of acetate- and propionate-units (Martin 1977), as was demonstrated by efficient incorporation of labelled acetate and propionate into nystatin and other polyene antibiotics during their biosynthesis (Birch et al. 1964; Manwaring et al. 1969; Linke et al. 1974). In the case of nystatin, incorporation of 16 acetate and 3 propionate units into its 38-carbon macrolactone ring was shown by Birch et al. (1964). The methyl side chains in the nystatin macrolactone ring apparently originate from incorporation of the propionate units.
The immediate precursors for the incorporation of acetate and propionate units in the macrolactone ring are malonyl-CoA and methylmalonyl-CoA, formed by carboxylation of acetyl-CoA and propionyl-CoA (Martin 1977). The level of carboxylation of acetate and propionate has been correlated with the level of nystatin biosynthesis in S. noursei (Roszkowski et al. 1972). Two enzymes, acetyl-CoA carboxylase and methylmalonyl-CoA carboxyltransferase, involved in formation of malonyl-CoA and methylmalonyl-CoA in S. noursei, have been characterised (Rafalski and Raczynska-Bojanowska 1975a). Comparative studies of low- and high-producing mutants demonstrated elevated activities of these enzymes (Rafalski and Raczynska-Bojanowska 1973) and an increased intracellular pool of acyl-CoAs in mutants producing high yields of nystatin (Rafalski and Raczynska-Bojanowska 1975b).
The negative effect of high levels of inorganic phosphate on the biosynthesis of secondary metabolites is frequently reported for Streptomyces, including those producing polyene macrolides (Martin and McDaniel 1977; Gil and Campelo-Diez 2003). Optimal conditions for production of polyenes occur at low levels (<5 mM) of inorganic phosphate (Martin and McDaniel 1977). Biosynthesis of the polyene candicidin in S. griseus was shown to be regulated by phosphate at the transcriptional level (Rebollo et al. 1989). Glucose is required for biosynthesis of polyenes, but high levels of glucose have a negative effect on production of polyene antibiotics (Martin and McDaniel 1977). Slow feeding of glucose to the fermentations was shown to give rise to a considerable increase in the yields of nystatin and other polyene macrolides (Tereshin 1976, Martin and McDaniel 1974). Recent studies in batch cultures with S. noursei wild-type strain demonstrated that the specific yield of nystatin (per biomass) is low under high glucose concentrations (Jonsbu et al. 2002), or in the presence of a rapidly metabolised nitrogen source such as ammonium (Jonsbu et al. 2000). The data indicated that growth rate is important in determining the specific rate of nystatin biosynthesis. This probably explains why complex nitrogen sources (e.g. soybean meal) are traditionally chosen for production of polyene antibiotics (Martin and McDaniel 1977).
Molecular cloning of the nystatin biosynthetic gene cluster
New insights into the enzymology of antibiotic biosynthesis can be obtained upon isolation and characterisation of the genes responsible for its biosynthesis. The chemical structure of nystatin shows features typical of macrolide antibiotics synthesised by polyketide synthase (PKS) type I (modular) enzymes, which perform condensation of small carboxylic acids into polyketide chains (Hopwood 1997). PKS I enzymes are composed of distinct modules, each of which is responsible for one condensation step (Donadio et al. 1991). PKS modules contain a minimal set of ketosynthase (KS), acyl transferase (AT), and acyl carrier protein (ACP) domains. The AT domain chooses a chain-building unit and transfers it to the ACP domain, from where it is condensed at the active site of the KS domain with the polyketide chain transferred from the ACP of a previous module. Beside these domains, PKS modules may possess up to three additional enzymatic activities, namely ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER). The presence or absence of these catalytic domains in a module determines the degree of reduction of the ketide unit introduced by the preceding module into the polyketide chain. Thus, the absence of these domains leads to the appearance of a keto group on the antibiotic molecule, while the presence of KR, KR + DH, and KR + DH + ER would yield hydroxyl, double bond, and saturated bond, respectively (Katz 1997).
Several strategies have been employed to clone the nystatin gene cluster, but only one that utilised a PKS type I-specific probe amplified by PCR from the S. noursei genome was successful (Brautaset et al. 2000). DNA sequencing and analysis of the nystatin biosynthesis genes revealed a cluster spanning ca. 125 kb of the DNA and containing 20 genes (Fig. 2). A combination of the in silico analysis of the nystatin biosynthetic enzymes, the chemical structure of nystatin, and analysis of analogs produced by certain mutants, have allowed a model for nystatin biosynthesis to be suggested (Fig. 3).

Organisation of the nystatin biosynthetic gene cluster in S. noursei ATCC 11455. The functions of the deduced gene products are indicated

Model for nystatin biosynthesis in S. noursei ATCC 11455
Synthesis of the macrolactone ring
Synthesis of the nystatin macrolactone ring apparently begins with the loading of a starter (di)carboxylic acid by a NysA protein comprising KS, AT, DH, and ACP domains. The chemical structure of nystatin implies an acetate unit as a starter for its biosynthesis, which could arise either directly from acetyl CoA, or after decarboxylation of malonyl CoA. In many PKS I systems, the loading module utilises malonyl CoA, which is decarboxylated before being brought into contact with the first condensing module; the “loading” KS seems to be crucial for decarboxylation (Bisang et al. 1999). The KS domain in NysA contains a Ser residue instead of the catalytic Cys present in the “condensing” KS domains, or the Gln implicated in decarboxylation normally found in the “loading” KS (Bisang et al. 1999). Site-specific mutagenesis of Ser to either Cys or Gln had no apparent effect on nystatin biosynthesis (Brautaset et al. 2003), suggesting either that this residue is unimportant for decarboxylation of malonyl CoA, or that acetyl CoA is used to prime nystatin biosynthesis. Data from further analysis, which involved construction of hybrid loading modules with an AT domain of “extender” type suggested that the natural AT in NysA can utilise both acetyl CoA and malonyl CoA as starters for initiation of nystatin biosynthesis (Brautaset et al. 2003).
After initiation, synthesis of the nystatin macrolactone ring continues via condensation of two propionate extender units with the acetate starter, the process being catalysed by the bimodular PKS NysB. The presence of an inactive DH domain in module 2 within NysB is apparently responsible for the appearance of a hydroxyl at C35 on the nystatin molecule. The NysC protein, a hexamodular PKS, adds six acetate extender units to the polyketide chain. Five out of six NysC modules contain KR-DH reductive loops, while an additional ER domain is present in module 5. The reductive domains in modules 3 and 4 are responsible for the appearance of the C32–C33 and C30–C31 conjugated double bonds, while the C26–C27, C24–C25, and C22–C23 double bonds are formed by modules 6, 7, and 8, respectively. Module 5, containing the full reductive loop, accounts for the saturated C28–C29 bond on the nystatin molecule that separates the two series of conjugated double bonds. Because of the ER domain in module 5, nystatin is synthesised as a tetraene. Interestingly, a hexamodular PKS AmphC involved in the synthesis of amphotericin in S. nodosus is highly similar to NysC, and also contains a full set of reductive domains in module 5 (Caffrey et al. 2001). However, the ER domain in AmphC seems to function poorly, most probably due to a shortened DH–ER interdomain linker, as both amphotericin A (tetrane) and B (heptaene) are synthesised by S. nodosus.
The next six rounds of condensation in the synthesis of the nystatin polyketide chain are performed by the hexamodular PKS NysI. The latter protein incorporates five acetate and one propionate extender units, and is responsible for the appearance of several features on the nystatin molecule that seem to be important for biological activity. The KR–DH reductive domain pair in module 9 of NysI provides for the fourth conjugated double bond, C20–C21, in the polyene region. The presence of an inactive DH domain in module 10 results in a C19 hydroxyl that is later used for the attachment of mycosamine moiety. Module 11 of NysI incorporates the propionate extender, providing an exocyclic methyl group at C16 that is later oxidised to a carboxyl. Finally, an inactive DH domain in module 11 together with an inactive KR domain in module 13 provide the means for formation of a C13–C17 hemiketalic ring upon interaction between the C13 keto and C17 hydroxy groups. Such a ring might assist in folding of the nystatin polyketide chain upon completion of its synthesis.
The trimodular NysJ and unimodular NysK PKS complete the synthesis of the nystatin polyketide chain via condensation of the last four acetate extender units. Inactive DH domains in NysJ modules 16 and 17 contribute to the appearance of the C7 and C5 hydroxy groups, respectively, in the polyol region of nystatin. Obviously, the C3 hydroxyl is formed due to the failure of NysK to reduce the keto group, although the DH domain on this PKS appears to be intact. There are some precedents for seemingly intact DH domains being inactive in vivo (Tang et al. 1998). A distinct thioesterase domain at the C-terminus of NysK is most probably responsible for cleavage of the thioester bond between the NysK ACP and the mature polyketide chain, and its cyclisation.
Yet another thioesterase is encoded by the nysE gene located downstream from nysC. NysE resembles type II thioesterases, which function as editing enzymes during polyketide biosynthesis (Kim et al. 2002). Since nystatin biosynthesis involves such a complex PKS system, it seems likely that an editing function provided by NysE is required to remove aberrant precursors and to make the process more efficient.
Post-PKS modifications and transport
From the structure of antibiotic, and from the organisation of the nystatin PKS, it can be deduced that at least three post-PKS modifications are required to convert the nystatin macrolactone ring into the fully decorated nystatin A1 molecule. These modifications include formation of a glycosidic bond at C19 between the aglycone and mycosamine, oxidation of a C16 methyl group to a carboxyl, and C10 hydroxylation. Although it was originally suggested that oxidation and hydroxylation precede the glycosylation step (Brautaset et al. 2000), the latest data generated in our lab suggest that oxidation of the C16 methyl comes first in a series of post-PKS modifications. This notion is supported by the fact that nystatin precursors lacking the carboxyl group at C16 but possessing a C10 hydroxyl and/or a mycosamine moiety have never been identified in extracts of S. noursei (Bruheim et al. 2004). In contrast, precursors lacking mycosamine or mycosamine + C10 hydroxyl can easily be detected.
Two cytochrome P450 monooxygenase genes, nysL and nysN, are located in the nystatin cluster, and it seems likely that one of them is responsible for the aforementioned oxidation of the C16 methyl group. Two other fully characterised polyene biosynthetic gene clusters, governing biosynthesis of pimaricin and amphotericin B, also contain similar genes (Aparicio et al. 2003). Inactivation of the nysL homolog, pimD, in the pimaricin producer, resulted in production of a pimaricin precursor lacking an epoxy group in its polyol region (Mendes et al. 2001). It seems likely therefore, that NysL is responsible for C10 hydroxylation, while NysN represents a C16 methyl oxidase. Interestingly, attempts to construct a mutant lacking an NysN homologue, AmphN, in the amphotericin producer have failed (Caffrey et al. 2001), and our own attempts to inactivate the nysN gene in S. noursei have so far been unsuccessful. Although the reason for the latter is still unclear, it is possible that the nystatin and amphotericin precursors lacking the C16 carboxyl are toxic for the producer organisms.
In nystatin biosynthesis, the attachment of the mycosamine moiety seems to precede the final hydroxylation at C10 (see below). It has been suggested that the mycosamine biosynthetic pathway is different from that of other deoxysugar components of macrolide antibiotics (Brautaset et al. 2000), and a model for mycosamine biosynthesis has recently been proposed (Aparicio et al. 2003). The glycosyltransferase NysDI presumably responsible for glycosylation of a nystatin aglycone at C19 is rather unique, as it resembles eukaryotic UDP-glucoronosyl transferases (Brautaset et al. 2000). Two other mycosamine biosynthetic enzymes, GDP-mannose dehydratase NysDIII and the aminotransferase NysDII are also encoded by the cluster, while genes for other enzymes required for mycosamine formation are missing. It seems plausible that the “missing” enzymes of the mycosamine biosynthetic pathway are recruited from primary metabolism.
Two putative ABC-type transporter proteins encoded by the nystatin gene cluster, NysH and NysG, were suggested to be responsible for the active transport of nystatin out of the producing organism. These proteins represent type III ABC transporters, which contain both ATPase and transmembrane domains within the same polypeptide (Mendez and Salas 2001). Most likely, NysH and NysG function as a heterodimer, as in-frame deletion of either of the corresponding genes in S. noursei had quantitatively the same negative effect on nystatin biosynthesis (unpublished data). Moreover, upon inactivation of either nysH or nysG, accumulation of the nystatin precursor lacking a C10 hydroxyl was observed. Interestingly, nysH and nysG mutants still produced and exported a considerable amount of nystatin, suggesting that other transport systems present in S. noursei may circumvent the NysH/NysG transporter defect (unpublished data).
Regulation of nystatin biosynthesis
Four regulatory genes found at the border of the nystatin gene cluster were shown to be directly involved in regulation of nystatin biosynthesis. Gene inactivation experiments demonstrated that three putative transcriptional activators NysRI, II, and III, and a putative transcriptional activator, NysRIV, with a PAS-like domain are required for efficient nystatin production in S. noursei (Sekurova et al. 2004). Surprisingly, individual expression of the regulatory genes did not lead to a dramatic increase in nystatin production, suggesting a requirement for co-ordinated expression. Interestingly, promoter-probe studies utilising an xylE reporter gene have shown that the promoters for the nysA gene encoding the nystatin PKS loading module, and for the nysH gene encoding a putative transporter, are the main targets of regulation. Cross-complementation studies helped establish a hierarchy of NysR regulators. It was shown that nysRIV can complement all the regulatory mutants, and thus presumably represents the actual transcriptional regulator for biosynthetic genes, while NysRI, II, and III regulate the expression of nysRIV (Sekurova et al. 2004). Besides the pathway-specific regulators, a pleiotropic regulatory locus affecting nystatin biosynthesis only under certain growth conditions has been identified in S. noursei (Sekurova et al. 1999).
Shunt products in nystatin biosynthesis
Interesting information on nystatin biosynthesis was obtained upon analysis of polyene macrolides produced by S. noursei wild-type strain (Bruheim et al. 2004). Beside nystatins A1 and A3, many other polyene macrolides could be detected in extracts of S. noursei culture using HPLC with a diode-array detector coupled to LC-MS. Structures for some of these compounds, namely 35-mycarosyl nystatin and 28,29-didehydro nystatin, have been solved (Fig. 1; Bruheim et al. 2004). Data on the molecular weight of the nystatin-related metabolites and their UV spectra could be correlated with the nystatin biosynthetic pathway, allowing identification of the branch points in the pathway that lead to accumulation of these analogues. The results obtained in such studies might provide important information for future engineered biosynthesis of novel polyene macrolides.
Perspectives for genetically engineered biosynthesis of nystatin analogues
The problems of toxicity and poor pharmacokinetics associated with polyene macrolides have prompted considerable effort towards producing analogues of these antibiotics, mainly amphotericin B, via chemical synthesis (Grzybowska et al. 1997; Szlinder-Richert et al. 2001). However, many chemical groups on polyene macrolide molecules are refractory to chemical modification, suggesting that interference with the biosynthetic pathway might be an alternative strategy for making novel analogues. Recent examples of successful engineered biosynthesis of novel polyenes, including production of 4,5-deepoxypimaricin upon inactivation of the pimD gene in S. natalensis (Mendes et al. 2001), and synthesis of deoxyamphotericins and deoxyamphoteronolides by engineered S. nodosus (Byrne et al. 2003), definitely demonstrate the utility of this approach. In our laboratory, we have produced a range of nystatin analogues upon engineering both nystatin PKS (Brautaset et al. 2002) and post-PKS modification steps (unpublished data). Since some of these analogues show increased antifungal activity, it seems plausible that these polyene macrolides could become lead compounds for further development of more efficient and less toxic antifungal drugs.
References
Abu-Salah KM (1996) Amphotericin B: an update. Br J Biomed Sci 53:122–133
Aparicio JF, Caffrey P, Gil JA, Zotchev SB (2003) Polyene antibiotic biosynthesis gene clusters. Appl Microbiol Biotechnol 61:179–188
Bisang C, Long PF, Cortes J, Westcott J, Crosby J, Matharu AL, Cox RJ, Simpson TJ, Staunton J, Leadlay PF (1999) A chain initiation factor common to both modular and aromatic polyketide synthases. Nature 401:502–505
Birch AJ, Holzapfel CW, Rickards RW, Djerassi C, Suzuki M, Westley J, Dutcher JD, Thomas R (1964) Nystatin. V. Biosynthetic definition of some structural features. Tetrahedron Lett 23:1485–1490
Borowski E (2000) Novel approaches in the rational design of antifungal agents of low toxicity. Farmaco 55:206–208
Brautaset T, Sekurova ON, Sletta H, Ellingsen TE, StrLm AR, Valla S, Zotchev SB (2000) Biosynthesis of the polyene antifungal antibiotic nystatin in Streptomyces noursei ATCC 11455: analysis of the gene cluster and deduction of the biosynthetic pathway. Chem Biol 7:395–403
Brautaset T, Bruheim P, Sletta H, Hagen L, Ellingsen TE, Strom AR, Valla S, Zotchev SB (2002) Hexaene derivatives of nystatin produced as a result of an induced rearrangement within the nysC polyketide synthase gene in S. noursei ATCC 11455. Chem Biol 9:367–373
Brautaset T, Borgos SE, Sletta H, Ellingsen TE, Zotchev SB (2003) Site-specific mutagenesis and domain substitutions in the loading module of the nystatin polyketide synthase, and their effects on nystatin biosynthesis in Streptomyces noursei. J Biol Chem 278:14913–14919
Brown R, Hazen EL, Mason A (1953) Effect of fungicidin (nystatin) in mice injected with lethal mixtures of aureomycin and Candida albicans. Science 117:609–610
Bruheim P, Borgos SEF, Tsan P, Sletta H, Ellingsen TE, Lancelin J-M, Zotchev SB (2004) Chemical diversity of polyene macrolides produced by Streptomyces noursei ATCC 11455 and recombinant strain ERD44 with genetically altered polyketide synthase NysC. Antimicrob Agents Chemother 48:4120–4129
Byrne B, Carmody M, Gibson E, Rawlings B, Caffrey P (2003) Biosynthesis of deoxyamphotericins and deoxyamphoteronolides by engineered strains of Streptomyces nodosus. Chem Biol 10:1215–1224
Caffrey P, Lynch S, Flood E, Finnan S, Oliynyk M (2001) Amphotericin biosynthesis in Streptomyces nodosus: Deductions from analysis of polyketide synthase and late genes. Chem Biol 8:713–723
Ciftci T, Borkman TA, McDaniel LE, Schaffner CP (1984) Comparative analysis of hexaene antibiotics. J Antibiot 37:876–884
Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L (1991) Modular organization of genes required for complex polyketide biosynthesis. Science 252:675–679
Ettlinger L, Corbaz R, Hutter R (1958) Zur systematik der actinomyceten.4. Eine arteinteilung der gattung Streptomyces Waksman et Henrici. Arch Mikrobiol 31:326–358
Gil JA, Campelo-Diez AB (2003) Candicidin biosynthesis in Streptomyces griseus. Appl Microbiol Biotechnol 60:633–642
Grzybowska J, Sowinski P, Gumieniak J, Zieniawa T, Borowski E (1997) N-methyl-–d-fructopyranosylamphotericin B methyl ester, new amphotericin B derivative of low toxicity. J Antibiot 50:709–711
Hazen EL, Brown RF, Mason A (1953) Protective action of Fungicidin (Nystatin) in mice against virulence enhancing activity of oxytetracycline on Candida albicans. Antibiot Chemother 3: 1125
Hopwood DA (1997) Genetic contributions to understanding polyketide synthases. Chem Rev 97:2465–2498
Jonsbu E, Ellingsen TE, Nielsen J (2000) Effects of nitrogen sources on cell growth and production of nystatin by Streptomyces noursei. J Antibiot 53:1354–1362
Jonsbu E, McIntyre M, Nielsen J (2002) The influence of carbon sources and morphology on nystatin production by Streptomyces noursei. J Biotechnol 95:133–144
Katz L (1997) Manipulation of modular polyketide synthases. Chem Rev 97:2557–2576
Kim BS, Cropp TA, Beck BJ, Sherman DH, Reynolds KA (2002) Biochemical evidence for an editing role of thioesterase II in the biosynthesis of the polyketide pikromycin. J Biol Chem 277:48028–48034
Lancelin J-M, Beau J-M (1989). Complete stereostructure of nystatin A1: a proton NMR study. Tetrahedron Lett 30:4521–4524
Linke HAB, Mechlinski W, Schaffner CP (1974) Production of amphotericin B-14C by Streptomyces nodosus fermentation, and preparation of the amphotericin B-14C-methyl-ester. J Antibiot 27:155–160
Manwaring DG, Rickards RW, Gaudiano G, Nicolella V (1969) The biosynthesis of the macrolide antibiotic lucensomycin. J Antibiot 22:545–550
Martin JF, McDaniel LE (1974) The submerged culture production of the polyene antifungal antibiotics candidin and candihexin. Dev Ind Microbiol 15:324
Martin JF, McDaniel LE (1977) Production of polyene macrolide antibiotics. Adv Appl Microbiol 21:2–52
Martin JF (1977) Biosynthesis of polyene macrolide antibiotics. Annu Rev Microbiol 31:13–38
Matsuoka M (1960) Biological studies on antifungal substances produced by Streptomyces fungicidicus. J Antibiot 13:121–124
Mechlinski W, Schaffner CP (1974) Separation of polyene antifungal antibiotics by high-speed liquid chromatography. J Chromatogr 99:619–633
Mendez C, Salas JA (2001) The role of ABC transporters in antibiotic-producing organisms: drug secretion and resistance mechanisms. Res Microbiol 152:341–350
Mendes MV, Recio E, Fouces R, Luiten R, Martin JF, Aparicio JF (2001) Engineered biosynthesis of novel polyenes: a pimaricin derivative produced by targeted gene disruption in Streptomyces natalensis. Chem Biol 8:635–644
Omura S, Tanaka H (1984) Production, structure, and antifungal activity of polyene macrolides. In: Omura S (ed) Macrolide antibiotics: chemistry, biology, and practice. Academic, New York, pp 351–405
Rafalski A, Raczynska-Bojanowska K (1973) Biochemical criteria in selection of high productive strains of Streptomyces noursei var polifungini. Acta Microbiol Pol 5:87
Rafalski A, Raczynska-Bojanowska K (1975a) Non-specific acetyl-CoA carboxylase and methylmalonyl-CoA carboxsyltransferase in Streptomyces noursei var. polifungini. Acta Biochim Pol 22:311–317
Rafalski A, Raczynska-Bojanowska K (1975b) Acyl-CoA pool and acyl-CoA thioesterase in Streptomyces noursei var. polifungini. Acta Biochim Pol 22:257–261
Rebollo A, Gil JA, Liras P, Asturias JA, Martin JF (1989) Cloning and characterization of a phosphate-regulated promoter involved in phosphate control of candicidin biosynthesis. Gene 79:47–58
Roder B, Plonka G, Bormann EJ, Grafe U (1985) Influence of nourseothricin on growth and secondary metabolism of Streptomyces noursei JA 3890b. J Basic Microbiol 25:175–186
Roszkowski J, Kotiuszko D, Rafalski A, Morawska H, Raczynska-Bojanowska K (1972) Characteristics of mutants of Streptomyces noursei var polifungini producing antibiotics. Acta Microbiol Pol 4:9–22
Sekurova O, Sletta H, Ellingsen TE, Valla S, Zotchev S (1999) Molecular cloning and analysis of a pleiotropic regulatory gene locus from the nystatin producer Streptomyces noursei ATCC 11455. FEMS Microbiol Lett 177:297–304
Sekurova ON, Brautaset T, Sletta H, Borgos SE, Jakobsen MOM, Ellingsen TE, Strom AR, Valla S, Zotchev SB (2004) In vivo analysis of the regulatory genes in the nystatin biosynthetic gene cluster of Streptomyces noursei ATCC 11455 reveals their differential control over antibiotic biosynthesis. J Bacteriol 186:1345–1354
Szlinder-Richert J, Mazerski J, Cybulska B, Grzybowska J, Borowski E (2001) MFAME, N-methyl-–d-fructosyl amphotericin B methyl ester, a new amphotericin B derivative of low toxicity: relationship between self-association and effects on red blood cells. Biochim Biophys Acta 1528:15–24
Tang L, Yoon YJ, Choi CY, Hutchinson CR (1998) Characterization of the enzymatic domains in the modular polyketide synthase involved in rifamycin B biosynthesis by Amycolatopsis editerranei. Gene 216:255–265
Tereshin IM (1976) Polyene antibiotics—present and future (E.R. Squibb lectures on chemistry of microbial products), University of Tokyo Press, Tokyo
Veiga M, Fabregas J (1983) Tetrafungin, a new polyene macrolide antibiotic. I. Fermentation, isolation, characterization, and biological properties. J Antibiot 36:770–775
Zielinski J, Golik J, Pawlak J, Borowski E, Falkowski L (1988) The structure of nystatin A3, a component of nystatin complex. J Antibiot 41:1289–1291
Zotchev SB (2003) Polyene macrolide antibiotics and their applications in human therapy. Curr Med Chem 10:211–223
Acknowledgements
This work was supported by the Research Council of Norway and Alpharma AS
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fjærvik, E., Zotchev, S.B. Biosynthesis of the polyene macrolide antibiotic nystatin in Streptomyces noursei. Appl Microbiol Biotechnol 67, 436–443 (2005). https://doi.org/10.1007/s00253-004-1802-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-004-1802-4